
Abstract
Compounds that can effectively inhibit the proteolytic activity of human neutrophil elastase (HNE) represent promising therapeutics for treatment of inflammatory diseases. We present here the synthesis, structure–activity relationship analysis, and biological evaluation of a new series of HNE inhibitors with a cinnoline scaffold. These compounds exhibited HNE inhibitory activity but had lower potency compared to N-benzoylindazoles previously reported by us. On the other hand, they exhibited increased stability in aqueous solution. The most potent compound, 18a, had a good balance between HNE inhibitory activity (IC50 value = 56 nM) and chemical stability (t1/2 = 114 min). Analysis of reaction kinetics revealed that these cinnoline derivatives were reversible competitive inhibitors of HNE. Furthermore, molecular docking studies of the active products into the HNE binding site revealed two types of HNE inhibitors: molecules with cinnolin-4(1H)-one scaffold, which were attacked by the HNE Ser195 hydroxyl group at the amido moiety, and cinnoline derivatives containing an ester function at C-4, which is the point of attack of Ser195.
Introduction
Human neutrophil elastase (HNE) is a serine protease belonging to the chymotrypsin family and is stored in azurophilic granules of neutrophils where, with other serine proteases, it participates in the oxygen-independent pathway of intracellular and extracellular pathogen destructionCitation1,Citation2. HNE is a small, basic, and soluble glycoprotein of about 30 kDa, containing 218 amino acid residues and four disulfide bridges. HNE utilizes a catalytic triad consisting of Ser195, His57, and Asp102Citation3. Due to its proteolytic activity against a variety of extracellular matrix proteins, such as elastin, fibronectin, collagen, proteoglycans, lamininCitation4, and some matrix metalloproteinases (i.e. MMP-2, MMP-3, and MMP-9)Citation5, HNE plays an important role in many physiological processes, such as blood coagulation, apoptosis, and inflammation, and is able to modulate cytokine and growth factors expressionCitation6. Under physiological conditions, the proteolytic activity of HNE is regulated by serpins, a family of endogenous inhibitors that includes α1-antitripsin (α1-AT), elafin, and secretory leucocyte protease inhibitor (SLPI)Citation7,Citation8.
Alterations in the balance between HNE activity and its regulatory inhibitors have been implicated in the development of a variety of diseases affecting the pulmonary system, such as chronic obstructive pulmonary disease (COPD)Citation9, cystic fibrosis (CF)Citation10,Citation11, acute respiratory distress syndrome (ARDS)Citation12, acute lung injury (ALI)Citation13, as well as other inflammatory disorders, such as psoriasis, dermatitis, atherosclerosis, and rheumatoid arthritisCitation14–16. HNE has also recently been implicated in the progression of various types of cancerCitation17,Citation18. In addition, HNE plays a central role in both acute pathogenesis and chronic functional recovery after brain traumatic injuryCitation19. Thus, it is evident that compounds able to modulate the proteolytic activity of HNE could represent promising therapeutic agents for the treatment of inflammatory diseases involving excessive HNE activityCitation20,Citation21.
Many examples of peptide and non-peptide HNE inhibitors have been reported in the literatureCitation20,Citation21. However, only two drugs are currently available for clinical useCitation22: Prolastin® (purified α1-AT), a peptide drug used for the treatment of α1-antitripsin deficiency (AATD)Citation23 and Sivelestat (Elaspol® 100), a low molecular weight non-peptide selective HNE inhibitor with an IC50 value of 44 nM (). However, the use of Sivelestat for ALI and ARDS is only approved in Japan and KoreaCitation24,Citation25. Additionally, the neutrophil elastase inhibitor AZD9668 (Alvelestat) () is currently under evaluation in clinical trials for patients with bronchiectasisCitation26. We recently discovered a new class of HNE inhibitors with an N-benzoylindazole scaffold (, structure A). The most active compounds had IC50 values in the low nanomolar range, and studies on the mechanism of action showed that these compounds were competitive and pseudo-irreversible HNE inhibitors, with an appreciable selectivity for HNE versus other proteasesCitation27,Citation28. An essential requirement for the activity of these compounds was a carbonyl group at position 1.
Figure 1. Structures of selected HNE inhibitors.
Further research in this field led us to investigate other nitrogenous bicyclic scaffolds, including cinnolines, which contain a pyridazine ring instead of a pyrazole. Using the connoline scaffold, we also evaluated those substituents that previously gave as the best results for modification of the N-benzoylindazoles in order to define if the requirements were similar for the two systems.
Materials and methods
All melting points were determined on a Büchi apparatus (New Castle, DE) and are uncorrected. Extracts were dried over Na2SO4, and the solvents were removed under reduced pressure. Merck F-254 commercial plates (Merck, Durham, NC) were used for analytical TLC to follow the course of reactions. Silica gel 60 (Merck 70–230 mesh, Merck, Durham, NC) was used for column chromatography. 1H NMR and 13C NMR spectra were recorded on an Avance 400 instrument (Bruker Biospin Version 002 with SGU, Bruker Inc., Billerica, MA). Chemical shifts (δ) are reported in ppm to the nearest 0.01 ppm using the solvent as an internal standard. Coupling constants (J values) are given in Hz and were calculated using TopSpin 1.3 software (Nicolet Instrument Corp., Madison, WI) and are rounded to the nearest 0.1vHz. Mass spectra (m/z) were recorded on an ESI-TOF mass spectrometer (Brucker Micro TOF, Bruker Inc., Billerica, MA), and reported mass values are within the error limits of ±5 ppm mass units. Microanalyses indicated by the symbols of the elements or functions were performed with a Perkin–Elmer 260 elemental analyzer (PerkinElmer, Inc., Waltham, MA) for C, H, and N, and the results were within ±0.4% of the theoretical values, unless otherwise stated. Reagents and starting material were commercially available.
Chemistry
(E)-Ethyl 3-oxo-3-(2-(pyrrolidin-1-yldiazenyl)phenyl)propanoate (2): To a suspension of diethylcarbonate (4.50 mmol) in anhydrous THF (23 mL) under nitrogen, 4.50 mmol of NaH were added. The suspension was stirred at 80 °C for 1 h, then a solution of 1Citation29 (2.95 mmol) in anhydrous THF (12 mL) was slowly added. The mixture was stirred at 80 °C for 3 h. After cooling, a saturated solution of NH4Cl was added, and the mixture was extracted with ethyl acetate (3 × 15 mL). The organic layer was evaporated in vacuo to obtain compound 2, which was purified by column chromatography using cyclohexane/ethyl acetate 2:1 as eluent. Yield = 95%; oil. 1H NMR (CDCl3) δ 1.21 (t, 3H, OCH2CH3, J = 6.8 Hz), 2.05–2.10 (m, 4H, 2 × CH2 pyrrolidine), 3.68–3.73 (m, 2H, CH2 pyrrolidine), 3.96–4.04 (m, 2H, CH2 pyrrolidine), 4.15 (q, 2H, OCH2CH3, J = 6.8 Hz), 4.17 (s, 2H, COCH2CO), 7.18 (t, 1H, Ar, J = 7.2 Hz), 7.44 (t, 1H, Ar, J = 6.8 Hz), 7.51 (d, 1H, Ar, J = 7.6 Hz), 7.69 (d, 1H, Ar, J = 8.0 Hz). ESI-MS calcd. for C15H19N3O3, 289.33; found: m/z 290.05 [M + H]+.
Ethyl 1-(cyclopropanecarbonyl)-4-oxo-1,4-dihydrocinnoline-3-carboxylate (4): To a cooled (0 °C) suspension of 3Citation30 (1.40 mmol) in anhydrous CH2Cl2 (2 mL), Et3N (0.1 mL) and 4.2 mmol of cyclopropanecarbonyl chloride were added. The mixture was stirred at 0 °C for 2 h and then at room temperature for an additional 2 h. The solvent was evaporated, cold water was added, and the mixture was neutralized with 0.5 N NaOH. The reaction mixture was extracted with CH2Cl2 (3 × 15 mL), the solvent was dried over sodium sulfate, evaporated in vacuo, and compound 4 was purified by column chromatography (eluent: cyclohexane/ethyl acetate 2:1). Yield = 13%; mp = 57–58 °C (EtOH/H2O). 1H NMR (CDCl3) δ 1.23–1.28 (m, 2H, cyclopropyl), 1.33–1.38 (m, 2H, cyclopropyl), 1.46 (t, 3H, OCH2CH3, J = 7.2 Hz), 3.20–3.25 (m 1H, cyclopropyl) 4.52 (q, 2H, OCH2CH3, J = 7.2 Hz), 7.57 (t, 1H, Ar, J = 7.6 Hz), 7.81 (t, 1H, Ar, J = 7.2 Hz), 8.38 (d, 1H, Ar, J = 9.6 Hz), 8.87 (d, 1H, Ar, J = 8.8 Hz). ESI-MS calcd. for C15H14N2O4, 286.28; found: m/z 287.10 [M + H]+.
