
Abstract
d-Serine is the co-agonist of NMDA receptors and binds to the so-called glycine site. d-Serine is synthesized by human serine racemase (SR). Over activation of NMDA receptors is involved in many neurodegenerative diseases and, therefore, the inhibition of SR might represent a novel strategy for the treatment of these pathologies. SR is a very difficult target, with only few compounds so far identified exhibiting weak inhibitory activity. This study was aimed at the identification of novel SR inhibitor by mimicking malonic acid, the best-known SR inhibitor, with a cyclopropane scaffold. We developed, synthesized, and tested a series of cyclopropane dicarboxylic acid derivatives, complementing the synthetic effort with molecular docking. We identified few compounds that bind SR in high micromolar range with a lack of significant correlation between experimental and predicted binding affinities. The thorough analysis of the results can be exploited for the development of more potent SR inhibitors.
Introduction
In the last 20 years, d-amino acids have been recognized to play functional roles in mammals, including humansCitation1,Citation2, and enzymes involved in their biosynthesis and metabolism are now emerging as interesting drug targetsCitation3,Citation4. Among the others, d-serine has drawn the attention of researchers since its role as N-methyl-d-aspartate receptor (NMDAR) co-agonist was demonstratedCitation5,Citation6. In mammals, d-serine is synthesized by serine racemase (SR), which catalyzes the conversion of l-serine into its enantiomerCitation7. NMDAR hyperfunction and dysregulation are associated with neuropathologies, such as Alzheimer's disease, amyotrophic lateral sclerosis, and neuropathic pain. So far, only a very small number of NMDAR modulators reached the clinical use with associated severe adverse effectsCitation8. Therefore, SR has emerged as new potential target for the treatment of neurological disordersCitation9–11. SR is a pyridoxal 5′-phosphate (PLP)-dependent enzyme, existing as a homodimer in the cytosol of mammalian cells. It belongs to the fold type-II family of PLP-dependent enzymes, characterized by two domains, a large one and a small oneCitation12–14. It is mainly expressed in the forebrain regions but it can also be found in hippocampus and other cortical regions as well as in the peripheral nervous systemCitation7,Citation15,Citation16. SR has also been detected in other tissues outside the nervous system, such as liver, kidneyCitation17 and skinCitation18. In addition to the reversible serine racemization, SR catalyzes the deamination of both l-serine and d-serine to pyruvate and ammonia through an α,β-elimination reactionCitation19. In cells transfected with SR the pyruvate/d-serine ratio was found to be around 4, thus indicating that the α,β-elimination reaction might play a physiological role in the control of d-serine concentrationCitation15. Crystal structures of eukaryotic SR were solved from different sources, human, ratCitation20 and Schizosaccharomyces PombeCitation21,Citation22, in holo form or in complex with the inhibitor malonate (). Despite the availability of structural data and the identification of a few inhibitorsCitation23–26, the development of specific and more potent SR inhibitors remains very challengingCitation13. While this work was in preparation, three papers aiming at the identification of novel SR inhibitors were published. Harty et alCitation26. designed substrate analogues as potential inhibitors, Vorlova et al.Citation25 synthesized a series of malonate analogues, and Mori et al.Citation24 identified novel potential inhibitors of SR by in silico ligand-based screening. All the compounds reported in these works exhibit very low inhibitory potencies, with inhibition constants in the high micromolar to millimolar range, with the only exception of 2,2-dichloromalonate, characterized by a Ki of 57 μM ()Citation25.
Figure 1. Malonate (1), 2-methylmalonate (2), 2,2-dichloromalonate (3), 1,2-cyclopropanedicarboxylate (4).
