
Abstract
Alanine racemase is a fold type III PLP-dependent amino acid racemase enzyme catalysing the conversion of l-alanine to d-alanine utilised by bacterial cell wall for peptidoglycan synthesis. As there are no known homologs in humans, it is considered as an excellent antibacterial drug target. The standard inhibitors of this enzyme include O-carbamyl-d-serine, d-cycloserine, chlorovinyl glycine, alafosfalin, etc. d-Cycloserine is indicated for pulmonary and extra pulmonary tuberculosis but therapeutic use of drug is limited due to its severe toxic effects. Toxic effects due to off-target affinities of cycloserine and other substrate analogs have prompted new research efforts to identify alanine racemase inhibitors that are not substrate analogs. In this review, an updated status of known inhibitors of alanine racemase enzyme has been provided which will serve as a rich source of structural information and will be helpful in generating selective and potent inhibitor of alanine racemase.
Introduction
Alanine racemase (Alr) also known as l-alanine racemase is ubiquitous prokaryotic enzyme providing the essential peptidoglycan precursor d-alanine. This enzyme belongs to the family of isomerases acting on amino acids and derivatives and participates in alanine and aspartate metabolism and d-alanine metabolism. The Alanine racemase utilises pyridoxal 5′-phosphate (PLP) to carryout racemisation of l-alanine, providing d-alanine for cell wall synthesisCitation1. The enzyme is generally absent in higher eukaryotes but is ubiquitous throughout bacteria, making alanine racemase an attractive target for antibacterial drug development. However, few similar enzymes have been found in eukaryotes, such as aquatic animalsCitation2, fungiCitation3,Citation4, fission yeastCitation5, and plantsCitation6.
In some bacteria alternate pathways are present for the synthesis of d-alanine, and hence alanine racemase would not be a viable target for antibacterial drug development. The genome sequences of methanogenic archaeon, Methanococcus maripaludis revealedCitation7 the presence of alanine dehydrogenase gene adjacent to genes for alanine racemase and alanine permease, apparently acquired from bacteria. In addition to its function in the utilisation of d-alanine, Alr exhibited role in protecting the system from inhibition by d-alanine, since growth of the Alr mutant on a mixture of D and l-alanine was compromised. Recently it has been shown that in the absence of genes coding for alanine racemase Alr and DadX homologs in Chlamydia pneumonia a serine hydroxymethyl transferase GlyA served as a source of d-AlaCitation8. Purified chlamydial GlyA also exhibited racemase activity on l-Ala in vitro and was inhibited by d-cycloserine. These results showed GlyA, besides d-Ala ligase MurC/Ddl, as an additional target of d-cycloserine in Chlamydiaceae. An alternative metabolic pathway which can supply essential d-alanine for peptidoglycan synthesis of Alr- and DaDX-deficient Escherichia coli mutants is reportedCitation9,Citation10 and provided evidence for significant alanine racemase coactivity of the E. coli cystathionine beta-lyase (MetC).
Bacterial Alr is one of the best-studied amino acid racemases. Some bacteria contain only one alanine racemase while others have two isozymes, encoded by the Alr and the DadB genesCitation11 belonging to the fold type III. The latter group includes E. coliCitation12 and Pseudomonas aeruginosaCitation13. DadB expression is induced by l-alanine to a level much greater than that of Alr and is probably responsible for the catabolism of d-AlaCitation14. Alr is constitutively expressed and seems to provide the d-alanine necessary to maintain cell growth. Bacterial species with two independent Alr genes make it difficult to target both genes for drug studies. However, knockout studies have shown that without the Alr being expressed, the bacteria would need an external source of d-alanine in order to surviveCitation15.
In fact, d-alanine, produced by the action of alanine racemase on l-alanine, is important to both Gram-positive and Gram-negative bacteria, since it is required for the synthesis of the peptidoglycan in the cell wall. Hence Alr can be considered an attractive target for the development of new antibacterial drugs. Many crystal structures of complexes between bacterial Alr and inhibitors have been reportedCitation16–19. In fact the crystal structure of Alr from Geobacillus stearothermophilus (formerly Bacillus) enzyme (PDB ID: 1SFT) (), one of the best-studied racemase reveals a homodimeric enzyme of 388 residuesCitation20 formed by a head-to-tail association of two monomers. Each monomer is composed of two folded domains: (i) an N-terminal domain formed by the portion 1-240 and (ii) a C-terminal domain with the remaining portion of the monomer (241-388). The N-terminal domain consists of an eight-stranded α/β-barrel, while the C-terminal domain is made up of β-strands. Molecular dynamics study revealed that a water molecule present in both active sites of the Alr homodimerCitation21 which plays an important role and should be taken into account for the design of specific Alr inhibitors. This water molecule can be utilised either as a bridging group or it be displaced with a suitable moiety.
Figure 1. Pictorial view of the Bacillus stearothermophilus Alr homodimer (PBD ID: 1SFT). PLP (pyridoxyl 5′-phosphate), ACT (acetate), Lys (lysine).
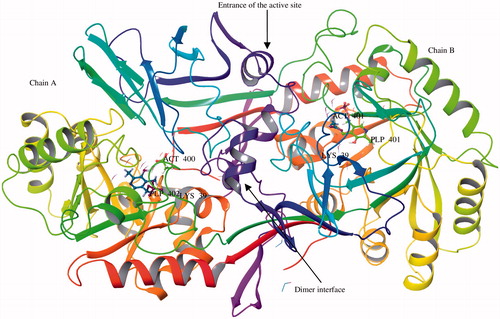
The active site in each monomer of Alr contains pyridoxal 5′-phosphate (PLP) binding residue covalently connected to the Lys39, and the amino acids in the immediate environment of the pyridoxal co-factor. The Lys39 is located at the C-terminal end of the first β-strand of the α/β-barrel and its side chain points toward the centre of the barrel. The catalytic domains of each monomer are inclined to each other at an angle of 125°. Therefore, the Lys39 residue also extends in a cleft formed by the two domains of the monomer whereas the PLP moiety positions itself in the approximate centre of the barrel, closer to the second domain of the other monomer. The phosphate group of PLP moiety forms several hydrogen bonds with residues of the same monomer and this renders the position of the phosphate group immobile. The Tyr265′, Asp313′, and Gln314′ residues which lie in the close proximity of PLP are part of a three-stranded antiparallel β-sheet in the second domain of the other monomerCitation22–24. These residues play important role in binding or catalysis. The racemisation of alanine by Alr is initiated via the initial transaldimination, during which the Lys39 residue is displaced by the incoming amino group of the alanine substrate. This is followed by the abstraction of the α-proton resulting in the formation of a “quinonoid” resonance-stabilised carbanion intermediate. The generated carbanion then undergoes one of a number of mechanistic fates including racemisation, transamination, and various elimination reactionsCitation25. Reprotonation on the opposite side of the α-carbon carbanion produces the opposite enantiomer of the alanine substrate whereas reprotonation on the original side of the carbanion by the protonated Lys39 ϵ-amino group yields the same enantiomer of alanine as that of the substrate. There are several reports describing either the role of Lys39, Arg219, and Tyr265′ residuesCitation24,Citation26–28 in the enzyme catalytic steps or substrate analogs used in structural studies to dateCitation20,Citation29–34. The structures of Alr with the covalent inhibitors alanine phosphonate and d-cycloserine showed that the inhibition of Alr was due to the formation of a stable covalent linkage with PLPCitation22,Citation35. The catalytic steps, abstraction of a proton and reprotonation, are also part of the mechanism of all other PLP-containing enzymesCitation36,Citation37.