Ethyl 1-methyl-4-oxo-1,4-dihydrocinnoline-3-carboxylate (5): A mixture of 0.23 mmol of 3Citation30, 0.34 mmol of Na2CO3, and 1.15 mmol of CH3I in 1 mL of anhydrous CH3CN was refluxed for 6 h. After cooling, the precipitate was filtered, the solvent was evaporated in vacuo, and compound 5 was obtained by crystallization with ethanol. Yield = 66%; mp = 185–190 °C (EtOH). 1H NMR (DMSO-d6) δ 1.31 (t, 3H, OCH2CH3, J = 7.2 Hz), 4.16 (s, 3H, CH3), 4.32 (q, 2H, OCH2CH3, J = 7.2 Hz), 7.62 (t, 1H, Ar, J = 7.6 Hz), 7.89 (d, 1H, Ar, J = 8.8 Hz), 7.95 (t, 1H, Ar, J = 8.4 Hz), 8.19 (d, 1H, Ar, J = 8.0 Hz). 13C NMR (DMSO-d6) δ 14.58 (CH3), 31.16 (CH), 44.94 (CH3), 61.47 (CH2), 117.85 (CH), 125.28 (CH), 126.34 (C), 127.0 (CH), 134.88 (CH), 138.84 (CH), 141.24 (C). IR: 1590 cm−1 (C = O ester), 1693 cm−1 (C = O). ESI-MS calcd. for C12H12N2O3, 232.24; found: m/z 233.09 [M + H]+.
Ethyl 1-(3-methylbenzyl)-4-oxo-1,4-dihydrocinnoline-3-carboxylate (6): A mixture of 3Citation30 (0.46 mmol), K2CO3 (0.7 mmol), and 3-methylbenzyl chloride (0.80 mmol) in 2 mL of anhydrous DMF was stirred at 80 °C for 1 h. After cooling, the mixture was diluted with cold water, and the precipitate was recovered by filtration. Yield = 61%: mp = 100–102 °C (EtOH). 1H NMR (CDCl3) δ 1.47 (t, 3H, OCH2CH3, J = 7.2 Hz), 2.33 (s, 3H, CH3), 4.52 (q, 2H, OCH2CH3, J = 7.2 Hz), 5.68 (s, 2H, CH2), 7.05 (s, 2H, Ar), 7.13 (d, 1H, Ar, J = 6.4 Hz), 7.24 (d, 1H, Ar, J = 8.0 Hz), 7.46 (t, 2H, Ar, J = 8.8 Hz), 7.66 (t, 1H, Ar, J = 7.2 Hz), 8.44 (d, 1H, Ar, J = 8.0 Hz). 13C NMR (CDCl3) δ 14.29 (CH3), 21.43 (CH3), 60.82 (CH2), 61.88 (CH2), 116.16 (CH), 123.65 (CH), 126.14 (CH), 126.67 (CH), 127.13 (CH), 129.03 (CH), 129.23 (CH), 134.04 (CH), 134.68 (C), 139.04 (C), 140.50 (C). IR: 1717 cm−1 (C = O ester), 1632 cm−1 (C = O). ESI-MS calcd. for C19H18N2O3, 322.36; found: m/z 323.14 [M + H]+.
Ethyl 1-(3-methylbenzoyl)-4-oxo-1,4-dihydrocinnoline-3-carboxylate (7): To a cooled (0 °C) suspension of the appropriate substrate 3Citation30 (0.40 mmol) in anhydrous CH2Cl2 (2 mL), Et3N (0.1 mL), and 1.15 mmol of m-toluoyl chloride were added. The solution was stirred at 0 °C for 2 h and then at room temperature for 2 h. After evaporation of the solvent, the residue was mixed with ice-cold water (20 mL) and neutralized with 0.5 N NaOH. Compound 7 was recovered by extraction with CH2Cl2 (3 × 15 mL) and was purified by column chromatography using cyclohexane/ethyl acetate 2:1 as eluent. Yield = 39%; mp = 81–83 °C (EtOH). 1H NMR (CDCl3) δ 1.34 (t, 3H, OCH2CH3, J = 7.2 Hz), 2.46 (s, 3H, CH3), 4.39 (q, 2H, OCH2CH3, J = 7.2 Hz), 7.41 (t, 1H, Ar, J = 7.6 Hz), 7.48 (d, 1H, Ar, J = 7.6 Hz), 7.60 (t, 1H, Ar, J = 8.0 Hz), 7.68 (d, 1H, Ar, J = 7.6 Hz), 7.74 (s, 1H, Ar), 7.83 (t, 1H, Ar, J = 7.5 Hz), 8.43 (t, 2H, Ar, J = 8.0 Hz). ESI-MS calcd. for C19H16N2O4, 336.34; found: m/z 337.11 [M + H]+.
1 -(3-Methylbenzoyl)-4-oxo-1,4-dihydrocinnoline-3-carboxylic acid (8): A mixture of 7 (0.15 mmol) and 6 N NaOH (5 mL) was stirred at 100 °C for 5 h. After cooling, the mixture was acidified with 6 N HCl, and the precipitate was recovered by suction and crystallized with ethanol. Yield = 11%; mp = 268–270 °C dec (EtOH). 1H NMR (CDCl3) δ 2.5 (s, 3H, CH3), 7.41 (m, 2H, Ar), 7.73 (m, 3H, Ar), 7.88 (d, 1H, Ar, J = 8.4 Hz), 8.02 (t, 1H, Ar, J = 7.8 Hz), 8.27 (d, 1H, Ar, J = 8.4 Hz). ESI-MS calcd. for C17H12N2O4, 308.29; found: m/z 309.08 [M + H]+.
General procedure for 10 b,c: To a cooled (0 °C) suspension of the appropriate substrate 9bCitation31 or 9cCitation32 (1.40 mmol) in conc. HCl (1.2 mL), 2.24 mmol of NaNO2 in 1 mL of H2O was slowly added. The mixture was stirred at 0 °C for 2 h and then at room temperature for 48 h. After concentration of the solvent, the solution was extracted with ethyl acetate (3 × 15 mL), and the organic layer was washed with brine (5 mL). Evaporation of the solvent resulted in final compounds 10b,c, which were purified by column chromatography using toluene/ethyl acetate 7:3 (for 10b) or cyclohexane/ethyl acetate 3:1 (for 10c) as eluents. 3-Cyclopropylcinnolin-4(1H)-one (10b): Yield = 12%; mp = 238–245 °C dec (EtOH). 1H NMR (CDCl3) δ 0.95–1.04 (m, 4H, 2 × CH2 cyclopropyl), 2.62–2.68 (m, 1H, CH cyclopropyl), 7.37 (t, 2H, Ar, J = 7.2 Hz), 7.67 (t, 1H, Ar, J = 7.6 Hz), 8.33 (d, 1H, Ar, J = 8.4 Hz), 10.50 (exch br s, 1H, NH). IR = 1718 cm−1 (C = O). ESI-MS calcd. for C11H10N2O, 186.21; found: m/z 187.02 [M + H]+.
