
Abstract
Two series of 1-substituted carbamoyl and thiocarbomoyl derivatives were prepared by either treating the corresponding pyrazole with the appropriate isocyanate and isothiocyanate respectively, or alternatively by condensing the appropriate diketone with the proper substituted semicarbazide or thiosemicarbazide. The structures of the prepared compounds were fully determined by analytical and spectral methods. Preliminary biological screening of the prepared compounds revealed significant antibacterial and cytotoxic activities for some compounds. Compounds 4a2 and 4a3 were found to be the most active against the human colon carcinoma HT29 (11.8 and 7.5 μg/mL, respectively) and human breast cancer MCF 7 (3.4 and 2.6 μg/mL, respectively) cell lines. The structure–activity relationship (SAR) and in silico drug relevant properties (HBD, HBA, tPSA, cLog P, molecular weight, % ABS, drug-likeness and drug score) further confirmed that the compounds are potential lead compounds for future drug discovery study.
Introduction
The worldwide-ongoing efforts of research on treatment of malignancy are focused on the discovery of novel potent and effective antineoplastic agents, particularly those interacting with novel biological targets. Nevertheless, in spite of the large number of available chemotherapeutic agents, the medical need is still largely unmet due to many factors among which the lack of selectivity of conventional drugs leading to toxicity, the metastatic spreading, and the intrinsic or acquired resistance to chemotherapy developed after few therapeutic cycles (multi-drug resistance; MDR). It is well known that the onset of drug resistance is one of the factors responsible for the failure of chemotherapeutic treatment of cancer patients. This also represents the main driver for the research of new lead molecules with anticancer activity able to overcome the known mechanisms of resistance. At the same time, random screening remains one of the essential means to discover new structure leads with antineoplastic activity.
Pyrazole compounds besides showing an array of biological activities have been characterized by their significant antiproliferative and antitumor activitiesCitation1–5. The discovery of the naturally occurring C-glycoside antibiotic pyrazofurin; 4-hydroxy-3-β-D-ribofuranosyl-1H-pyrazole-5-carboxamideCitation6, however, has provided a basis for a rational design of new pyrazoles as potential antimicrobialCitation7,Citation8, antiviralCitation9,Citation10 and anticancer agentsCitation8,Citation11–13. It has also been reported that isoquinoline-1-carboxaldehyde thiosemicarbazone and its several congeners which possess the N-N-S or O-N-S tridentate ligand system, exhibit substantial antineoplastic activityCitation14,Citation15. Furthermore, the azomethine linkage in this class that plays significant role in certain biological transformations which includes vitamin B6-catalysed reactionsCitation16,Citation17 paved the way to study pyrazoline derivatives having a carbamoyl or thiocarbamoyl moiety at position-1. This modification carries two fold objective of preparing compounds with possible antineoplastic and antimicrobial activities. In the present work we tried to optimize this chemical series by introducing carbamoyl and thiocarbamoyl moieties at position-1 in the pyrazole ring. The substituent effect is further extended by introducing substituted phenyl groups at nitrogen of carbamoyl and thiocarbamoyl moieties.
Furthermore, molecular descriptors, such as partition coefficient (log P), molecular weight (MW), and hydrogen bond acceptors/donors are usually associated with molecular properties such as membrane permeability and oral bioavailability. Using these descriptors, LipinskiCitation18 formulated a rule for drug design which states that the compounds are more likely to be orally bioavailable if they obey the following criteria: log p ≤ 5, molecular weight ≤500, hydrogen bond acceptors ≤10, and hydrogen bond donors ≤5. We therefore, decided to carry out studies in silico for Lipinski parameters, topological polar surface area (tPSA) and the percentage of absorption (% ABS). Furthermore, according to Veber et al., good bioavailabilityCitation19 is more likely for compounds with ≤10 rotatable bonds and TPSA of ≤140 Å2. For this reason we have also included the measurement of the number of rotatable bonds in this series of compounds, drug-likeness and drug scores in order to rationalize how physicochemical properties influence the molecular behavior in vivo.