It is evident from the crystal structure of Alr from B. stearothermophilus enzyme (PDB ID: 1SFT) that catalytic pocket is compact and located deep near the PLP moleculeCitation1,Citation20. The α/β-barrel of enzyme is approximately 45 Å in diameter and the packing of the dimers is extremely dense. The dimer is associated with a very small number of narrow solvent channels and there is no evidence of any other channels in any other direction. However, there are contacts between different dimers in the crystal but there is no evidence of any hydrogen bond or salt bridge interactions between different dimers. Catalytic site is too small, only small enough to accommodate small molecules. Moreover, it is difficult for these molecules to reach the compact active site because of a constriction at the entrance of corridor formed near the PLP binding residue. The majority of substrate analogs, like d-cyloserine, engage the PLP and lack target specificity; this is a major limitation of this class of inhibitors. Potential side effects due to off-target affinities of cycloserine and other substrate analogs have prompted renewed research efforts to identify Alr inhibitors that are not substrate analogs and that act through different mechanisms of enzyme inhibition. Structure-based strategies have been applied for the identification of novel inhibitors against B. stearothermophilus Alr. However, these efforts have not yet produced strong inhibitors with potent antibacterial activityCitation21a,b. What could be the strategies for design of AlaR inhibitors with an improved therapeutic index compared with those that currently are rather promising? One strategy may be designing inhibitors that bind the dimer interface and block dimerisation. Another approach may be placing an inhibitor at the entrance of catalytic pocket thereby blocking the substrate entry. Perhaps the promising strategy may be the in silico fragment-based design or extended pharmacophore modelling of small inhibitors in the catalytic pocket to target the Lys39 and Tyr265′ residues, important for the catalytic activity. Several systematic reviews on Alr have appeared in literaturesCitation38–40 covering the structure and catalytic mechanisms. In this review, we report the most significant examples of Alr inhibitors that exhibit antibacterial activity reported in the literature.
Inhibitors of alanine racemase
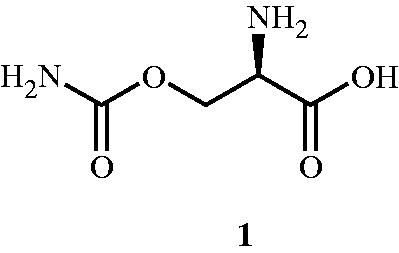
The quest for identifying the inhibitors for Alr enzyme began in 1966 when Lynch and NeuhausCitation41 explained the mechanism of action of O-carbamyl-d-serine 1 () in Streptococcus faecalis. In this study, O-carbamyl-d-serine was found to be an effective inhibitor of Alr and was supported by the enzyme kinetic study (Km of 4.8 × 10−4 and 6.8 × 10−3 M for d-alanine and l-alanine, respectively). The study emphasised on establishing the primary site of action of O-carbamyl-d-serine on the Alr compared with the other sites of actions such as d-ala-d-ala synthetase, d-ala-d-ala adding enzyme, and phospho-NAc-muramyl-pentapeptide translocase. The determination of primary site of action was based on the observed accumulation of UDP-NAc-muramyl-l-ala-d-glu-l-lys and on the absence of d-ala-O-carbamyl-d-serine accumulation. It was also observed that d-isomer 1 had no effect on the strains Streptococcus lactis, Lactobacillus arabinosus, and E. coli at 200 μg mL−1 (GI50 = 1.3 × 10−3 M) and these results were also in agreement with those reported earlierCitation42.
Figure 2. Structure of Alr inhibitor O-carbamyl-d-serine 1.
In the year 1972, Neuhaus and LambertCitation43 explained the inhibitory activity of d-cycloserine 2 towards the E. coli Alr. They also proved l-cycloserine 3 to be an effective competitive inhibitor of the enzyme which was earlier provedCitation44 to be inefficient for the inhibition of Staphylococcus aureus Alr due to the lack of conformation required for the molecule to bind to the substrate site. The competitive inhibitory activity of both cycloserine conformations () was explained with the aid of Alr enzyme kinetics. The Alr enzyme had Michaelis–Menten constants (Km) of 4.6 × 10−4 and 9.7 × 10−4 M for d- and l-alanine, respectively. The ratio of Vmax in the l- to d-direction to the Vmax in the d- to l-direction was 2.3 and the equilibrium constant calculated from the Haldane relationship was 1.11 + 0.15. Both d- and l-cycloserines were found to be competitive inhibitors with inhibitory constants (Ki) of 6.5 × 10−4and 2.1 × 10−3 M, respectively (Km d-alanine/Ki d-cycloserine: 0.71; Km l-alanine/Ki l-cycloserine: 0.46).
Figure 3. Structures of Alr inhibitor d-cycloserine 2 and l-cycloserine 3.
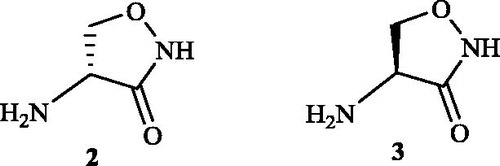
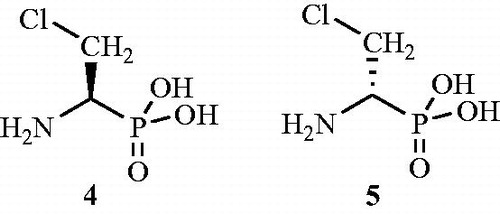
The inhibitory activity (90–95% inhibition) of β-choro-d-alanine 4 () against E. coli and Bacillus subtilis Alrs was studied by Manning et al.Citation45. Authors also carried out in vivo study for the same inhibitor in mice and found it an effective antibacterial agent against Diplococcus pneumoniae, Streptococcus pyogenes, and E. coli. The study also proposed the possibility of the inhibitor 4 to be incorporated into the cell wall for effecting inhibitory activity towards cell wall synthesis. The inhibitory activity of the other enantiomer, i.e. β-chloro-l-alanine 5 was also established and was found to be effective in the inhibition of bacterial growth like that of the d-isomer. But the l-isomer was found to have less specificity towards the concerned Alr enzymes due to its inhibitory activity towards decarboxylase and transaminases. This resulted in the blockage of the production of essential l-amino acids with a loss of viability of bacterial and mammalial cells.
Figure 4. Enantiomers of β-chloroalanine as Alr inhibitor 4 and 5.
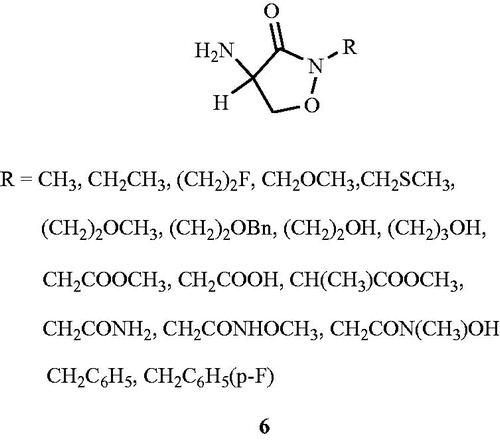
Kim et al.Citation46 investigated the importance of N(2)-structural site in cyloserine for bioactivity. Eighteen derivatives of N(2)-substituted d,l-cycloserine 6 () were synthesised from d,l-cycloserine. N(2)-Substitution of carboxybenzyl-protected derivatives of d,l-cycloserine proceeded smoothly with the requisite alkyl halide in the presence of potassium tert-butoxide in dimethylformamide. The synthesised compounds were evaluated for their inhibitory activity against purified Alrs (Alr gene product) from E. coli, S. aureus and M. tuberculosis. Structural modification at the N(2) position resulted in reduced activity in the enzyme assay and underscore the importance of structural modification at N(2)-position of cycloserine. Compound with CH2CONHOCH3 substituent at (N)-2 position exhibited modest inhibitory activity against purified Alr enzyme from M. tuberculosis (Ki = 0.36 mM), E. coli (Ki = 0.47 mM) and S. aureus (Ki = 1.16 mM) compared with the d,l-cycloserine (M. tuberculosis Ki = 0.0086 mM, E. coli Ki = 0.0138 mM, S. aureus Ki = 0.0073 mM). Rest of the compounds exhibited weak inhibitory activity against the tested Alr enzymes. All synthesised compounds showed an MIC value of > 2.5 mM against E. coli and S. aureus compared with the d,l-cycloserine (E. coli MIC value 0.3 mM, S. aureus MIC value 0.2 mM).