General procedure for 11a–c: Compounds 11a–c were obtained by following the same procedure performed for compound 7, starting from precursors 10a–c [10aCitation33]. Compounds 11a–c were recovered by extraction with CH2Cl2 (3 × 15 mL) and were purified by column chromatography using toluene/ethyl acetate 8:2 (for 11a,b) or cyclohexane/ethyl acetate 3:1 (for 11c) as eluents. 1-(3-Methylbenzoyl)-3-phenylcinnolin-4(1H)-one (11a): Yield = 22%; mp = 119–121 °C (EtOH). 1H NMR (CDCl3) δ 2.48 (s, 3H, CH3), 7.42–7.55 (m, 4H, Ar), 7.54 (d, 1H, Ar, J = 7.6 Hz), 7.81 (t, 1H, Ar, J = 7.6 Hz), 7.88–7.97 (m, 2H, Ar), 8.03 (m, 4H, Ar), 8.71 (d, 1H, Ar, J = 8.4 Hz). ESI-MS calcd. for C22H16N2O2, 340.37; found: m/z 341.12 [M + H]+.
3-[(Tetrahydro-2H-pyran-2-yloxy)methyl]cinnolin-4(1H)-one (12): To a mixture of ammonium cerium(IV) nitrate (CAN) (0.09 mmol) in anhydrous CH3CN (5 mL), 4.50 mmol of substrate 10dCitation33 and 4.50 mmol of 3,4-dihydro-2H-pyran (commercially available) were added, and the suspension was stirred at room temperature for 24 h. After evaporation of the solvent, 20 mL of H2O was added, and the mixture was extracted with CH2Cl2. The organic layer was evaporated in vacuo, resulting in crude 12, which was purified by column chromatography using CH2Cl2/MeOH 9:1 as eluent. Yield = 42%; oil. 1H NMR (CDCl3) δ 1.55–1.67 (m, 4H, cC5H9O), 1.76–1.86 (m, 2H, cC5H9O), 3.57–3.62 (m, 1H, OCH-H), 4.02 (t, 1H, OCH-H, J = 9.6 Hz), 4.80 (d, 1H, CH-H, J = 12.4 Hz), 4.90–5.00 (m, 2H, CH cC5H9O + CH-H), 7.39 (t, 1H, Ar, J = 7.6 Hz), 7.53–7.58 (m, 1H, Ar), 7.70 (t, 1H, Ar, J = 8.8 Hz), 8.29 (d, 1H, Ar, J = 8.0 Hz). ESI-MS calcd. for C14H16N2O3, 260.29; found: m/z 261.04 [M + H]+.
1-(3-Methylbenzoyl)-3-[(tetrahydro-2H-pyran-2-yloxy)methyl]cinnolin-4(1H)-one (13): Compound 13 was obtained following the same procedure performed for compound 7 but starting from precursor 12. Compound 13 was recovered by extraction with CH2Cl2 (3 × 15 mL) and was purified by column chromatography using toluene/ethyl acetate 8:2 as eluent. Yield = 55%; oil. 1H NMR (CDCl3) δ 1.44–1.65 (m, 6H, cC5H9O), 2.46 (s, 3H, CH3), 3.45–3.50 (m, 1H, OCH-H), 3.85–3.90 (m, 1H, OCH-H), 4.70–4.80 (m, 3H, CH cC5H9O + CH2), 7.37–7.47 (m, 2H, Ar), 7.56 (t, 1H, Ar, J = 8.0 Hz), 7.58–7.68 (m, 2H, Ar), 7.81 (t, 1H, Ar, J = 8.8 Hz), 8.38 (d, 1H, Ar, J = 9.6 Hz), 8.53 (d, 1H, Ar, J = 8.8 Hz). ESI-MS calcd. for C22H22N2O4, 378.42; found: m/z 379.16 [M + H]+.
3-(Hydroxymethyl)-1-(3-methylbenzoyl)cinnolin-4(1H)-one (11d): A mixture of 13 (0.11 mmol), trifluoroacetic acid (0.3 mL), and CH2Cl2 (1.7 mL) was stirred at room temperature for 3 h. Evaporation of the solvent resulted in compound 11d, which was purified by column chromatography using CH2Cl2/MeOH 9.5:0.5 as eluent. Yield = 62%; oil. 1H NMR (CDCl3) δ 2.40 (s, 3H, CH3), 5.58 (s, 2H, CH2), 7.31–7.46 (m, 4H, Ar), 7.74 (t, 1H, Ar, J = 7.6 Hz), 7.91 (d, 2H, Ar, J = 8.0 Hz), 8.34 (d, 1H, Ar, J = 8.8 Hz). 13C NMR (CDCl3) δ 21.35 (CH3), 59.17 (CH2), 116.83(CH), 124.47 (CH), 124.94 (CH), 127.23 (CH), 128.24 (CH), 130.34 (CH), 133.82 (CH), 134.09 (CH), 141.72 (C), 171.20 (C). ESI-MS calcd. for C17H14N2O3, 294.30; found: m/z 295.10 [M + H]+.
General procedure for 16a–c and 17a–c: Compounds 16a–c and 17a–c were obtained following the same procedure performed for compound 7 but starting from precursors 15a–cCitation30,Citation34. Compounds 16a,c and 17a,c were recovered by extraction with CH2Cl2 (3 × 15 mL), while the crude 16b and 17b were recovered by vacuum filtration. Final 16a–c and 17a–c were purified by column chromatography using cyclohexane/ethyl acetate in the following different ratios as eluents: 3:1 for 16a and 17a; 6:1 for 16b and 17b; 5:1 for 16c and 17c. 3-Chloro-1-(3-methylbenzoyl)cinnolin-4(1H)-one (16a). Yield = 33%; mp = 128–135 °C (EtOH). 1H NMR (CDCl3) δ 2.48 (s, 3H, CH3), 7.43 (t, 1H, Ar, J = 7.6 Hz), 7.49 (d, 1H, Ar, J = 7.6 Hz), 7.59–7.65 (m, 2H, Ar), 7.70 (s, 1H, Ar), 7.85 (t, 1H, Ar, J = 8.0 Hz), 8.43 (d, 1H, Ar, J = 8.0 Hz), 8.49 (d, 1H, Ar, J = 9.2 Hz). IR = 1663 cm−1 (C=O amide), 1723 cm−1 (C=O). ESI-MS calcd. for C16H11ClN2O2, 298.72; found: m/z 300.05 [M + H]+. 3-Chlorocinnolin-4-yl 3-methylbenzoate (17a): Yield = 17%; mp = 115–123 °C (EtOH). 1H NMR (CDCl3) δ 2.53 (s, 3H, CH3), 7.52 (t, 1H, Ar, J = 7.8 Hz), 7.60 (d, 1H, Ar, J = 7.6 Hz), 7.83 (t, 1H, Ar, J = 7.6 Hz), 7.90–7.95 (m, 2H, Ar), 8.15 (s, 2H, Ar), 8.64 (d, 1H, Ar, J = 8.4 Hz). IR = 1744 cm−1 (C=O ester). ESI-MS calcd. for C16H11ClN2O2, 298.72; found: m/z 300.05 [M + H]+.
General procedure for 18a–g: Compounds 18a–g were obtained following the same procedure as performed for compound 7 but starting from precursor 14Citation30. Compounds 18a,e,g were recovered by extraction with CH2Cl2 (3 × 15 mL), while crude 18b–d and 18f were recovered by vacuum filtration. Final compounds 18a,b,e,g were purified by column chromatography using cyclohexane/ethyl acetate 4:1 (for 18a) or 1:1 (for 18b and 18g) or toluene/ethyl acetate 9:1 (for 18e) as eluents, or by crystallization from ethanol (for 18c,d,f).
1-(3-Methylbenzoyl)cinnolin-4(1H)-one (18a): Yield = 55%; mp = 138–139 °C (EtOH). 1H NMR (CDCl3) δ 2.47 (s, 3H, CH3), 7.40–7.50 (m, 2H, Ar), 7.56–7.66 (m, 3H, Ar), 7.76–7.86 (m, 2H, Ar), 8.36 (d, 1H, Ar, J = 9.6 Hz), 8.50 (d, 1H, Ar, J = 9.2 Hz). 13C NMR (CDCl3) δ 21.35 (CH3), 118.77 (CH), 125.54 (CH), 126.82 (CH), 127.90 (CH), 128.24 (CH), 131.15 (CH), 133.99 (CH), 134.61 (CH), 140.11 (CH), 171.15 (C), 171.25 (C). IR = 1645 cm−1 (C=O amide), 1709 cm−1 (C=O). ESI-MS calcd. for C16H12N2O2, 264.28; found: m/z 265.09 [M + H]+.