Experimental
Chemistry
Melting points were determined on a Gallenkamp melting point apparatus (London, UK) and are uncorrected. The infrared (IR) spectra were recorded on Shimadzu FT-IR 8400S infrared spectrophotometer (Tokyo, Japan) using the KBr pellet technique. 1H and 13C NMR spectra were recorded on a Bruker DPX-400 FT NMR spectrometer (Bruker BioSpin AG, Fällanden, Switzerland) using tetramethylsilane as the internal standard and a mixture of CDCl3 and DMSO-d6 as a solvent (Chemical shifts in δ, ppm). Splitting patterns were designated as follows: s: singlet; d: doublet; m: multiplet. Elemental analyses were performed on a 2400 Perkin Elmer Series 2 analyzer (Waltham, MA) and the found values were within ±0.4% of the theoretical values. Follow up of the reactions and checking the homogeneity of the compounds were made by TLC on silica gel-protected aluminum sheets (Type 60 F254, Merck, Darmstadt, Germany) and the spots were detected by exposure to UV-lamp at λ 254. The starting chalcones 2 were prepared according to a literature procedureCitation20.
3-(Indol-3-yl)-1-substituted propenones (2a–c)
A solution of the indole-3-carboxaldehyde 1 (1.31 g, 0.01 mol) in ethanol (20 mL) was added to a solution of the appropriate ketone (0.01 mol) in 20% ethanolic KOH (20 mL) with stirring. The stirring was continued at room temperature for 6 h. The reaction mixture was then poured onto water (200 mL) and left overnight. The precipitated solid product was collected by filtration, washed with water, dried and recrystallized from ethanol as pale yellow needles. IR (cm−1): 1654–1660(conjugated C=O), 1604–1611(C=C).
2a: 1H NMR (DMSO-d6/CDCl3): δ 6.67 (d, (J = 10 Hz), 1H, H-α), 7.54 (d (J = 10 Hz), 1H, H-β), 7.02–7.65 (m, 8H, Ar-H), 10.1 (s, NH); 13C NMR: 129.3 (C-α), 142.8 (C-β), 102.4, 111.2, 120.3, 121.2, 122.3, 127.4, 128.2, 128.5, 135.6, 135.8, 144.4, 155.5 (Ar-C), 180.7 (C=O).
2b: 1H NMR (DMSO-d6/CDCl3): δ 6.45 (d, (J = 10 Hz), 1H, H-α), 7.34 (d (J = 10 Hz), 1H, H-β), 6.64–7.72 (m, 8H, Ar-H), 9.98 (s, NH); 13C NMR: 128.6 (C-α), 141.5 (C-β), 100.6, 110.1, 119.8, 121.6, 122.5, 124.5, 127.0, 128.4, 131.5, 136.6, 148.8, 153.2 (Ar-C), 175.2 (C=O).
2c: 1H NMR (DMSO-d6/CDCl3): δ 6.45 (d, (J = 10 Hz), 1H, H-α), 7.34 (d, (J = 10 Hz), 1H, H-β), 6.64–7.72 (m, 8H, Ar-H), 9.98 (s, NH); 13C NMR: 128.6 (C-α), 141.5 (C-β), 108.4, 112.3, 118.9, 120.4, 121.9, 124.7, 127.2, 128.3, 132.1 135.6, 151.3, 156.8 (Ar-C), 175.0 (C=O).
3-(3-Substituted-4,5-dihydro-1H-pyrazol-5-yl)-1H-indole (3a--c)
Hydrazine hydrate (0.015 mol, 99%) was added to a cooled solution of the appropriate chalcone (0.01 mol) in ethanol (25 ml) over 15 min. The mixture was then refluxed for 1 h on a boiling water-bath. The resulting reaction mixture was concentrated and allowed to attain room temperature. The precipitated product was filtered off, dried and recrystallized from ethanol as colorless needles.
1-Substituted thiocarbamoyl-4,5-dihydro-1-H-pyrazole derivatives (4a--c)
Method A
A mixture of the corresponding pyrazoline (3a–c, 0.05 mol), anhydrous K2CO3 (0.1 mol) in dry acetone (20 ml) was stirred at room temperature. Subsequently, a solution of the appropriate isothiocyanate (0.07 mol) in dry acetone (20 ml) was added dropwise to the reaction mixture. After stirring and refluxing the mixture for 6 h, acetone was removed under reduced pressure. The resulting solid residue which was precipitated upon acidification with 20% hydrochloric acid was purified by recrystallization from ethanol as colorless/pale needles (4a–c).