Figure 5. Structures of d,l-cycloserine analogs 6 as Alr inhibitors.
Alafosfalin 7, a synthetic l-alanine analog (), was discovered by Atherton et al.Citation47. Compound was found to be a selective inhibitor of peptidoglycan biosynthesis in both Gram-positive and Gram-negative bacteria. It increased the accumulation of uridine diphosphate-N-acetyl-muramyl-tripeptide in Gram-positive organisms and significantly reduced the intracellular pool levels of d-alanine. In the bacterial cells, alafosfalin was found to be transported by stereospecific peptide permeases and was finally hydrolyzed to 1-aminoethyl phosphonic acid by the action of intracellular amino peptidases. The target site for this hydrolyzed product was identified to be the Alr enzyme which was reversibly and competitively inhibited in Gram-negative E. coli (Ki = 0.03 mM; Km = 0.93 mM) and was irreversibly inhibited in a time-dependent manner in Gram-positive S. aureus and S. faecalis (Ki = 0.4 mM; Km = 9 mM). Mechanism of alafosfalin activity was further explored and authorsCitation22 declared that alafosfalin formed an external aldimine with the bound PLP cofactor, but was neither racemised nor efficiently hydrolyzed and upon formation of the external aldimine, the phosphonate group interacted with putative catalytic residues and thereby rendered them unavailable for catalysis.
Figure 6. Structure of alafosfalin 7.
In comparison with known inhibitors d-cycloserine 2 and d-chloroalanine 4, alafosfalin 7 was found to be more effective in reducing d-alanine pool levels and this was attributed to the rapid initial rate of alafosfalin uptake (10 nmoL mg−1 of cell dry weight min−1 at 100 μM) in comparison with d-cycloserine and d-alanineCitation48. One of the major disadvantages associated with alafosfalin is its altered and variable activity towards Gram-positive and Gram-negative bacteria. The authors disclosed multiple factors associated with this behaviour which include rate of uptake, rate of intracellular cleavage, size of the alanine pool, formation of metabolite and the mechanism of racemase inhibition. The low activity observed against certain strains of Streptococci and PseudomonasCitation49 was, however, almost entirely due to poor transport.
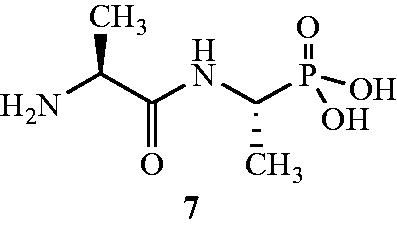
The potency and antibacterial spectrum of dipeptide, synthetic analogs and oligopeptide variants of alafosfalin 8Citation50 is also reported. Some of the synthesised compound exhibited better in vitro antibacterial activity compared with alafosfalin when administered orally into the infected rodents, but majority of the phosphonodipeptides was found to be more effective on parenteral administration. The leucine 8a and methionine 8b analogs () showed significant in vitro antibacterial activity against E. coli (MIC values of 0.25 µg mL−1 in both cases), Klebsiella aerogenes (MIC value 0.12 µg mL−1 in both cases), Enterobacter (MIC values of 0.5 and 1.0 µg mL−1), Serratia marcescens (MIC value 2.0 µg mL−1 in both cases), Salmonella typhimurium (MIC values of 8.0 µg mL−1 in both cases), S. faecalis (MIC values of 2.0 and 1.0 µg mL−1) and S. aureus (MIC values of 8.0 and 1.0 µg mL−1). However, synthetic analogs 2-aminopentanoyl 8c and 2-aminohexanoyl 8d exhibited moderate inhibitory activity against P. aeruginosa with MIC values of 32 and 64 μg mL−1, respectively. The 2-aminodecanoyl analog 8e showed potent activity against S. typhimurium and Proteus mirabilis (MIC values 0.5 and 16 µg mL−1, respectively). In case of oligopeptide analogs, several phosphonotetrapeptides with N-terminal sarcosyl residues exhibited higher levels of activity against Haemophilus influenzae and S. faecalis. The corresponding tetra- and higher phosphono-oligopeptides were also found to be active against Streptococcus pneumoniae and S. pyogenes.
Figure 7. Derivatives of alafosfalin 8a–e as Alr inhibitors.
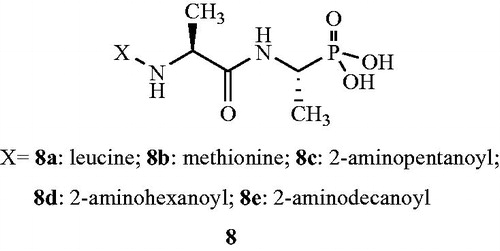
Yet in another publication, Atherton et al.Citation51 investigated the in vitro and in vivo activities of phosphonopeptides 9 having aminomethylphosphonic acid as C-terminal residue and linked to the l-amino acids. The structural analogs belonging to this particular series were shown to have less prominent Alr inhibitory effect as compared with the alafosfalin 7. The inhibitor was found to be associated with active transport of the peptide mimetics into the bacterial cells, followed by intracellular release of high concentrations of aminomethylphosphonic acid which inhibited bacterial cell wall biosynthesis. Aminomethylphosphonic acid had the ability to mimic both d- and l-alanine and inhibited d-Ala-d-Ala synthetase, Alr, and UDP-N-acetylmuramyl-l-alanine synthetase. As a part of their study, the authors investigated the Alr inhibitory activity of phosphonopeptides with alternative aminoalkylphosphonic acid residues. The phosphonopeptides combined with 1-aminomethyl phosphonic acid was considered for further study (). It was brought into light that the dipeptides of the 1-aminomethyl phosphonic acid series were less potent antibacterial agents than were the corresponding alafosfalin analogs 8. But significant inhibitory activity was observed in case of arginine analog 9a against Proteus and Staphylococci isolates. On the contrary, 2-aminopentanoyl 9b analog exhibited significant in vivo activity against Staphylococci. Finally authors concluded that the mechanism of action of dipeptides in case of 1-aminomethyl phosphonic acid series differs from analogs based on alafosfalin 8 in that the aminomethylphosphonic acid moiety can inhibit enzymes which utilise both d- and l-amino acids as substrates.
Figure 8. Aminomethylphosphonopeptide analogs 9 as Alr inhibitors.
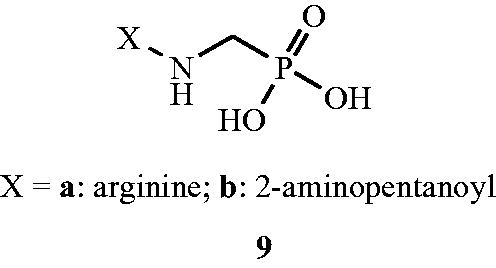
Inactivation by 1-aminoethylphosphonic acid () enantiomers (10,11) towards S. faecalis Alr was studied by Badet and WalshCitation52. The purified Alr enzyme from mutant S. faecalis which was O-carbamyl-d-serine resistant was inhibited in a time-dependent manner by both l-10 and d-(1-aminoethyl)phosphonates 11. The valuable proof for irreversible inhibition of Alr enzyme by these inhibitors was investigated with [1-2H]-, [1-3H]- and [1,2-l4C] labelling with concerned 1-aminoethylphosphonic acid. The 1-aminoethylphosphonic acid isomers are slow binding inhibitors of Alr and extrapolated limiting Kinact for l isomer is 5.3 min−1, and an extrapolated tl/2 for regain of enzyme activity is about 19 d, suggested a ratio of 106 in favour of isomerisation of an initial, rapidly formed complex to an isomerised tight complex with very slow dissociation properties. No such slow-binding, time-dependent inhibition was detected with pure Alr from Gram-negative bacteria.