(4-Hydroxy-3,4-dihydrocinnolin-1(2H)-yl)(m-tolyl)meyhanone (19): Compound 18a (0.189 mmol) was subjected to catalytic reduction in EtOH (15 mL) for 4 h with a Parr instrument (Parr Instrument Company, Moline, IL) using 0.094 mmol of 10% Pd/C as a catalyst and a constant pressure at 30 PSI. The catalyst was filtered, the solvent was evaporated under vacuum, and the crude product was purified by column chromatography using cyclohexane/ethyl acetate 1:1 as eluent. Yield = 42%; mp = 187–190 °C dec (EtOH). 1H NMR (DMSO-d6) δ 2.35 (s, 3H, CH3), 2.95–3.01 (m, 1H, CH), 3.08–3.15 (m, 1H, CH), 4.45 (s, 1H, C-H), 5.44 (exch br s, 1H, NH), 5.71–5.76 (m, 1H, OH), 7.17 (t, 1H, Ar, J = 7.4 Hz), 7.24–7.31 (m, 3H, Ar), 7.41 (d, 2H, Ar, J = 8.4 Hz), 7.49 (d, 1H, Ar, J = 7.2 Hz), 7.83 (d, 1H, Ar, J = 8.4 Hz). 13C NMR (DMSO-d6) δ 21.41 (CH3), 53.49 (CH2), 61.94 (CH), 122.08 (CH), 124.60 (CH), 125.80 (CH), 127.16 (CH), 127.76 (CH), 129.05 (CH), 130.34 (CH), 130.59 (CH), 130.76 (CH), 137.07 (C), 137.66 (C), 138.52 (C), 171.62 (C). IR = 1644 cm−1 (C=O amide), 3266 cm−1 (NH), 3431 cm−1 (OH). ESI-MS calcd. for C16H16N2O2, 268.31; found: m/z 269.12 [M + H]+.
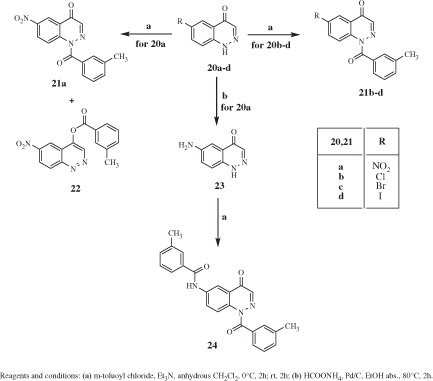
General procedure for 21a–d, 22, and 24: Compounds 21a–d, 22, and 24 were obtained following the same procedure as for compound 7 but starting from precursors 20a–dCitation34–37 and 23Citation38. Compounds 21a–d, 22, and 24 were recovered by extraction with CH2Cl2 (3 × 15 mL) and were purified by column chromatography using cyclohexane/ethyl acetate 3:1 (for 21a,b,d and 22), 2:1 (for 21c), or 1:6 (for 24) as eluents.
1-(3-Methylbenzoyl)-6-nitrocinnolin-4(1H)-one (21a): Yield =18 %; mp = >300 °C dec (EtOH). 1H NMR (CDCl3) δ 2.48 (s, 3H, CH3), 7.45 (t, 1H, Ar, J = 7.6 Hz), 7.52 (d, 1H, Ar, J = 7.6 Hz), 7.65 (d, 1H, Ar, J = 8.0 Hz), 7.68 (s, 1H, Ar), 7.87 (s, 1H, Ar), 8.58–8.64 (m, 2H, Ar), 9.20 (d, 1H, Ar, J = 2.4 Hz). IR = 1376–1525 cm−1 (NO2), 1654 cm−1 (C=O amide), 1723 cm−1 (C=O). ESI-MS calcd. for C16H11N3O4, 309.28; found: m/z 310.08 [M + H]+. 6-Nitrocinnolin-4-yl 3-methylbenzoate (22): Yield = 18%; mp = 145–147 °C (EtOH). 1H NMR (CDCl3) δ 2.55 (s, 3H, CH3), 7.55 (t, 1H, Ar, J = 7.0 Hz), 7.62 (d, 1H, Ar, J = 7.6 Hz), 8.15 (s, 2H, Ar), 8.66 (d, 1H, Ar, J = 9.6 Hz), 8.84 (d, 1H, Ar, J = 9.6 Hz), 9.03 (s, 1H, Ar), 9.73 (s, 1H, Ar). IR = 1376–1583 cm−1 (NO2), 1752 cm−1 (C=O). ESI-MS calcd. for C16H11N3O4, 309.28; found: m/z 310.08 [M + H]+.
HNE inhibition assay
Compounds were dissolved in 100% DMSO at 5 mM stock concentrations. We confirmed that the final concentration of DMSO in the reactions (1%) had no effect on enzyme activity. The HNE inhibition assay was performed in black flat-bottom 96-well microtiter plates, as described previouslyCitation39. Briefly, buffer A (200 mM Tris-HCl, pH 7.5, 0.01% bovine serum albumin, and 0.05% Tween-20) plus 20 mU/mL of HNE (Calbiochem, New Orleans, LA) was added to wells containing different concentrations of each compound. After a 5-min pre-incubation, the reaction was initiated by addition of 25 μM elastase substrate (N-methylsuccinyl–Ala–Ala–Pro–Val-7-amino-methyl-coumarin; Calbiochem) in a final reaction volume of 100 μL/well. Kinetic measurements were obtained every 30 s for 10 min at 25 °C using a Fluoroskan Ascent FL fluorescence microplate reader (Thermo Electron, Franklin, MA) with excitation and emission wavelengths at 355 and 460 nm, respectively. For all compounds tested, the concentration of inhibitor that caused 50% inhibition of the enzymatic reaction (IC50) was calculated by plotting % inhibition versus logarithm of inhibitor concentration (at least six points). The data are presented as the mean values of at least three independent experiments with relative standard deviations of <15%.
For selected lead compounds, the inhibition constant (Ki) values were determined using Dixon plots of three to four different concentrations of the substrate, as described previouslyCitation39. At each substrate concentration, rates were determined with five different inhibitor concentrations, and the inverse of the velocities was plotted against the final inhibitor concentration. Ki was determined from the intersection of the plotted lines.
Reactivation of the HNE−inhibitor complex was investigated, as described previouslyCitation40. Briefly, HNE (20 mU/mL) was incubated with excess inhibitor at a final concentration of 50 μM in 1 mL of Tris bufer (200 mM Tris-HCl, pH 7.5). After 20 min, a 50 μL aliquot was removed and assayed to verify complete inhibition of HNE enzymatic activity. Excess inhibitor was removed via Vivaspin 10 000 MWCO PES (Sartorius, Goettingen, Germany) filtration by centrifuging at 10 000 g for 5 min, adding 0.5 mL of the Tris buffer to the retentate, and centrifuging again under the same conditions. This process was repeated again, and the final retentate was suspended in 0.5 mL of buffer A at 25 °C. At the indicated times, 50 μL aliquots were removed and added to microplate wells containing 25 μM of the fluorogenic HNE substrate dissolved in 50 μL of buffer A, and enzymatic activity was monitored, as described above. A control containing HNE (20 mU/mL) and 0.25% DMSO was run under the same conditions.
Analysis of compound stability
Spontaneous hydrolysis of selected derivatives was evaluated at 25 °C in 0.05 M phosphate buffer, pH 7.3. Kinetics of hydrolysis were monitored by measuring changes in absorbance spectra over time using a SpectraMax Plus microplate spectrophotometer (Molecular Devices, Sunnyvale, CA). Absorbance (At) at the characteristic absorption maxima of each compound was measured at the indicated times until no further absorbance decreases occurred (A∞)Citation41. Using these measurements, we created semilogarithmic plots of log(At–A∞) versus time, and k′ values were determined from the slopes of these plots. Half-conversion times were calculated using t1/2 = 0.693/k′, as described previouslyCitation27,Citation39.