Method B
A solution of the corresponding chalcone 2 (0.05 mol) in ethanol (30 ml) was treated with the appropriate thiosemicarbazide (0.05 mol). The mixture was heated on a boiling water-bath for 1 h. The crude product thus separated on cooling, was purified by recrystallization from ethanol as colorless/pale needles of compounds 4a–c.
1-Substituted carbarmoyl-4,5-dihydro-1-H-pyrazole derivatives (5)
Method A
A mixture of the corresponding pyrazolines (0.05 mol), anhydrous K2CO3 (0.1 mol) in dry acetone (20 ml) was refluxed with stirring. To this refluxing reaction mixture a solution of the appropriate isocyanate (0.075 mol) in dry acetone (20 ml) was added dropwise. After stirring and refluxing the reaction mixture overnight, acetone was removed under reduced pressure. The crude product was separated on cooling which was purified by recrystallization from ethanol-benzene (2:1) as needles.
Method B
A solution of the appropriate chalcone 2 (0.05 mol) in ethanol (30 ml) was treated with the corresponding semicarbazide (0.05 mol). The mixture was allowed to condense at moderate temperature on a steaming water-bath for 1 h. The crude product thus separated on cooling, was purified by recrystallization from ethanol:benzene (2:1) as needles.
Method C
A mixture of the appropriate thiocarbamoyl derivative (1.05 mol), yellow HgO (0.1 mol) in ethanol (50 ml) was first stirred for few minutes and then refluxed for 3 h. The resulting reaction mixture was filtered. The filtrate was concentrated and allowed to crystallize. The crude crystals formed was filtered and recrystallized from ethanol:benzene (2:1) as needles. The products obtained following this technique were identical with those prepared by methods A and B as confirmed by their m.p., mixed m.p. and TLC.
Biological evaluation
Procedure for anti-microbial activity
The preliminary anti-microbial activities of the synthesized chalcones, pyrazoline carbamoyl and thiocarbamoyl derivatives 2–5 were measured in a concentration of 50 µg/mL by disc diffusion methodCitation21,Citation22. The prepared compounds were tested for their antimicrobial activity against two types of bacteria: Staphylococcus aureus (ATCC 25923) as Gram positive and Escherichia coli (ATCC 25922) as Gram negative bacteria. The antifungal activity was tested using the pathogenic yeast strain Candida albicans (ATCC 10231) and Aspergillus niger (ATCC 16404). DMSO was used as a solvent control and the standard drugs used were AmpicillinCitation23 and GriseofulvinCitation24. The disc diffusion method was performed using Muller–Hinton agar (Hi-Media) medium. The inhibition zones were measured in mm at the end of an incubation period of 24 h at 37 °C for bacteria and 72 h at 24° C for fungi.
Procedure for anti-microbial activity using UV (366 nm) light
This test was performed as mentioned before but the Petri-discs containing micro-organisms and the testing compounds were subjected to UV light (366) for 3 h before transferring to the incubation period.
In vitro MTT cytotoxicity assay
All the following procedures were done in a sterile area using a Laminar flow cabinet biosafety class II level (Baker, SG403INT, Stanford, ME). Cells were batch cultured for 10 days, then seeded at concentration of 10 × 103 cells/well in fresh complete growth medium in 96-well microtiter plastic plates at 37 °C for 24 h under 5% CO2 using a water jacketed carbon dioxide incubator (Sheldon, TC2323, Cornelius, OR). Media was aspirated, fresh medium (without serum) was added and cells were incubated either alone (negative control) or with different concentrations of the test compounds to give a final concentration of 100, 50, 25, 12.5, 3.125, 1.56, 0.78 μg/mL. DMSO was employed as a vehicle for dissolution of the tested compounds and its final concentration on the cells was less than 0.2%. Cells were suspended in RPMI 1640 medium (for HePG2 and HT29 cell lines) and DMEM (for MCF 7 cell line), 1% antibiotic-antimycotic mixture (10 000 IU/mL penicillin potassium, 10 000 μg/mL streptomycin sulfate and 25 μg/mL amphotericin B), and 1% l-glutamine in 96-well flat bottom microplate at 37 °C under 5% CO2.