Figure 9. Structures of 1-aminoethylphosphonic acid enantiomers 10a–g and 11 as Alr inhibitors.
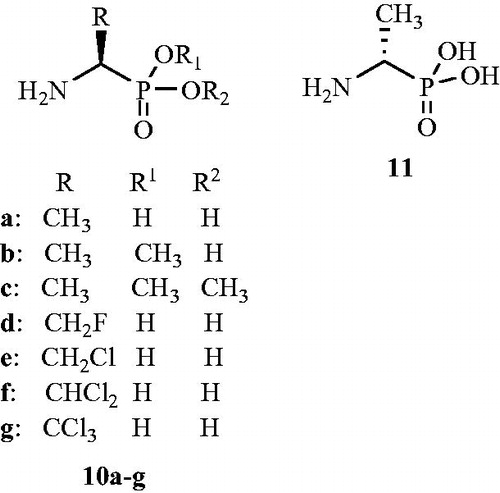
In another reportCitation53, inactivation of the dadB S. typhimurium Alr by β-l-chloroalanine 4, β-d-chloroalanine 5, β-d-fluoroalanine 12, β-l-fluoroalanine 13, and O-acetyl-d-serine 14 () was described. Enzyme released a labile pyridoxal phosphate-inactivator adduct on denaturation. The structure of this adduct suggested that the enzyme was inactivated by trapping the coenzyme in a ternary adduct with inactivator and the active site lysine. Under denaturing conditions, facile α,β-elimination occurred, releasing the aldol adduct of pyruvate and pyridoxal phosphate. Reduction of the ternary enzyme adduct blocked this elimination pathway. Authors also declared the overall mechanism of racemase inactivation in light of these results.
Figure 10. Structures of β-d-fluoroalanine 12, β-l-fluoroalanine 13 and O-acetyl-d-serine 14 as Alr inhibitors.
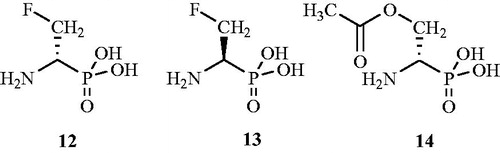
The role of fluorinated aminophosphonic acid analogs as potent Alr inhibitors was established by Flynn et al.Citation54 in 1985. Compounds β-monofluoro-15, β,β-difluoro-16 and β,β,β-trifluoro aminoethane phosphonic acids 17 () were synthesised and evaluated for their antibacterial activity. All three tested compounds were found to be time-dependent inactivators of Alr with the monofluoro analog being most potent antibacterial agent.
Figure 11. Structures of fluorinated 1-aminoethyl phosphonic acid analogs 15–17 as Alr inhibitors.
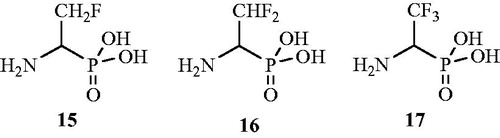
In another publication, d,l-(1-amino-2-propeny1)phosphonic acid 18 () is reported as an inhibitor of AlrCitation55. Tested vinylglycine analog was found to be potent inhibitor of Alr enzymes from P. aeruginosa and S. faecalis. The in vitro assay revealed competitive inhibition of P. aeruginosa Alr (Km = 14.2 mM; Ki = of 8.5 mM) by compound 18. The same compound also exhibited significant inhibition of S. faecalis Alr (Km = 7 mM; Ki = 3.5 mM) but unfortunately in in vitro studies, it did not show antibacterial activity against both Gram-positive and Gram-negative micro-organisms and this may be due to the inability of compound to penetrate into the bacterial cell.
Figure 12. Structures of (β-chloro-α-aminoethyl)phosphonic acid derivatives 18–20 as Alr inhibitors.
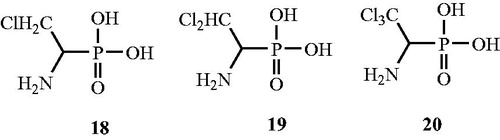
In continuation of the above work, Quang et al.Citation56 synthesised (β-chloro-α-aminoethyl)phosphonic acid derivatives () such as β-chloro-19, β,β-dichloro- 20 and (β,β,β-trichloro-α-aminoethy1)phosphonic acids 21 and henceforth evaluated in vitro for their Alr inhibitory activity. The strong inhibitory activity against P. aeruginosa and S. faecalis Alr enzymes was observed for the monochloro 19 and dichloro 20 derivatives of phosphonic acids but they were not found to be suicidal substrates. The researchers postulated that the antibacterial activity of these compounds can be enhanced by the peptide-transport strategy.
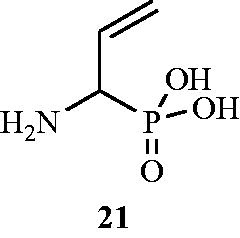
Figure 13. Structure of dl-(1-amino-2-propeny1)phosphonic acid 21 as an Alr inhibitor.
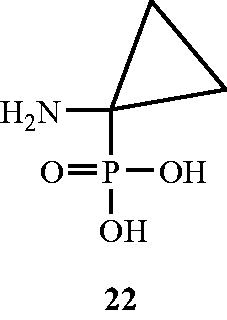
In another approach, Erion and WalshCitation57 established the ability of 1-aminocyclopropanephosphonate 22 () as an inhibitor of Alr enzyme from B. stearothermophilus strain. Compound was found to be a potent inhibitor with a Km/Ki ration of 2000. Inhibition by the tested compounds towards the concerned enzyme was characterised by slow binding (second-order rate constants <150 M−1 S−1) slow-dissociating behaviour. The pre-steady-state-kinetic studies revealed that a kinetically determined intermediate complex of the inhibitor being responsible for inhibitory mechanism towards the enzyme. The authors claimed 22 as a slow-binding and slow-dissociating potent inhibitor of Alr enzyme.
Figure 14. Structure of 1-aminocyclopropanephosphonate 22 as an Alr inhibitor.
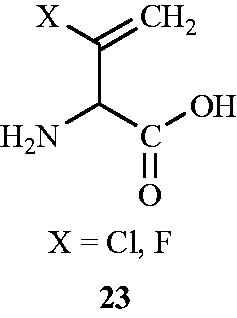
The role of halovinylglycine 23 as potent mechanism-based irreversible inactivators of Alr isolated from E. coli was investigated by a group of researchersCitation58 in 1987. d-, l- and d,l-3-Chlorovinylglycine were synthesised from the corresponding N-(benzyloxycarbony1)-vinylglycine methyl ester which in turn was obtained from the corresponding d-, l- or methionine. 3-Fluorovinylglycine was synthesised from 2-fluoroacrolein by a modified Strecker synthesisCitation59. Incubation of homogeneous E. coli Alr with d-chlorovinylglycine, l-chlorovinylglycine and DL-fluorovinylglycine resulted in an irreversible inactivation of the enzyme as demonstrated by the inability of the enzyme to regain catalytic activity after prolonged dialysis. The study confirmed superior antibacterial activity of d-chlorovinylglycine 20 () (MIC values of 32 and 64 µg mL−1 towards S. aureus and S. faecalis, respectively) as compared with other halovinylglycines.