The stability of selected compounds in human serum and buffer was also evaluated by analytical RP-HPLC. Human serum was obtained by allowing fresh human blood to coagulate at room temperature for 1 h. To 500 μL of human serum or phosphate buffer (0.05 M, pH 7.3), 10 μL of the 10 mM compound stock solution (in DMSO) was added, and the mixture was incubated at 25 °C. At the indicated times, aliquots (50 μL) were removed, instantly mixed with 200 μL acetonitrile to precipitate proteins in serum samples, and centrifuged for 5 min at 10 000g. The samples were analyzed on an automated HPLC system (Shimadzu, Torrance, CA) with a Phenomenex Jupiter C18 300 A column (Phenomenex Inc., Torrance, CA) (5 μm, 25 cm ×0.46 cm) eluted with acetonitrile/water (65%/35%, v/v) containing 0.1% (v/v) TFA at a flow rate of 1 mL/min at 40 °C. The elution was monitored using a diode array detector (Shimadzu SPD-M10A VP, Shimadzu Corporation, Kyoto, Japan) set to spectrum max plot (wavelength at which the highest absorbance occurs) in the region of 200−300 nm. The amount of compound that remained in serum was calculated from the area under the eluted peak.
Molecular modeling
Initial structures of the compounds were generated with HyperChem 8.0 (Shimadzu Corporation, Kyoto, Japan) and optimized by the semi-empirical PM3 method. Docking of the molecules was performed with the use of Molegro Virtual Docker (CLC Bio, København, Denmark) (MVD), version 4.2.0 (CLC Bio, København, Denmark), as described previouslyCitation28. The structure of HNE complexed with a peptide chloromethyl ketone inhibitorCitation42 was used for the docking study (1HNE entry of the Protein Data Bank). The search area for docking poses was defined as a sphere with 10 Å radius centered at the nitrogen atom in the five-membered ring of the peptide chloromethyl ketone inhibitor. After removal of this peptide and co-crystallized water molecules from the program workspace, we set side chain flexibility for the 42 residues closest to the center of the search area (His40, Phe41, Cys42, Gly43, Ala55, Ala56, His57, Cys58, Val59, Ala60, Tyr94, Pro98, Asn99A, Leu99B, Asp102, Trp141, Gly142, Leu143, Leu167, Arg177, Val190, Cys191, Phe192, Gly193, Asp194, Ser195, Gly196, Ser197, Ala213, Ser214, Phe215, Val216, Arg217A, Gly218, Gly219, Cys220, Ser222, Leu223, Tyr224, Asp226, Ala227, and Phe228). Fifteen docking runs were performed for each compound, with full flexibility of a ligand around all rotatable bonds and side chain flexibility of the above-mentioned residues of the enzyme. Parameters used within Docking Wizard of Molegro program were as described previouslyCitation28.
The docking poses corresponding to the lowest-energy binding mode of each inhibitor were evaluated for the ability to form a Michaelis complex between the hydroxyl group of Ser195 and the carbonyl group in the amido moiety of an inhibitor. For this purpose, values of d1 [distance O(Ser195)·C between the Ser195 hydroxyl oxygen atom and the inhibitor carbonyl carbon atom closest to O(Ser195)] and α [angle O(Ser195)···C=O, where C=O is the carbonyl group of an inhibitor closest to O(Ser195)] were determined for each docked compoundCitation43. In addition, we estimated the possibility of proton transfer from Ser195 to Asp102 through His57 (the key catalytic triad of serine proteases) by calculating distances d2 between the NH hydrogen in His57 and carboxyl oxygen atoms in Asp102, as described previouslyCitation28. The distance between the hydroxyl proton in Ser195 and the pyridine-type nitrogen in His57 is also important for proton transfer. However, because of easy rotation of the hydroxyl about the C–O bond in Ser195, we measured distance d3 between the oxygen in Ser195 and the basic nitrogen atom in His57. The effective length L of the channel for proton transfer was calculated as L = d3 + min (d2).
Results and discussion
Chemistry
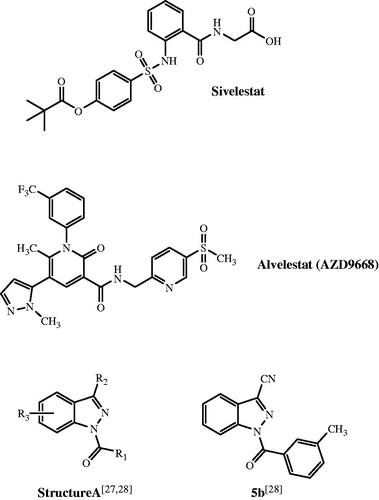
All final compounds were synthesized as reported in Schemes 1–4, and the structures were confirmed on the basis of the analytical and spectral data. Scheme 1 depicts the synthetic pathway for the 3-carbethoxy compounds 4–7 with different substitutions at N-1. Compound 3, which was previously describedCitation30, is the key intermediate for final compounds 4–7. It was synthesized starting from precursor 1Citation29, which was treated with diethylcarbonate and NaH (2) and then transformed into the 4-oxo-1,4-dihydro-cinnoline 3 (30) by cyclization with trifluoracetic acid. According to the literature, the 4-oxo tautomer is predominant for the 4-hydroxycinnoline nucleusCitation44,Citation45. In any case, treatment of intermediate 3Citation30 with different halides led to the corresponding N-1 derivatives (products 4–7). Furthermore, hydrolysis of the ethyl ester of compound 7 with NaOH resulted in compound 8.
Scheme 1. Synthesis of cinnoline derivatives 4–8.
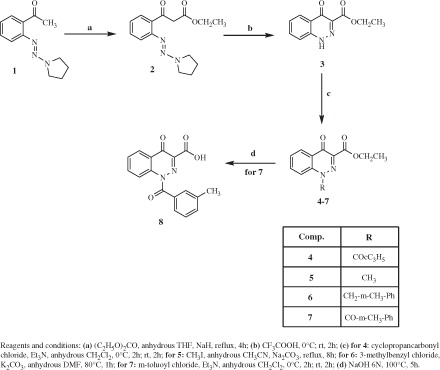
Keeping m-methylbenzoyl at position N-1, we next inserted various substituents at position 3 of cinnoline, as shown in Scheme 2. Starting from precursors 9a–dCitation31,Citation32, which were synthesized following the Sonogoshira reactionCitation46, we performed the cyclization to 4-oxo-1,4-dihydrocinnoline with HCl and NaNO2 to form intermediates 10a–d [10a,dCitation33]. Compounds 10a–c were then treated with m-toluoyl chloride and Et3N in CH2Cl2 to obtain 11a–c. To obtain 11d, it was necessary to protect 10d with 3,4-dihydro-2H-pyran (12), insert the benzoyl fragment at N-1 (13), and finally remove the protecting group with trifluoroacetic acid (11d).
Scheme 2. Synthesis of cinnoline derivatives 11a–d.
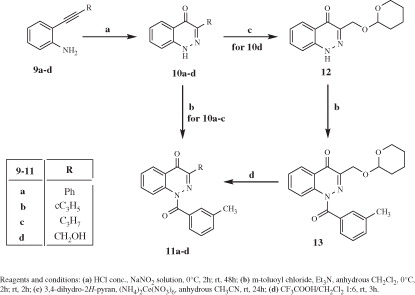
The introduction of halogens at position 3 is shown in Scheme 3. 1H-cinnolin-4-one [14Citation30] was treated with N-chlorosuccinimide (NCS), N-iodosuccinimide (NIS), or bromine to obtain 15a–cCitation30,Citation34, which, in turn, were reacted with m-toluoyl chloride. The presence of a halogen at position 3 shifts the prototropic tautomer composition, and in the reaction mixture it is possible to recover in analog ratio the amide derivatives 16a–c and the ester derivatives 17a–c from the attack at N-1 and at OH of position 4, respectively. Scheme 3 also shows synthesis of the 3-unsubsituited cinnolines 18a–g, which were obtained following the same procedure used for 16a–c. The 4-carbonyl group of 18a was further reduced to obtain compound 19.
Scheme 3. Synthesis of cinnoline derivatives 16a–c, 17a–c, and 19.