After 24 h of incubation, the medium was aspirated; 40 mL of MTT salt (2.5 mg/mL) was added to each well and incubated for further 4 h at 37 °C under 5% CO2. To stop the reaction and dissolve the formed crystals, 200 mL of 10% sodium dodecyl sulfate in deionized water was added to each well and incubated overnight at 37 °C. The absorbance was then measured using a microplate multiwell reader (Bio-Rad Laboratories Inc., model 3350, Hercules, CA) at 595 nm and a reference wavelength of 620 nm. A statistical significance was tested between samples and negative control (cells with vehicle) using independent t-test by SPSS 11 program. DoxorubicinCitation25 is used as positive control cytotoxic agent. The results of LC50 (mM), which is the lethal concentration of the compound causing death of 50% of the cells in 24 h, are presented in .
Results and discussion
Chemistry
The starting compounds in this study are 3-(Indol-3-yl)-1- substituted propenones 2a–c could be synthesized via Claisen-Schmidt condensation of indole-3-carboxaldehyde with different heterocyclic ketones 1. These chalcones were reacted with hydrazine hydrate to afford the desired 4,5-dihydro-1-H-pyrazole derivatives 3a–c (Scheme 1). Their IR spectra revealed two absorption bands at 1594–1602 cm−1 and 3365–3388 cm−1 corresponding to C=N and NH functions, respectively. The 1H-NMR exhibited besides the aromatic protons at δ 6.01–7.41 ppm, three multiplets (each of one proton intensity) at δ 5.25–5.42, 3.49–3.55 and 3.78–3.89. The low-field multiplet is assigned to H-5 of the pyrazoline while the other two multiplets are due to H-4 (). The structures were further confirmed from their 13C NMR spectral data (). The new 1-thiocarbamoyl and carbamoyl derivatives 4 and 5 were prepared by either treating the corresponding pyrazole with the appropriate isothiocyanate or isocyanate respectively or by condensing the corresponding chalcone with the proper substituted semicarbazideCitation15 or thiosemicabazideCitation26 (). Furthermore, mild oxidation of the thiocarbamoyl derivative 4 with yellow mercuric oxide afforded the corresponding carbamoyl derivative 5. The IR spectra of the carbamoyl derivative 5 exhibited distinguishable carbonyl absorption at 1668–1672 cm−1, whereas compound 4 revealed distinct thiocarbonyl band at 1060–1155 cm−1. Their structures were further confirmed from their 1H-NMR and 13C NMR spectral data ( and ).
Scheme 1. Synthesis of compounds 2–5.
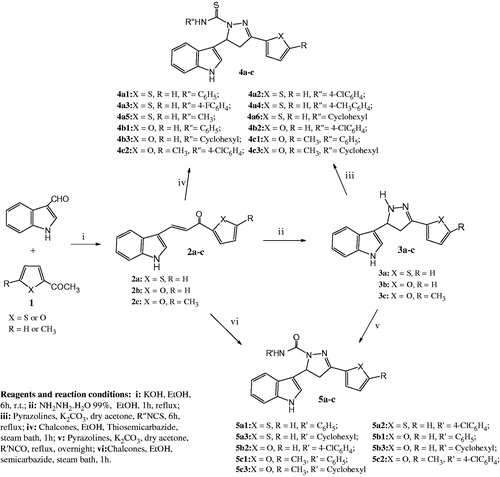
Table 1. Physical and analytical data of compounds 2–5.
Table 2. 1H NMR spectral data (δ/ppm)a of compounds 3–5.
Table 3. 13C NMR spectral data (δ/ppm)a of compounds 2–5.
In silico calculations of molecular properties
Molecular descriptors represent the combined physicochemical, pharmacokinetic and pharmacodynamic effects of the synthesized compounds 2–5 in order to verify that these compounds exhibit good (theoretical) oral bioavailability potential. Lipinski rule of five is considered predictive for oral bioavailability; however 16% of oral drugs violate at least one of the criteria and 6% fail in two or moreCitation27. The lipophilicity (log P) and topological polar surface area (tPSA) were calculated using the online software MolinspirationCitation28, while the aqueous solubility, drug-likeness, drug score and toxicity risk assessment (TRA) were calculated using the OSIRIS property explorer softwareCitation29. Molinspiration uses sophisticated Bayesian statistics to compare structures of representative ligands active on the particular target with structures of inactive molecules and to identify substructure features (which in turn determine physicochemical properties) typical for active molecules. For the study of drug-likeness, the OSIRIS program uses a list of 5300 molecular fragments, where the frequency of occurrence of each fragment is determined based on a collection of 3300 drugs and 15 000 commercially available chemicals (Fluka AG, Buchs, Switzerland) that are not drugs. tPSA was used to calculate the percentage of absorption (%ABS) according to the equation: %ABS = 109 − 0.345 × TPSA, as reported by Zhao et alCitation30. Furthermore, according to Veber et al., good bioavailabilityCitation19 is more likely for compounds with ≤10 rotatable bonds and TPSA of ≤140 Å2. Reduced molecular flexibility, as measured by the number of rotatable bonds, and low polar surface area or total hydrogen bond count (sum of donors and acceptors) are found to be important predictors of good oral bioavailability, independent of molecular weight. The calculation data are shown in .