Figure 15. Structure of halovinylglycines 23 as Alr inhibitors.
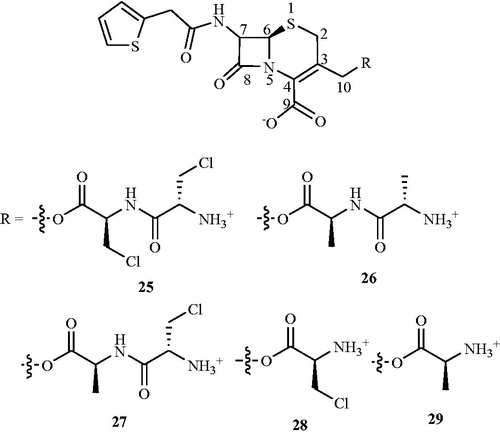
In search of Alr inhibitors, Mobashery and JohnstonCitation60−62 prepared and investigated the reactions of the peptidyl (24–26) and amino acid (27, 28) () C10-esters of deacetylcephalothin with purified enzymes in vitro. Compound 27 showed time-dependent inactivation of E. coli Alr in a process that requires O-lactamase for the initial liberation of β-chloro-l-alanine from the cephalosporin. The purified E. coli Alr experienced time-dependent loss of activity on incubation with a 10 mM solution of 27 that had been previously treated with E. coli TEM O-lactamase (t1/2 – 12 min). Inactivation by 10 mM/3-β-LAla 4 alone gave inhibition with a t1/2 = 8 min but in the absence of the lactamase compound 27 showed only 10% inactivation of the racemase in 30 min. Alanine racemase is similarly inactivated by compound 24, but this inhibition required the sequential action of both β-lactamase and alanine aminopeptidase. However, when the racemase was exposed to a 10 mM solution of 24 that had been preincubated with both the TEM lactamase and alanine aminopeptidase, time-dependent loss of enzyme activity was observed. The half-time for inactivation in this case was approximately 7 min. Inactivation of purified E. coli Alr by 20 mM β-Cl-l-alanine alone occurs with t1/2 = 7.5 min. The dialanylcephem ester 25 incubated with Alr showed no enzyme inactivation. Moreover, only a 5% loss of racemase activity was observed in 30 min when the enzyme was exposed to a 10 mM solution of compound 25 that had been treated, like compound 24, with both β-lactamase and alanine aminopeptidase.
To enhance the transport by peptide conjugates, Patchett et al.Citation63 further synthesised halovinylglycine compounds and their l-norvalyl peptide derivatives. Synthesised compounds were screened for their antibacterial activity against a panel of Gram-positive and Gram-negative bacteria including MSSA and MRSA. Among all the synthesised compounds, l-norvalyl-l-chlorovinylglycine 24 () displayed maximum inhibitory activity against Gram-positive bacteria S. aureus and S. faecalis (MIC values of 0.5 and 4.0 μg mL−1, respectively). Tested compound 29 also exhibited significant inhibitory activity (an MIC value of 4.0 μg mL−1) against MRSA and found to be more active than d-cycloserine 2 against many of the tested virulent cultures. However, it was less active than d-cycloserine 2 against Bacillus cereus, Erysipelothrix rhusiopathiae, and Listeria monocytogenes.
Figure 16. Structure of l-norvalyl-l-chlorovinylglycine 24 as an Alr inhibitor.
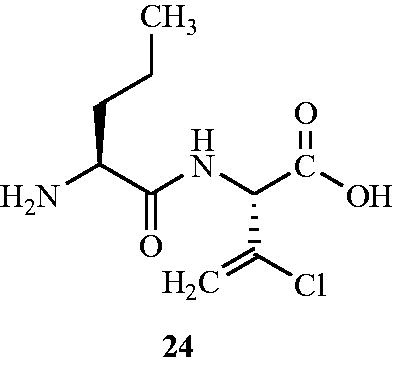
Figure 17. Structures of peptidyl and amino acid C10-esters of deacetylcephalothin 25–29 as Alr inhibitors.
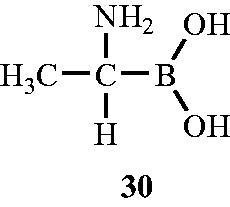
In the year 1989, Duncan et al.Citation64 discovered the Alr inhibitory activity of (1-aminoethyl) boronic acid towards Alr enzyme isolated from B. stearothermophilus. (1-Aminoethy1)boronic acid (Ala-B) 30 () an structural analog of alanine in which the carboxyl group was substituted by boronic acid showed significant inhibitory activity against Alr enzyme. The nature of inhibition was found to be time dependent and slow binding due to generation of long-lived, slow dissociating complexes. The test compound was found to have Ki value of 20 mM and Kinact of 0.15–0.35 min−1. Kinetic analysis of (1-aminoethyl) boronic acid suggested that it can compete with d-alanine at the d-alanine binding sites, and on inactivation with Ala-B, labelled d-alanine, and labelled ATP, the inactive enzyme has stoichiometric amounts of d-alanine, ADP, Pi and Ala-B bound. The mechanistic approach of Ala-B towards Alr enzyme involved loss of 420 nm chromophore suggested that the inactive complex was not a protonated Schiff base. Unlike other usual Alr inhibitors reported, which operates by binding with the PLP cofactor, Ala-B incubation leads to only transient incubation as activity was restored upon dilution of the enzyme-Ala-B mixture (t1/2 – 60–70 min). Authors concluded that Ala-B was less potent in action than Ala-P in terms of both Ki and the lifetime of the corresponding E–I′ complex formation and the two analogs probably act by distinct microscopic interactions.
Figure 18. Structure of (1-aminoethy1)boronic acid (Ala-B) 30 as an Alr inhibitor.
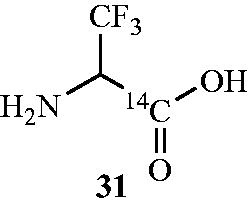
In another work, the mechanism of inactivation of Alr by [14C] β,β,β-trifluoroalanine 31 () was investigated. The compound was synthesised and screened for its Alr inhibitory activity from the Gram-negative organism S. typhimurium and Gram-positive organism B. stearothermophilusCitation65. Incubation of trifluoroalanine 31 with the Alr from S. typhimurium and B. stearothermophilus resulted in time-dependent inhibition and Ki values were found to be very high (Ki = 65 ± 10 and >100 mM, respectively; Kinact = 0.08 ± 0.02 and >2.0 min−1, respectively) indicative of poor binding. Authors declared that this may be due to either steric interference of the trifluoromethyl group at the active site or low concentration of zwitterionic trifluoroalanine, which may be important in binding. Upon inactivation, a change in the absorption spectrum of the native enzyme from 418 nm to a broad 460–490 nm was observed with concomitant loss in enzyme activity. This suggested that the inactive enzyme species was different than the inactive enzyme complex formed upon inactivation of Alr with the haloalaninesCitation53, in which the 418 nm chromophore was replaced by a 325-nm chromophore, indicative of an unconjugated pyridoxyl group.
Figure 19. Structure of Alr inhibitor β,β,β-trifluoro alanine 31.