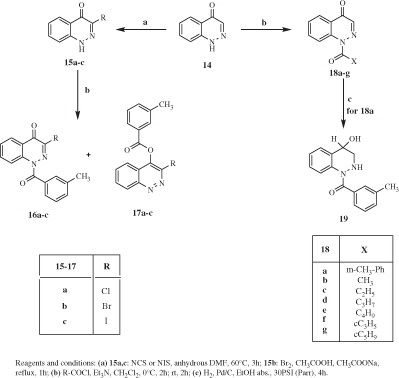
Scheme 4 shows the synthetic routes for the 3-substituted derivatives 21a–d, 22, and 24. The intermediates 20a–dCitation34–37 were treated with m-toluoyl chloride following the same conditions as described previously to obtain compounds 21a–d. The potent electron withdrawing effect of the nitro group in compound 20a, making possible the existence of the two tautomers, led to a mixture of the 1-amido 21a and the 4-ester derivative 22 in a 1:1 ratio. The 6-nitro compound 20aCitation34 was also transformed into the corresponding 6-amino 23Citation38 by reduction with ammonium formate and Pd/C as a catalyst. Further treatment of 23 with m-toluoyl chloride resulted in a double addition final compound 24.
Scheme 4. Synthesis of cinnoline derivatives 21a–d, 22. and 24.
Structure–activity relationship analysis and kinetic features of cinnoline derivatives
All compounds were evaluated for their ability to inhibit HNE, and the results are reported in . Since this new series was designed as an elaboration of the potent N-benzoylindazoles previously synthesized by our research groupCitation27,Citation28, the choice of substituents that were inserted into the cinnoline nucleus was made with the goal of evaluating the similarity and differences between the two scaffolds. Thus, we inserted the functional groups and substituents that produced the best results in the previous series, such as nitro and bromine at position 6, ethyl ester at position 3, and m-methylbenzoyl at position 1. We also evaluated if COOH and H at position 3 resulted in a similar loss of activity, as was observed for the N-benzoylindazoles. Moreover, we synthesized two compounds lacking the amide function at position 1 in order to understand if, like the N-benzoylindazole derivatives, the carbonylic group at N-1 was also the point of attack of Ser195 for this series.
Table 1. HNE inhibitory activity of cinnolone derivatives 4–8, 11a–d, 16a–c, and 18a–g.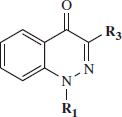
Table 2. HNE Inhibitory activity of cinnolone derivatives 21a–d and 24.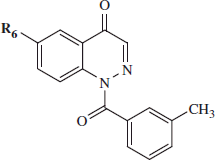
Table 3. HNE inhibitory activity of cinnoline derivatives 17a–c, 22, and 19.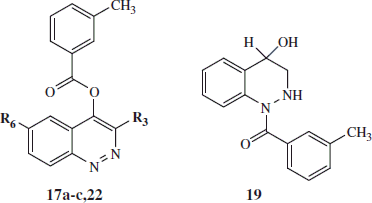
Among the 3-carbethoxy derivatives (), only compounds 4 and 7 containing an amide group at position 1 exhibited submicromolar HNE inhibitory activity (IC50 = 0.58 and 0.21 μM, respectively). As expected, elimination of the carbonyl group led to completely inactive derivatives (5 and 6), confirming that the C=O at N-1 was essential for HNE inhibitory activity, which was also evident in our previous studies with indazole-based HNE inhibitors. While keeping the m-methylbenzoyl fragment, which was found to be the best substituent for the series of indazolesCitation28, we modified and eliminated the groups at position 3. The introduction of a phenyl (compound 11a), hydroxymethyl (compound 11d), or COOH (compound 8) group led to the loss of activity. The activity was also very low for compound 11c (IC50 = 8.10 μM) containing a propyl group at position 3. In contrast, HNE inhibitory activity was maintained with the insertion of an halogen (compounds 16a–c) or (cyclo)alkyl group (11b) (IC50 = 0.43–1.69 μM). Starting from this result and maintaining an unsubstituted position 3, we evaluated effects of the replacement at position 1 of the phenyl with (cyclo)alkyl fragments (compounds 18b–g) (), but the activity of this series was one order of magnitude lower than for 18a.
We next introduced at position 6 (R6) of 18a a variety of substituents that were found to be favorable for activity of the reference N-benzoylindazolesCitation28. However, none of these substituents led to increased potency, and these compounds had IC50 values in the micromolar/submicromolar range (compounds 21a–d and 24, IC50 value = 0.26–6.30 μM) ().
In , we report HNE inhibitory activity of compounds 17a–c and 22, which are the ester isomers of the N-1 benzoyl derivatives of 16a–c and 21a. Interestingly, all compounds, although lacking the amidic function at N-1, exhibited moderate inhibitory activity. Moreover, compounds 17c (R3 = I) and 22 (R6 = NO2) had IC50 values comparable with their amidic isomers 16c and 21a (IC50 values = 2.70 and 1.30 μM for 17c and 16c, respectively, and IC50 values = 0.59 and 0.26 μM, for 22 and 21a, respectively). Thus, these results suggest that ester derivatives 17a–c and 22 probably interact with the enzyme differently from the amide derivatives. Finally, the 3,4-dihydrocinnoline 19 was completely devoid of activity.
Based on our SAR analysis, we can highlight the similarities and differences between the new scaffold and N-benzoylindazoles of the previous series. Considering the cinnoline core, our data suggest that the carbonyl group at N-1 is the one involved in the Ser195 attack, and the best substituent at this position is 3-methylbenzoyl, which was observed for the N-benzoylindazolesCitation28. Position 3 of the cinnolines should not be substituted because inclusion of various groups or atoms led to a decrease in activity, which was not observed with the indazole series. The same effect on activity occurred for position 6, probably because the insertion of a carbonyl to expand the pentatomic ring influenced the interaction of the molecule with the catalytic site. In addition, the aromatic compounds showed an appreciable HNE inhibitory activity, suggesting that the carbonyl ester may be involved in the catalysis.
Inhibition constants (Ki) were determined for the two compounds with the highest HNE inhibitory activity (18a and 18e). Although compounds 18a and 18e were quite potent, with Ki values of 75 and 110 nM, respectively, their activity was lower that our reference N-benzoylindazole HNE inhibitor (Ki = 27 nM), which was previously reported as compound 5b (1-(3-methylbenzoyl)-1H-indazole-3-carbonitrile)Citation28. In addition, double-reciprocal Lineweaver–Burk plots of substrate hydrolysis by HNE in the absence and presence of compounds 18a and 18e showed that they were competitive inhibitors ( shows a representative plot for 18a).
Figure 2. Kinetics of HNE inhibition by cinnoline derivative 18a. Representative double-reciprocal Lineweaver–Burk plot of substrate hydrolysis by HNE in the absence and presence of the compounds 18a is shown. The representative plot is from three independent experiments.
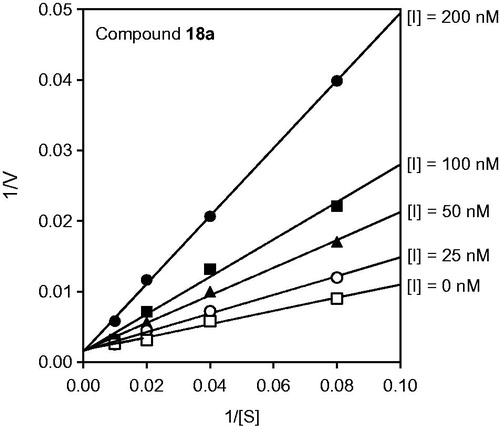
Compound and HNE-inhibitor complex stability
The most active cinnoline derivatives with an IC50 < 1 µM were evaluated for chemical stability in aqueous buffer using spectrophotometry to assess compound hydrolysis. As an example, the hydrolysis of compound 21a is shown in Figure S1. The absorbance maxima at 325 nm decreased over time, indicating that this compound was hydrolyzed almost completely after 60 min in aqueous buffer with a t1/2 of 33 min. The other compounds had t1/2 values from 38.5 to 233 min (), indicating that the tested cinnoline derivatives were more stable than our previously described HNE inhibitors with the N-benzoylindazole scaffoldCitation27,Citation28. Indeed, cinnoline derivatives possess an endocyclic carbonyl group, which participates in the total conjugation system of the molecule. Acceptor character of this group causes destabilization of the carbocation intermediate that emerges on protonation of the amide oxygen atom and may lead to lowered hydrolysis rates of cinnolines as compared to their indazole analogues.