Table 4. Molecular Properties Calculationa: calculated absorption (% ABS), polar surface area (PSA), LogS, Lipinski’s parameters, drug-likeness and drug score of the compounds.
The calculation results show that more than half of compounds meet the Lipinski rules of the five, suggesting that these compounds theoretically would not have problems with oral bioavailability. Compounds which had scores of less than 5 for lipophilicity, ranging from 2.72 to 4.99 for 15 compounds (2a–c, 3a–c, 4a1, 4a5, 4b1, 4b2, 4c1, 5a1, 5b1, 5b2 and 5c1) and 12 compounds (4a2–4, 4a6, 4b3, 4c2, 4c3, 5a2, 5a3, 5b3, 5c2 and 5c3) had scores more than 5 ranging from 5.00 to 5.83. Most of the compounds also showed a tPSA much less than 140 Å2, indicating a good permeability of the drug in the cellular plasma membrane. All compounds have shown very high percentage of absorption (% ABS) calculated ranged from 83.60 to 97.66%, which is an indication of good bioavailability by oral route (). shows that all compounds have Log S value less than −4.00, ranging between −4.05 and −6.66.
The toxicity risk assessment (TRA) indicators, namely, mutagenic effect (MUTA), tumorigenic effect (TE), irritant effect (IRR) and reproductive effect (RE) are the predictors for potential toxicity risks. These calculations are based on a precomputed set of structural fragment(s) that give rise to toxicity alerts in case they are encountered in the structures currently undertaken. The TRA suggested that compounds 2a–c, 3a–c and 5a did not show any toxicity risk profile. However, compounds 4a–c showed the possibility of mutagenic and reproductive effects and compounds 5b–c have shown the possibility of mutagenic effect. None of the compounds have shown the risk of tumorigenic or irritant effects ().
A positive value for drug-likeness indicates that the compound contains predominantly fragments that are often present in most currently used drugs. The drug score combines drug-likeness, lipophilicity, solubility, molecular weight and the risk of toxicity into a single numerical value that can be used to predict a global value for each compound as a potential new drug candidate. The results in the calculations show that most of the compounds gave values for drug-likeness between 1.38 and 7.29. Compounds 4a6 and 5b3 have shown drug-likeness 0.02 and 0.38, respectively. However, only three compounds, namely, 4b3, 4c3 and 5c3 have displayed negative values of druglike-ness (−1.54, −2.56 and −0.72 respectively). For comparisons we have done similar calculations of reference drugs as well.
All compounds showed positive values in the drug score calculation, the values ranged from 0.09 to 0.80. Even those compounds which have shown negative druglike-ness have displayed positive drug scores (). The results show that these compounds, especially compounds 2, 3 and some members from 4 and 5, have potential as new drug candidates. Moreover, non-violations of Lipinski’s “Rule-of-Five” and Veber’s “criteria for good bioavailability” also confirm the suitability of these compounds to be used as a template for the design of dual-target–directed drugs.