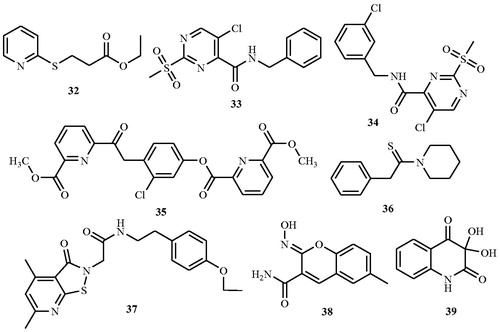
In search of Alr inhibitors with an innovative mechanism of action, derivatives of (1-aminoethyl)phosphonate (32a–h) () were synthesised and screened for their inhibitory activity against Gram-positive bacterium B. stearothermophilus Alr at pH 7.4 in 100 mM HEPESCitation66. A time-dependent inhibition by compound 32 a was observed with a Ki of 1.25 µM; however, Alr from Gram-negative S. typhimurium was found to be nonsusceptible to time-dependent inhibition. It was observed that the substitution of the methyl group in 32a by fluoromethyl (32e, Ki = 1.4 µM) or monochloromethyl (32f, Ki = 28.5 µM) retains inactivating properties but the substitution of more bulky dichloromethyl 32 g or trichloromethyl 32 h groups resulted in loss of activity. Authors declared that phosphonate dianion is crucial for stable formation of an isomerised long-lived enzyme-(1-aminoethyl)phosphonate-inhibited complex.
Figure 20. Structures of non-substrate Alr inhibitors 32–39.
To understand the structural basis of the inactivation of enzyme by (1-aminoethyl)phosphonate 32 a, Stamper et al.Citation23 determined the crystal structure of the complex between compound 32 a and B. stearothermophilus Alr at 1.6 Å resolution. The inhibitor 32a was shown to form an external aldimine with the bound PLP cofactor, although it was neither racemised nor efficiently hydrolyzed. Authors declared that 32a is an effective inhibitor of Alr because, upon formation of the external aldimine, the phosphonate group interacts with putative catalytic residues, thereby rendering them unavailable for catalysis. Furthermore, this aldimine appears to be inappropriately aligned for efficient Cα proton abstraction. The combinations of these effects probably lead to a stable aldimine derivative and potent inactivation of Alr by this compound. It was also observed that the tetrahedral geometry of the phosphonate positioned the α-carbon of the l-Ala-P in a manner different than that expected for a productive substrate. In this position, the α-carbon is not in the plane of the PLP ring, which may make activation of the Cα-H bond more difficult. Hence, the enzyme appears to be “locked” as an unreactive external aldimine, leading to the unusually long half-life. The involvement of Lys39 and Tyr265 in mechanistic action of inhibitor at enzyme active site was also studied.
Anthony et al.Citation67 identified a novel series of Alr inhibitors, developed by high-throughput screening (53 000 small molecule) against M. tuberculosis as non-substrate analogs having different mechanism of enzyme inhibition. Seventeen novel non-substrate Alr inhibitors that are structurally different than any known enzyme inhibitors were identified. Seven of these were active against M. tuberculosis with minimal cytotoxic effect against mammalian cells. Sixteen inhibitors were evaluated for their in vitro inhibitory activity by the microbroth dilution method against M. tuberculosis at a concentration of 50 and 100 μg mL−1 and results were compared with the standard drug d-cycloserine 2. Eight compounds 33–40 () inhibited growth at both the concentrations with minimal inhibitory concentrations (MICs) from <1.56 to 12.5 μg mL−1 and several of which were lower than the MIC value of cycloserine (6.5 μg mL−1). Enzyme activity was measured at 15 different inhibitor concentrations (0.01–0.98 mM) and at five different substrate concentrations (0.5–2.5 mM). The Ki values of the eight compounds ranged from 0.038 to 0.927 mM. The cytotoxic study on the seven selected inhibitors was also carried out and the TC50 value of each compound was determined. The values of TC50 for the seven compounds ranged between 33 and 406 μM while the TC50 of cycloserine was 203 μM. From the study, authors concluded that with the exception of compound 40 which was cytotoxic at slightly above its MIC value (Ti = 1), the remaining compounds did not show cytotoxicity at their respective MICs, suggesting their suitability as potential leads.
In the year 2012, Ciustea et al.Citation68 discovered thiadiazolidinone 41, as a new class of Alr inhibitor by the means of high-throughput screening. Compound 41 was investigated for the mode of action, antimicrobial activities, and mammalian cell cytotoxicity in order to assess its potential for development as a therapeutic agent against MRSA. Compound 41 was evaluated for its Alr inhibitory activity in a NADH-based fluorescent-coupled time-dependent enzyme assay and the result was compared with those of known irreversible inhibitor, d-cycloserine 2 and the reversible inhibitor, O-acetyl-d-serine 14. Results suggested irreversible Alr inhibition which was greater than 90%. Antibacterial activity of compound 41 was examined against 10 strains of S. aureus, including five MRSA strains with MIC values of 12.5–32 μg mL−1 that were uniform against all 10 strains, and similar to those of cycloserine (12.5 μg mL−1). Encouraged by the above finding nine closely related analogs 42a–i (), each with an intact thiadiazolidinone core, but containing various substitutions of the N-linked groups, was investigated. Among them IC50 value of the nine analogs ranged from 0.36 to 6.4 µM with the majority being better than or comparable to the IC50 (2.3 µM) of 41. On one hand, structural examination revealed that the nature of substitutions on the fourth position of the thiadiazolidinone ring did not markedly effect Alr inhibition as illustrated by the relatively low IC50 values (1.5–6.5 μM) of analogs carrying smaller alkyl groups (methyl or cyclopropylmethyl) in place of the parafluorobenzyl group in 41. On the other hand, substitutions on second position of the thiadiazolidinone ring resulted in a significant impact on Alr inhibition. An electron-poor aromatic group was found to be preferred at this position, as shown by lower IC50 values of the nitro- and trifluoromethyl-substituted analogs (0.36–0.79 μM). Compounds 42a–i inhibited the growth of S. aureus, including MRSA strains, with MICs ranging from 6.25 to 100 μg mL−1. The antibacterial activity showed selectivity against Gram-positive bacteria and fungi, but not Gram-negative bacteria. Tested compounds 42a–i also inhibited human HeLa cell proliferation.
Figure 21. Structure of thiadiazolidinones 40 and 41a–k as Alr inhibitors.
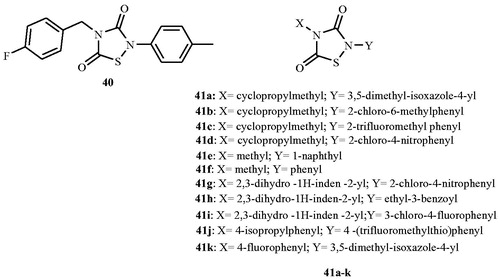
In 2013, Lee et al.Citation69 explored the role of thiadiazolidinones (41a–k) () in the inhibition of Mycobacterial Alr. Compounds were evaluated for the inhibition of M. tuberculosis and Mycobacterium smegmatis Alr activity and bacterial growth. Tested compounds inhibited M. tuberculosis and M. smegmatis Alrs to different extents with an IC50 value ranging from <0.03 to 28 μM and 23 to >150 μM, respectively. Compounds also inhibited the growth of these bacteria, including MDR strains of M. tuberculosis. The MIC for drug-susceptible M. tuberculosis and M. smegmatis ranged from 6.25 μg mL−1 to 100 μg mL−1, and from 1.56 to 6.25 μg mL−1 for drug-resistant M. tuberculosis. Eleven of the 12 members of the thiadiazolidinone family showed potent activity (an IC50 value <10 mM) against the M. tuberculosis enzyme, but their activity against the M. smegmatis enzyme was greatly diminished. Authors declared that investigated compounds are unlikely to interact with the enzyme-bound PLP cofactor or to occupy the substrate-binding site due to spatial constraints within the active site and likely blocked Alr activity by a mechanism other than active site binding.