Table 4. Half-life (t1/2) for the spontaneous hydrolysis of selected cinnolinone derivatives.
To test compound stability in serum, the two most potent cinnoline HNE inhibitors (18a and 18e) were incubated in human serum for various times at 25 °C, and then the samples were analyzed by analytical RP-HPLC to quantify the percent of initial compound remaining. Note that compound stability in phosphate buffer analyzed by this method was consistent with the results obtained by conventional spectrophotometry (Figure S1). We found that following 5 min of incubation in human serum, <3% of the initial amounts remained. After 10 min in serum, there were essentially no detectable compounds (Figure S2).
Two relatively stable compounds (11b and 18b) were evaluated for reversibility of HNE inhibition over time in comparison with HNE inhibitor 5bCitation28 (). As shown in Figure S3, an inhibition was maximal during the first 2 h after treatment with 10 µM 18b, but inhibition was soon reversed and recovery of HNE activity was observed. Although HNE inhibitory activity of 11b was lower than reference compound 5b, both compounds were equally active over the 10-h incubation period (Figure S3).
The stability of HNE–inhibitor complex was also evaluated by treating the most potent cinnoline derivatives (18a and 18e) at a relatively high concentration (25 μM), removing free inhibitor by ultrafiltration of the enzyme/inhibitor mixture, and then monitoring recovery of HNE activity over time. We found that inhibition of HNE by the selected inhibitors was fully reversible, whereas the reference compound 5b completely inhibited activity of HNE even after a 6-h incubation (data not shown).
Molecular modeling
The geometric characteristics of molecule orientations and arrangements in the catalytic triad (Ser195, His57, and Asp102) were in general agreement with the HNE inhibitory activities of the compounds (). The key residues were oriented in a fashion favorable for proton transfer within the oxyanion hole and formation of the Michaelis complex important for inhibition. Both cinnoline derivative 18a and reference N-benzoylindazole 5bCitation28 () were H-bonded with Gly193 and Ser195, but had somewhat different poses within the receptor cavity ( and S4). Particularly, the poses differed in the orientation of the C=O bond of the 3-methylbenzoyl fragment, although this fragment was oriented similarly for the poses of 5bCitation28 and 18a and is directed deep into the receptor binding site. In addition, these molecules with different scaffolds had different values of angle α. The angle magnitude of 105.2° for compound 5bCitation28 was close to the middle of the optimum interval (80–120°)Citation28, while the α value of 18a lies near the lower boundary of the interval (). A hypothetical model for the Ser195 nucleophilic attack at the carbonyl group of molecule 18a, accompanied by synchronous proton transfer from Ser195 to Asp102 via the catalytic triad and according to the mechanism characteristic of serine proteasesCitation40,Citation47 is shown in .
Figure 3. Superimposed docking poses of peptide chloromethyl ketone and novel small-molecule HNE inhibitors. Co-crystallized peptide chloromethyl ketone inhibitor is shown in yellow. Residues within 5 Å of this ligand are visible. Panel (A) Docking poses of reference compound 5bCitation28 (dark-green) and compound 18a (blue). Panel (B) Docking poses of compounds 16b (violet) and 17b (brown).
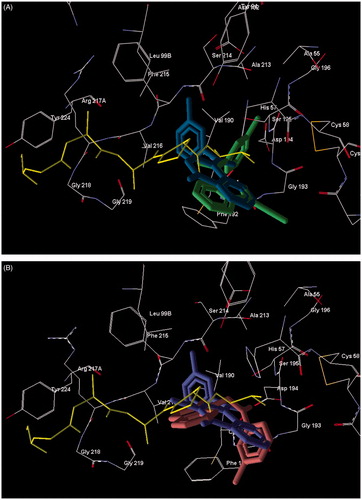
Figure 4. Hypothetical model for the nucleophilic attack of Ser195 at the carbonyl group of cinnoline derivative (18a) accompanied by synchronous proton transfer from Ser195 to Asp102 via the catalytic triad. The key angle α is indicated (see text for details). The model is based on the proposed mechanism of synchronous proton transfer from the oxyanion hole in serine proteasesCitation40,Citation47.
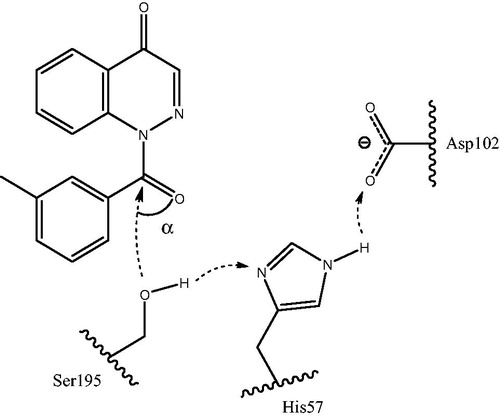
Table 5. Geometric parameters of the enzyme–inhibitor complexes predicted by molecular docking.
Comparison of docking poses for isomeric compounds 16b and 17b showed that 16b is bonded with Gly193 and Ser195, whereas 17b forms additional H-bonds with Asp194 and Val216. All these molecules form H-bonds with participation of the carbonyl oxygen atom in the 3-methylbenzoyl fragment, further supporting the importance of this molecular moiety for the inhibitory activity. The main moieties of molecules 16b and 17b are oriented differently within the binding site (, S5, and S6). However, carbonyl functional groups of both isomers are located in the same area near Ser195 in positions favorable for ligand–receptor interaction specific for serine proteases. Note that 17b interacts with Ser195 by an ester carbonyl group and is anchored by H-bonds between Val216 and two endocyclic nitrogen atoms.
Conclusions
These results confirm that the transformation of the indazole into the cinnoline nucleus though the enlargement of the pyrazole ring leads to compounds that retain some HNE inhibitory activity, but with lower potency and with different structure–activity relationships as compared with N-benzoylindazoles. Compounds 18a and 18e were the most potent, with Ki values of 75 and 110 nM, respectively. Analysis of reaction kinetics revealed that these cinnoline derivatives are reversible competitive inhibitors of HNE. Studies of chemical stability, carried out on selected active compounds, showed that this series is more stable than the N-benzoylindazoles, with 18f (IC50 value = 0.2 μM) being the most stable (t1/2 = 233 min). Finally, molecular modeling studies show that we synthesized two different types of HNE inhibitors: (1) compounds with a cinnolin-4(1H)-one scaffold, where Ser195-OH attacks the N1 C=O and (2) cinnoline derivatives bearing an ester function at C-4, which is the attack point of Ser195-OH. Both types of the nucleophilic attack of Ser195 on a carbonyl carbon atom can be accompanied by proton transfer from the serine hydroxyl group via His57 to Asp102. Our results indicate that the geometric features of docking poses and orientations of side chains for the catalytic triad (Ser195, His57, and Asp102) are favorable for accomplishment of this mechanism. Thus, the novel HNE inhibitors reported here represent potential starting points for future manipulation and optimization.
Supplementary material available online
Supplementary material, Figures S1-S6.
IENZ_1057718_Supp.pdf
Download PDF (403.9 KB)Declaration of interest
This work was supported in part by NIH IDeA Program COBRE Grant GM110732 (MTQ) and a USDA National Institute of Food and Agriculture Hatch project, and the Montana State University Agricultural Experiment Station.