Anti-microbial activity
Compounds 2–5 were screened in vitro for their antimicrobial and antifungal activity against Escherichia coli, Staphylococcus aureus, Aspergillus niger and Candida albicans. The zones of inhibition formed for the compounds against bacteria and fungi are summarized in . All compounds were found to possess mild to moderate activity. Compounds 3a, 3b, 4a2, 4a3, 4b2, 4c2, 5a2, 5b2, 5c2 were however, significantly active when compared with rest of the series. Moreover, after using UV-Visible light, most of the tested compounds showed an additional activity especially towards Escherichia coli and Candida albicans (). The anti-microbial activity of compounds 3a, 3b, 4a2, 4a3, 4b2, 4c2, 5a2, 5b2 and 5c2 was further corroborated by their high values of drug-likeness (4.98, 3.18, 6.28, 4.44, 4.75, 3.69, 7.29, 5.87 and 4.76 respectively) and significant drug scores (0.75, 0.80, 0.12, 0.14, 0.14, 0.12, 0.37, 0.26 and 0.23 respectively). All test data in and were of average values from triplicate runs and the test compounds showed reduced antimicrobial activities when compared with their respective standards. Compounds containing thiocarbamoyl part showed enhanced activity in the present series. The most active compound 4a3 which contains 4-fluorophenyl substituent at the thiocarbamoyl end showed best antimicrobial activity () and this was further supported by the value of its drug-likeness (4.44) and a quite favorable percentage of absorption (94.02%) (). In compounds 4a6, 4b3, 4c3, 5a3, 5b3 and 5c3 where the aromatic ring was replaced by a cyclohexyl ring, a marked reduction in their antimicrobial activity were seen and this was further substantiated by the values of their drug-likeness which was found to be negative in compounds 4b3, 4c3 and 5c3.
Table 5. Anti-bacterial and anti-fungal data of compounds 2–5.
Table 6. Cytotoxic effects (LC50; μg/mL)a of the active compounds on some human tumor cell lines using the MTT assay.
In vitro MTT cytotoxicity assay
Eighteen analogs namely; 3a–c, 4a1, 4a2, 4a3, 4a4, 4a6, 4b1, 4b2, 4c1, 4c2, 5a1, 5a2, 5b1, 5b2, 5c1 and 5c2 were selected to be evaluated for their in vitro cytotoxic effect via the standard MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) methodCitation31,Citation32 against a panel of three human tumor cell lines, namely, Caucasian breast adenocarcinoma MCF7, hepatocellular carcinoma HePG2 and colon carcinoma HT29. The results are presented in as LC50 (μg/mL) which is the lethal concentration of the compound that causes death of 50% of the cells in 24 h.
The obtained data revealed that, the three tested human tumor cell lines exhibited variable degree of activity profiles towards 11 of the tested compounds, namely 3a,b, 4a2, 4a3, 4a6, 4b1, 4b2, 4c2, 5a2, 5b2, 5c2. Among these, the human colon carcinoma HT29 cell line showed pronounced sensitivity against compounds 4a2 and 4a3 with LC50 values of 11.8 and 7.5 μg/mL, respectively. Moreover, a significant cytotoxic potential was displayed by compound 4b2 against the same cell line (14.2 μg/mL). However, compounds 3a and 3b revealed an obvious cytotoxicity profile against colon carcinoma HT29 with LC50 values 20.5 and 18.4 μg/mL, respectively. Compounds 4b1, 4c2, 5a1 and 5b2 were able to exhibit moderate activity against the same cell line with LC50 values range of 37.7-62.3 μg/mL. Mild activity was displayed by compound 4a6 (LC50, 80.2 μg/mL) against human colon carcinoma (HT29). Furthermore, the growth of the human hepatocellular carcinoma HePG2 cell line was found to be moderately inhibited by nine of the active compounds, namely 3a–b, 4a2, 4a3, 4b1, 4b2, 4c2, 5a2 and 5b2, with LC50 values in the range of 21.3–50.7 μg/mL. Among these, the highest cytotoxic activity was displayed by compounds 3b, 4a2 and 4a3 which were almost equipotent with LC50 values 25.2, 22.9 and 21.3 μg/mL, respectively. On the other hand, human breast cancer MCF 7 was proved to be the least sensitive among the cell lines tested as it was affected by only seven of the test compounds. An outstanding growth inhibition potential was shown by compounds 4a2 and 4a3 as evidenced from their LC50 values (3.4 and 2.6 μg/mL, respectively), however compound 4b2 displayed appreciable activity. The rest four active compounds namely 3a, 3b, 4c2 and 5b2 showed moderate to mild activity against the same cell line with LC50 values of 45.5, 36.3, 54.6 and 66.0 μg/mL, respectively. Further interpretation of the results revealed that, compounds 3a, 3b, 4a2, 4b2 and 4a3 showed considerable broad spectrum of cytotoxic activity against the three tested human tumor cell lines. In particular, compounds 4a2 and 4a3 proved to be the most active members in this study with a broad spectrum of activity against the tested cell lines, with special effectiveness against the human colon carcinoma HT29 and human breast cancer MCF 7 cell lines (). The results in the molecular properties calculations showed that most of the compounds gave favorable values for drug-likeness between 1.38 and 7.29. These values were in total agreement with the tested values for compounds 4a3 and 4a4 which were found to be most active against the human colon carcinoma HT29 and human breast cancer MCF 7 cell lines with favorable drug-likeness values (6.28 and 4.44 respectively). A close examination of the structure of the active compounds showed that the presence of electron-withdrawing groups (X = Cl or F) in the thiocarbamoyl moiety (4a2, 4a3, 4b2, 4c2) play a crucial role in enhancing the activity, whereas, the presence of fluorine atom in compound 4a3 was responsible for the high activity displayed by this analogs. Moreover, it is clear from the results that the 1-carbamoyl derivatives (5) are much less active than the corresponding 1-thiocarbamoyl analogs (4) and the parent pyrazole compounds (3). No compound with a cyclohexyl group at the thiocarbamoyl end could display any cytotoxic activity indicating a negative role of this substituent.