Conclusion
Alanine racemase, a pyridoxal 5′-phosphate (PLP)-dependent enzyme, is an important target for antibacterial drug design as it is a key enzyme in peptidoglycan biosynthesis and bacterial cell wall formation. Including natural analogs, many synthetic moieties for inhibitory action were brought into light with efficient mechanistic action and minimal side effects in comparison with the standard drugs. d-Cycloserine has been developed commercially for the treatment of pulmonary tuberculosis, but its clinical utility is limited due to toxicity issues arising from lack of target-specificity. The cause of toxicity is probably connected to the activity of d-cycloserine as a partial agonist of the glycine site of NMDA receptors and as an inhibitor of PLP-dependent enzymes (serine, arginine and aspartate racemases) involved in the biosynthesis and metabolism of the neurotransmitter GABA. Other inhibitors which are not used clinically such as alanine phosphonate and propionate also target other PLP-dependent enzymes and thereby suffer from the same lack of specificity. This emphasises the need for the development of new inhibitors for Alr with greater specificity, which may translate into less toxicity to humans. Future goal should be focused on the discovery of inhibitors characterised by a high selectivity for the Alr enzyme.
Declaration of interest
The authors report that they have no conflicts of interest. We would like to thank University Grants Commission (UGC), Government of India, for the financial support (No. F1-17.1/2014-15/RGNF-2014-15-SC-KER-61880).
References
- Watanabe A, Yoshimura T, Mikami B, et al. Reaction mechanism of alanine racemase from Bacillus stearothermophilus. X-Ray crystallographic studies of the enzyme bound within-(5′-phosphopyridoxyl)alanine. J Biol Chem 2002;277:19166–72
- Abe H, Yoshikawa N, Sarower MG, Okada S. Physiological function and metabolism of free d-alanine in aquatic animals. Biol Pharm Bull 2005;28:1571–7
- Hoffmann K, Schneider-Scherzer E, Kleinkauf H, Zocher R. Purification and characterization of eucaryotic alanine racemase acting as key enzyme in cyclosporin biosynthesis. J Biol Chem 1994;269:12710–14
- Cheng YQ, Walton JD. A eukaryotic alanine racemase gene involved in cyclic peptide biosynthesis. J Biol Chem 2000;275:4906–11
- Uo T, Yoshimura T, Tanaka N, et al. Functional characterization of alanine racemase from Schizosaccharomyces pombe: a eucaryotic counterpart to bacterial alanine racemase. J Bacteriol 2001;83:2226–33
- Ono K, Yanagida K, Oikawa T, et al. Alanine racemase of alfalfa seedlings (Medicago sativa L.): first evidence for the presence of an amino acid racemase in plants. Phytochemistry 2006;67:856–60
- Moore BC, Leigh JA. Markerless mutagenesis in Methanococcus maripaludis demonstrates roles for alanine dehydrogenase, alanine racemase, and alanine permease. J Bacteriol 2005;187:972–9
- Benedetti SD, Buhl H, Gaballah A, et al. Characterization of serine hydroxymethyltransferase GlyA as a potential source of d-alanine in Chlamydia pneumonia. Front Cell Infect Microbiol 2014;4:19
- Kang L, Shaw AC, Xu D, et al. Upregulation of MetC Is essential for d-alanine-independent growth of an alr/dadX-deficient Escherichia coli strain. J Bacteriol 2011;193:1098–106
- Wild J, Hennig J, Lobocka M, et al. Identification of the dadX gene coding for the predominant isozyme of alanine racemase in Escherichia coli K12. Mol Gen Genet 1985;198:315–22
- Walsh CT. Enzymes in the d-alanine branch of bacterial cell wall peptidoglycan assembly. J Biol Chem 1989;64:2393–6
- Lobocka M, Hennig J, Wild J, Klopotowski T. Organization and expression of the Escherichia coli K-12 dad operon encoding the smaller subunit of d-amino acid dehydrogenase and the catabolic alanine racemase. J Bacteriol 1994;176:1500–10
- Strych U, Huang HC, Krause KL, Benedik MJ. Characterization of the alanine racemases from Pseudomonas aeruginosa PAO1. Curr Microbiol 2000;41:290–4
- McFall E, Newman EB. Alanine racemase enzyme inhibitors as antibacterial agents: a review. In: Neidhardt FC, ed. Escherichia coli and Salmonella: cellular and molecular biology. Washington DC: ASM Press; 1996:358–79
- Strych U, Benedik MJ. Mutant analysis shows that alanine racemases from Pseudomonas aeruginosa and Escherichia coli are dimeric. J Bacteriol 2002;184:4321–5
- LeMagueres P, Im H, Dvorak A, et al. Crystal structure at 1.45 Å resolution of alanine racemase from a pathogenic bacterium, Pseudomonas aeruginosa, contains both internal and external aldimine forms. Biochemistry 2003;42:14752–61
- LeMagueres P, Im H, Ebalunode J, et al. The 1.9 Å crystal structure of alanine racemase from Mycobacterium tuberculosis contains a conserved entryway into the active site. Biochemistry 2005;44:1471–81
- Priyadarshi A, Lee EH, Sung MW, et al. Structural insights into the alanine racemase from Enterococcus faecalis. Biochim Biophys Acta 2009;1794:1030–40
- Im H, Sharpe ML, Strych U, et al. The crystal structure of alanine racemase from Streptococcus pneumoniae, a target for structure-based drug design. BMC Microbiol 2011;11:116
- Shaw JP, Petsko GA, Ringe D. Determination of the structure of alanine racemase from Bacillus stearothermophilus at 1.9 A° resolution. Biochemistry 1997;36:1329–42
- (a) Mustata GI, Soares TA, Briggs JM. Molecular dynamics studies of alanine racemase: a structural model for drug design. Biopolymers 2003;70:186–200. (b) Mustata GI, Briggs JM. A structure-based design approach for the identification of novel inhibitors: application to an alanine racemase. J Comput Aided Mol Des 2002;16:935–53.