References
- Korkmaz B, Moreau T, Gauthier F. Neutrophil elastase, proteinase 3 and cathepsin G: physicochemical properties, activity and physiopathological functions. Biochimie 2008;90:227–42
- Korkmaz B, Horwitz MS, Jenne DE, Gauthier F. Neutrophil Elastase, Proteinase 3, and Cathepsin G as therapeutic targets in human diseases. Pharmacol Rev 2010;62:726–59
- Bode W, Meyer E, Powers JC. Human leukocyte and porcine pancreatic elastase: X-ray crystal structures, mechanism, suelaabstrate specificity, and mechanism-based inhibitors. Biochemistry 1989;28:1951–63
- Chua F, Laurent GJ. Neutrophil elastase: mediator of extracellular matrix destruction and accumulation. Proc Am Thorac Soc 2006;3:424–7
- Geraghty P, Rogan MP, Greene CM, et al. Neutrophil elastase up regulates cathepsin B and matrix metalloproteinase-2 expression. J Immunol 2007;178:5871–8
- Bergin DA, Greene CM, Sterchi EE, et al. Activation of the epidermal growth factor receptor (EGFR) by a novel metalloprotease pathway. J Biol Chem 2008;283:31736–44
- Tremblay GM, Janelle MF, Bourbonnais Y. Anti-inflammatory activity of neutrophil elastase inhibitors. Current Opin Investig Drugs 2003;4:556–65
- Heutinck KM, ten Berge IJ, Hack CE, et al. Serine proteases of the human system in health and disease. Mol Immunol 2010;47:1943–55
- O’Donnell R, Peebles A, Ward C, et al. Relationship between peripheral airway dysfunction, airway obstruction and neutrophil inflammation in COPD. Thorax 2004;59:837–42
- Voynow JA, Fisher BM, Zheng S. Proteases and cystic fibrosis. Int J Biochem Cell B 2008;40:1238–45
- Gifford AM, Chalmers JD. The role of neutrophils in cystic fibrosis. Curr Opin Hematol 2014;21:16–22
- Lee WL, Downey GP. Leukocyte elastase: physiological functions and role in acute lung injury. Am J Respir Crit Care Med 2001;164:896–904
- Tsushima K, King LS, Aggarwal NR, et al. Acute lung injury. Intern Med 2009;48:621–30
- Meyer-Hoffert U, Wingertszahn J, Wiedow O. Human leukocyte elastase induces keratinocyte proliferation by epidermal growth factor receptor activation. J Invest Dermatol 2004;123:338–45
- Hilbert N, Schiller J, Arnhold J, Arnold K. Cartilage degradation by stimulated human neutrophils: elastase is mainly responsible for cartilage damage. Bioorg Chem 2002;30:119–32
- Henriksen PA, Sallenave JM. Human neutrophil elastase: mediator target in atherosclerosis. In J Biochem Cell B 2008;40:1095–100
- Sato T, Takahashi S, Mizumoto, T, et al. Neutrophil elastase and cancer. Surg Oncol 2006;15:217–22
- Moroy G, Alix AJ, Sapi J, et al. Neutrophil elastase as a target in lung cancer. Anticancer Agents Med Chem 2012;12:565–79
- Semple BD, Trivedi A, Gimlin K, Noble-Haeusslein LJ. Neutrophil elastase mediates acute pathogenesis and is a determinant of long-term behavioral recovery after traumatic injury to the immature brain. Neurobiol Dis 2015;74:263–80
- Groutas WC, Dou D, Alliston KR. Neutrophil elastase inhibitors. Expert Opin Ther Pat 2011;21:339–54
- Henriksen PA. The potential of neutrophil elastase inhibitors as anti-inflammatory therapies. Curr Opin Hematol 2014;21:23–8
- Sjö P. Neutrophil elastase inhibitors: recent advances in the development of mechanism based and nonelectrophilic inhibitors. Fut Med Chem 2012;4:651–60
- Bayer Corp: Prolastin. Company World Wide Web Site. Available from: http://www.bayerdirect.com/pro.htm/ [last accessed 24 Mar 2002]
- Kawata K, Suzuki M, Sugitani M, et al. ONO-5046, a novel inhibitors of human neutrophil elastase. Biochem Biophys Res Commun 1991;177:814–20
- Iwata K, Doi A, Ohji G, et al. Effect of neutrophil elastase inhibitors (Sivelestat sodium) in the treatment of acute lung injury (ALI) and acute respiratory distress (ARDS): a systematic review and meta-analysis. Intern Med 2010;49:2423–32
- Stockley R, De Soyza A, Gunawardena K, et al. Phase II study of a neutrophil elastase inhibitor (AZD9668) in patients with bronchiectasis. Respir Med 2013;107:524–33
- Crocetti L, Giovannoni MP, Schepetkin IA, et al. Design, synthesis and evaluation of N-benzoylindazole derivatives and analogues as inhibitors of human neutrophil elastase. Bioorg Med Chem 2011;19:4460–72
- Crocetti L, Schepetkin IA, Cilibrizzi A, et al. Optimization of N-benzoylindazole derivatives as inhibitors of human neutrophil elastase. J Med Chem 2013;56:6259–72
- Nan G, Ren F, Luo M. Suzuki-Miyaura cross-coupling reaction of 1-aryl-triazenes with arylboronic acids catalyzed by a recyclable polymer-supported N-heterocyclic carbine-palladium complex catalyst. Beilstein J Org Chem 2010;6:1–6
- Muthupplaniappan M, Margal S, Sukeerthi K, et al. Novel cannabinoid receptor ligands, pharmaceutical compositions containing them, and process for their preparation. PTC Int Appl 2009. WO 2009053799 A1 20090430
- Qiuping D, Wu J. A facile route to 2,4-Dihydro-1H-benzo[d][1,3]thiazines via silver-catalyzed tandem addition-cyclization reaction. J Comb Chem 2008;10:541–5
- Chakraborty A, Sinha S. Synthesis of 3-[2-(1,3-butadienyl)]-1H-indoles en route to murrapanine analogue. Tetrahedron Lett 2011;52:6635–8
- Dey R, Ranu BC. A convenient and efficient protocol for the synthesis of 4(1H)-cinnolones, 1,4-dihydrocinnolines, and cinnolines in aqueous medium: application for detection of nitrite ions. Tetrahedron 2011;67:8918–24
- Haider N, Holzer W. Product class 9: cinnolines. Sci Synth 2004;16:251–313
- Glówka ML, Iwanicka I, Olczak AJ. Crystal structure communications. Acta Crystallogr C 1994;C50:1770–2
- Woods KW, Fischer JP, Claiborne A, et al. Synthesis and SAR of indazole-pyridine based protein kinase B/Akt inhibitors. Bioorg Med Chem 2006;14:6832–46
- Leonard NJ, Boyd SN. Cinnolines. 2. Synthesis of 4-hydroxycinnolines. J Org Chem 1946;11:419–28
- Tong Y, Penning TD, Florjanuc A, et al. Preparation of substituted fused tricycles as inhibitors of kinase for treating cancer. U.S. Pat Appl Publ 2012. US 20120220572 A1 20120830
- Schepetkin IA, Khlebnikov AI, Quinn MT. N-Benzoylpyrazoles are novel small-molecule inhibitors of human neutrophil elastase. J Med Chem 2007;50:4928–38
- Groutas WC, Kuang R, Venkataraman R, et al. Structure-based design of a general class of mechanism-based inhibitors of the serine proteinases employing a novel amino acid-derived heterocyclic scaffold. Biochemistry 1997;36:4739–50
- Forist AA, Weber DJ. Kinetics of hydrolysis of hypoglycemic 1-acyl 3,5-dimethylpyrazoles. J Pharm Sci 1973;62:318–19
- Navia MA, McKeever BM, Springer JP, et al. Structure of human neutrophil elastase in complex with a peptide chloromethyl ketone inhibitor at 1.84 Å resolution. Proc Natl Acad Sci USA 1989;86:7–11
- Peters MB, Merz KM. Semiempirical comparative binding energy analysis (SE-COMBINE) of a series of trypsin inhibitors. J Chem Theory Comput 2006;2:383–99
- Holzer W, Eller GA, Schönberger S. On the tautomerism of cinnolin-4-ol, cinnoline-4-thiol, and cinnolin-4-amine. Heterocycles 2008;75:77–86
- Stanovnik B, Tišler M, Katritzky AR, Denisko OV. The tautomerism of heterocycles: substituent tautomerism of six-membered ring heterocycles. Adv Heterocycl Chem 2006;91:1–134
- Bhattacharya S, Sengupta S. Palladium catalyzed alkynylation of aryl halides (Sonogoshira reaction) in water. Tetrahedron Lett 2004;45:8733–6
- Vergely I, Laugaa P, Reboud-Ravaux M. Interaction of human leukocyte elastase with a N-aryl azetidinone suicide substrate: conformational analyses based on the mechanism of action of serine proteinases. J Mol Graphics 1996;14:158–67