Conclusion
The results obtained from OSIRIS property explorer and MOLINSPIRATION showed that the synthesized series of carbamoyl and thiocarbamoyl derivatives of pyrazole as potential drug molecules with a drug score values ranged from 0.09 to 0.80. All compounds have exhibited good (theoretical) oral bioavailability potential calculated ranged from 83.60 to 97.66%. All compounds have shown number of rotatable bonds much less than 10 (between 2 and 5) which is an indication of good bioavailability by oral route as shown in . The results in the calculations showed that most of the compounds gave favorable values for drug-likeness between 1.38 and 7.29. Compounds 3a, 3b, 4a2, 4a3, 4b2, 4c2, 5a2, 5b2 and 5c2 have shown mild to moderate antimicrobial activity. Furthermore compounds 4a2 and 4a3 were found to be most active against the human colon carcinoma HT29 and human breast cancer MCF 7 cell lines. The antimicrobial and cytotoxic activities of these compounds were in total agreement with their favorable drug-likeness, drug-scores and oral bioavailability potential as calculated from OSIRIS property explorer and MOLINSPIRATION programs.
Declaration of interest
This work was supported by the Deanship of Scientific Research (DSR), King Abdulaziz University, Jeddah, under grant no. (130-045-D1434). The authors, therefore, acknowledge with thanks DSR technical and financial support. Sincere gratitude is conveyed to the staff members of the Department of Health and Human Services, National Cancer Institute (NCI), Bethesda, Maryland, USA, for carrying out the anticancer screening of the newly synthesized compounds. The authors wish also to thank the Department of Biology, Faculty of Science, Alexandria University for carrying out the microbiological screening.
References
- Insuasty B, Tigreros A, Orozco F, et al. Synthesis of novel pyrazolic analogues of chalcones and their 3-aryl-4-(3-aryl-4,5-dihydro-1H-pyrazol-5-yl)-1-phenyl-1H-pyrazole derivatives as potential antitumor agents. Bioorg Med Chem 2010;18:4965–74
- Labbozzetta M, Baruchello R, Marchetti P, et al. Lack of nucleophilic addition in the isoxazole and pyrazole diketone modified analogs of curcumin, implications for their antitumor and chemosensitizing activities. Chem Biol Interact 2009;181:29–36
- Ludwig S, Planz O, Sedlacek HH, et al. Use of caspase inhibitors as antiviral agents, and test system for their discovery. PCT Int Appl 2004; Patent No. WO 2004085682 A220041007
- Perchellet EM, Ward MM, Skaltsounis AL, et al. Antiproliferative and proapoptotic activities of pyranoxanthenones, pyranothioxanthenones and their pyrazole-fused derivatives in HL-60 cells. Anticancer Res 2006;26:2791–804
- Pevarello P, Brasca MG, Amici R, et al. 3-Aminopyrazole inhibitors of CDK2/Cyclin A as antitumor agents. 1: lead finding. J Med Chem 2004;47:3367–80
- Comber RN, Gray RJ, Secrist JA. Acyclic analogues of pyrazofurin: syntheses and antiviral evaluation. Carbohydr Res 1991;216:441–52
- El-Gaby MSA, Atalla AA, Gaber AM, et al. Studies on aminopyrazoles: antibacterial activity of some novel pyrazolo[1,5-a]pyrimidines containing sulfonamido moieties. Il Farmaco 2000;55:596–602
- Baraldi PG, Pavani MG, Nunez MC, et al. Antimicrobial and antitumor activity of n-heteroimmine-1,2,3-dithiazoles and their transformation in triazolo-, imidazo-, and pyrazolopirimidines. Bioorg Med Chem 2002;10:449–56