- Stamper GF, Morollo AA, Ringe D. Reaction of alanine racemase with 1-aminoethylphosphonic acid forms a stable external aldimine. Biochemistry 1998;37:10438–45
- Sun S, Toney MD. Evidence for a two-base mechanism involving tyrosine-265 from arginine-219 mutants of alanine racemase. Biochemistry 1999;38:4058–65
- Watanabe A, Yoshimura T, Mikami B, Esaki N. Tyrosine 265 of alanine racemase serves as a base abstracting a hydrogen from l-alanine: the counterpart residue to Lys39 specific to d-alanine. J Biochem 1999;126:781–6
- John RA. Pyridoxal phosphate-dependent enzymes. Biochim Biophys Acta 1995;1248:81–96
- Ondrechen MJ, Briggs JM, McCammon JA. A model for enzyme–substrate interaction in alanine racemase. J Am Chem Soc 2001;123:2830–4
- Watanabe A, Yoshimura T, Mikami B, Esaki N. Role of tyrosine 265 of alanine racemase from Bacillus stearothermophilus. J Biochem 1999;125:987–90
- Rubinstein A, Major DT. Understanding catalytic specificity in alanine racemase from quantum mechanical and molecular mechanical simulations of the arginine 219 mutant. Biochemistry 2010;49:3957–64
- Rubinstein A, Major DT. Catalyzing racemizations in the absence of a cofactor: the reaction mechanism in proline racemase. J Am Chem Soc 2009;131:8513–21
- Toney MD. Reaction specificity in pyridoxal phosphate enzymes. Arch Biochem Biophys 2005;433:279–87
- Davis E, Hutchinson ES, Reading HO, et al. The structure of alanine racemase from Acinetobacter baumannii. Acta Cryst 2014;F70:1199–205
- Strych U, Davlieva M, Longtin JP, et al. Purification and preliminary crystallization of alanine racemase from Streptococcus pneumonia. BMC Microbiol 2007;7:40
- Yoshimura T, Goto M. d-Amino acids in the brain: structure and function of pyridoxal phosphate-dependent amino acid racemases. FEBS J 2008;275:3527–37
- Morollo AA, Petsko GA, Ringe D. Structure of a Michaelis complex analogue: propionate binds in the substrate carboxylate site of alanine racemase. Biochemistry 1999;38:3293–301
- Fenn TD, Stamper GF, Morollo AA, Ringe D. A side reaction of alanine racemase: transamination of cycloserine. Biochemistry 2003;42:5775–83
- Olson GT, Mengmeng F, Lau S, et al. An aromatization mechanism of inactivation of d-aminobutyric acid aminotransferase for the antibiotic l-cycloserine. J Am Chem Soc 1998;120:2256–67
- Almo SL, Smith DL, Danishefsky AT, Ringe D. Structure of E. coli aspartate transaminase. Prot Eng 1994;7:405–12
- Lin YL, Gao J, Rubinstein A, Major DT. Molecular dynamics simulations of the intramolecular proton transfer and carbanion stabilization in the pyridoxal 5′-phosphate dependent enzymes l-dopa decarboxylase and alanine racemase. Biochim Biophys Acta 2011;814:1438–46
- Yoshimura T, Esaki N, Soda K. Structure and function of alanine racemase. Bull Inst Chem Res Kyoto Univ 1992;70:378–84
- Conti P, Tamborini L, Pinto A, et al. Drug discovery targeting amino acid racemases. Chem Rev 2011;111:6919–46
- Lynch JL, Neuhaus FC. On the mechanism of action of the antibiotic o-carbamyl-d-serine in Streptococcus faecalis. J Bacteriol 1966;91:449–60
- Skinner CG, Mccord TJ, Ravel JM, Shive W. O-Carbamyl-l-serine, an inhibitory analog of l-glutamine. J Am Chem Soc 1956;78:2412–14
- Lambert MP, Neuhaus FC. Mechanism of d-cycloserine action: alanine racemase from Escherichia coli W. J Bacteriol 1972;110:978–87
- Roze U, Strominger JL. Alanine racemase from Staphylococcus aureus: conformation of its substrates and its inhibitor, d-cycloserine. Mol Pharmacol 1966;2:92–4
- Manning MJ, Merrifield NE, Jones WM, Gotschlich EC. Inhibition of bacterial growth by 3-chloro-d-alanine. Proc Natl Acad Sci 1974;71:417–21
- Kim MG, Strych U, Krause K, et al. N(2)-substituted d,l-cycloserine derivatives: synthesis and evaluation as alanine racemase inhibitors. J Antibiot (Tokyo) 2003;56:160–8
- Atherton FR, Hall MJ, Hassall CH, et al. Phosphonopeptides as antibacterial agents: mechanism of action of alaphosphin. Antimicrob Agents Chemother 1979;15:696–705
- Wargel RJ, Shadur CA, Neuhaus FC. Mechanism of d-cycloserine action: transport mutants for d-alanine, d-cycloserine, and glycine. J Bacteriol 1971;105:1028–35
- Allen JG, Atherton FR, Hall MJ, et al. Phosphonopeptides as antibacterial agents: alaphosphin and related phosphonodipeptides. Antimicrob Agents Chemother 1979;15:684–95
- Atherton FR, Hall MJ, Hassall CH, et al. Phosphonopeptide antibacterial agents related to alafosfalin: design, synthesis, and structure–activity relationships. Antimicrob Agents Chemother 1980;18:897–905
- Atherton FR, Hall MJ, Hassall CH, et al. Antibacterial activity and mechanism of action of phosphonopeptides based on aminomethylphosphonic acid. Antimicrob Agents Chemother 1982;22:571–8
- Badet B, Walsh C. Purification of an alanine racemase from Streptococcus faecalis and analysis of its inactivation by (1-aminoethyl)phosphonic acid enantiomers. Biochemistry 1985;24:1333–41
- Badet B, Roise D, Walsh CT. Inactivation of the dadB Salmonella typhimurium alanine racemase by d and l isomers of beta-substituted alanines: kinetics, stoichiometry, active site peptide sequencing, and reaction mechanism. Biochemistry 1984;23:5188–94
- Flynn GA, Beight DW, Bohme EHW, Metcalf BW. The synthesis of fluorinated aminophosphonic acid inhibitors of alanine racemase. Tetrahedron Lett 1985;26:285–8
- Quang YV, Carniato D, Quang LV, et al. (1-Amino-2-propeny1) phosphonic acid, an inhibitor of alanine racemase and d-alanine–alanine ligase. J Med Chem 1986;29:579–81
- Quang YV, Carniato D, Quang LV, et al. (β-Chloro-α-aminoethyl)phosphonic acids as inhibitors of alanine racemase and d-alanine: d-alanine ligase. J Med Chem 1986;29:148–51
- Erion MD, Walsh CT. 1-Alanine racemase aminocyclopropanephosphonate: time-dependent inactivation of 1-aminocyclopropanecarboxylate deaminase and Bacillus stearothermophilus alanine racemase by slow dissociation behavior. Biochemistry 1987;26:3417–25
- Thornberry NA, Bull HG, Taub D, et al. 3-Halovinylglycines efficient irreversible inhibitors of Escherichia coli. J Am Chem Soc 1987;109:1543–4
- Baldwin JE, Haber SB, Hoskins C, Kruse LI. Synthesis of beta, gamma-unsaturated amino acids. J Org Chem 1977;42:1239–41
- Mobashery S, Johnston M. Reactions of Escherichia coli TEM β-lactamase with cephalothin and with C10-dipeptidyl cephalosporin esters. J Biol Chem 1986;261:7879–87
- Mobashery S, Lerner SA, Johnston M. Conscripting β-lactamase for use in drug deliver. Synthesis and biological activity of a cephalosporin C10-ester of an antibiotic dipeptide. J Am Chem Soc 1986;108:1685–6
- Mobashery S, Johnston M. Inactivation of alanine racemase by β-chloro-l-alanine released enzymatically from amino acid and peptidyl C10-esters of deacetylcephalothin. Biochemistry 1987;26:5878–84
- Patchett AA, Taub D, Weissberger B, et al. Antibacterial activities of fluorovinyl-and chlorovinylglycine and several derived dipeptides. Antimicrob Agents Chemother 1988;32:319–23
- Duncan K, Faraci WS, Matteson DS, Walsh CT. (1-Aminoethyl) boronic acid: a novel inhibitor for Bacillus stearothermophilus alanine racemase and Salmonella typhimurium d-alanine: d-alanine Ligase (ADP-Forming). Biochemistry 1989;28:3541–9
- Faraci WS, Walsh CT. Mechanism of inactivation of alanine racemase by β,β,β-trifluoroalanine. Biochemistry 1989;28:431–7
- Badet B, Inagaki K, Soda K, Walsh CT. Time-dependent inhibition of Bacillus stearothermophilus alanine racemase by (1-aminoethyl)phosphonate isomers by isomerization to noncovalent slowly dissociating enzyme-(1-aminoethyl)phosphonate complexes. Biochemistry 1986;25:3275–82
- Anthony KG, Strych U, Yeung KR, et al. New classes of alanine racemase inhibitors identified by high-throughput screening show antimicrobial activity against Mycobacterium tuberculosis. PLoS One 2011;6:e20374
- Ciustea M, Mootien S, Rosato AE, et al. Thiadiazolidinones: a new class of alanine racemase inhibitors with antimicrobial activity against methicillin-resistant Staphylococcus aureus. Biochem Pharmacol 2012;83:368–77
- Lee Y, Mootien S, Shoen C, et al. Inhibition of mycobacterial alanine racemase activity and growth by thiadiazolidinones. Biochem Pharmacol 2013;86:222–30