- Larsen JS, Zahran MA, Pedersen EB, et al. Synthesis of triazenopyrazole derivatives as potential inhibitors of HIV-1. Monatsh Chem 1999;130:1167–73
- Moukha-chafiq JO, Taha ML, Lazrek HB, et al. Synthesis and biological activity of some 4-substituted 1-[1-(2,3-dihydroxy-1-propoxy)methyl-1,2,3-triazol-(4 & 5)-ylmethyl]-1H-pyrazolo[3,4-d]pyrimidines. Il Farmaco 2002;57:27–32
- Ishida J, Ohtsu H, Tachibana Y, et al. Antitumor agents. Part 214: synthesis and evaluation of curcumin analogues as cytotoxic agents. Bioorg Med Chem 2002;10:3481–7
- Daidone G, Maggio B, Raffa D, et al. Synthesis and in vitro antileukemic activity of new 4-triazenopyrazole derivatives. Il Farmaco 2004;59:413–17
- Aydin S, Basu NK, Turan SÖ, et al. Synthesis of 1-aroyl-3,5-dimethyl-1H-pyrazoles as anti-HCV and anticancer agents. Lett Drug Design Discov 2014;11:121–31
- Brockman RW, Thomson JR, Bell MJ, et al. Observations on the antileukemic activity of pyridine-2-carboxaldehyde thiosemicarbazone and thiocarbohydrazone. Cancer Res 1956;16:167–70
- Garg NG, Sharma RA. Potential antineoplastics. I: 2-amino-4,6-dimethyl-5-arylazopyrimidines and 1-thiocarbamoyl-3,5-diphenyl-4-arylazopyrazoles. J Med Chem 1969;12:1122–4
- Metzler DD, Ikawa M, Snell LE. A general mechanism for vitamin B6-catalyzed reactions. J Am Chem Soc 1954;76:648–52
- Snell EE. Summary of known metabolic functions of nicotinic acid, riboflavin and vitamin B6. Physiol Rev 1953;33:509–24
- Lipinski CA, Lombardo F, Dominy BW, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 1997;23:3–25
- Veber DF, Johnson SR, Cheng HY, et al. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 2002;45:2615–23
- Hassan SY, Basaif SA, Faidallah HM. Synthesis of novel indan [1,2-c] pyrazole benzenesulfonylureas, thioureas and their cyclized derivatives. Egyptian J Chem 1999;42:213–20
- Kirkpatrick WR, Turner TM, Fothergill AW, et al. Fluconazole disk diffusion susceptibility testing of Candida species. J Clin Microbiol 1998;36:3429–32
- Premkumar T, Govindarajan S. Antimicrobial study of pyrazine, pyrazole and imidazole carboxylic acids and their hydrazinium salts. World J Microbiol Biotechnol 2005;21:479–80
- Acred P, Brown DM, Turner DH, et al. Pharmacology and chemotherapy of ampicillin—a new broad-spectrum penicillin. Brit J Pharmacol Chemother 1962;18:356–69
- Goldman L, Schwarz J, Preston RH, et al. Current status of griseofulvin: report on one hundred seventy-five cases. J Am Med Assoc 1960;172:532–8
- Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol 2013;65:157–70
- Wright JB, Dulin WE, Markillie JH. The antidiabetic activity of 3,5-dimethylpyrazoles. J Med Chem 1964;7:102–5
- Bickerton GR, Paolini GV, Besnard J, et al. Quantifying the chemical beauty of drugs. Nat Chem 2012;4:90–8
- Cheminformatics on the web. Available from: http://www.molinspiration.com/ [last accessed 2 Jan 2015]
- Organic Chemistry Portal. Available from: http://www.organic-chemistry.org/prog/peo/ [last accessed 2 Feb 2015]
- Zhao MY, Abraham MH, Le J, et al. Rate-limited steps of human oral absorption and QSAR studies. Pharm Res 2002;19:1446–57
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63
- Denizot F, Lang R. Rapid colorimetric assay for cell growth and survival modification to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods 1986;89:271–7