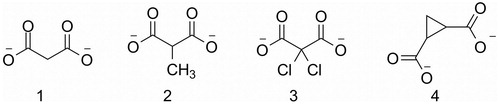
Inspired by SR inhibitors based on the structure of malonateCitation23, we investigated the possibility of developing a new series of potential SR inhibitors based on the cyclopropane scaffold. The rationale for our choice was the overall similarity between cis 1,2-cyclopropanedicarboxylic acid and malonic acid. As can be noticed in , a three-dimensional superposition of the two molecules shows how the cyclopropane ring allows the right orientation of carboxylic groups to superimpose with those of malonate. Moreover, given the apparent difficulty to identify potent inhibitors, we reasoned that low molecular weight compounds may increase the probability to find reasonable hits, which can then be further optimized without stepping away from developability metrics such as the Lipinski rule of fiveCitation27.
Figure 2. Superposition of malonate (cyan) and cis-1,2-cyclopropanedicarboxylate (green).
In the present study, also on the basis of our expertise in the synthesis of cyclopropane derivativesCitation28,Citation29, we present our efforts to develop a new series of non-covalent competitive SR inhibitors based on the cyclopropane scaffold (), together with synthetic routes, chemical characterization, experimental tests, and a rational explanation for the observed results.
Table 1. Compounds synthesized and tested for SR inhibitory activity.
Methods
Synthetic chemistry
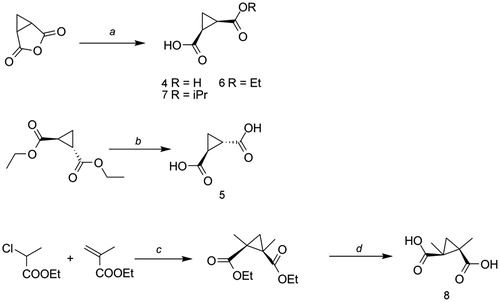
The synthesis of cis-cyclopropane dicarboxylic acid 4 and the alkyl-ester derivatives 6 and 7 started from commercially available 3-oxabicyclo[3.1.0]hexane-2,4-dione, which was hydrolyzed either in water at room temperature (4), or ethyl alcohol (6) and isopropyl alcohol (7) at reflux, affording the title compounds in good overall yields (). The hydrolysis of diethyl (1R,2R)-cyclopropane-1,2-dicarboxylate in the presence of an excess of potassium hydroxide, followed by acidification of the medium with HCl 1N, led to the synthesis of the trans-1,2-cyclopropanedicarboxylic acid 5. Finally, the synthesis of 8 was achieved through basic hydrolysis of intermediate 1,2-diethyl-1,2-cyclopropanedicarboxylate, obtained according to the reported McCoy procedureCitation30,Citation31, reacting the corresponding methylacrylate and the α-chloro ester. Detailed experimental procedures are available in the Supporting information.
Biochemical assays
The six selected compounds were assayed in vitro on recombinant human SR. The affinity of compounds was measured by spectrofluorimetry, exploiting the fluorescence emission properties of the cofactor at 500 nm upon excitation at 412 nm using a Spex Fluoromax-2 fluorimeter (HORIBA Jobin Yvon, Kyoto, Japan)Citation11. Briefly, a solution containing 2.5 μM SR, expressed in E. coli and purified as previously describedCitation32 in 50 mM TEA buffer pH 8, 10% DMSO, was titrated by addition of ligand at 20 °C. The dependence of the fluorescence emission intensity of the cofactor on ligand concentration was fitted by a binding isotherm to calculate the dissociation constant (Kd; ). For competitive inhibitors, Kd calculated with this method is equal to Ki. The IC50 value of selected compounds was measured by enzymatic assays. Since racemization and elimination reactions are known to share the same enzymatic active site, it is possible to study SR activity by analyzing either reaction. In this case, the β-eliminase activity of hSR in the presence of selected inhibitors was measured using a coupled assay with lactate dehydrogenase. The reaction kinetics was measured at 37 °C following the disappearance of NADH at 340 nm using a Varian CARY400 spectrophotometer (Agilent Technologies, Santa Clara, CA). The reaction mixture contained 0.4 μM hSR in 50 mM TEA, pH 8, 5–10 % DMSO, and a concentration of l-Ser equal to the KM of the enzyme in the absence of DMSO. IC50 value was calculated, for a competitive inhibition mechanism as
(1)
Table 2. Available SR crystal structures: structural information and water molecules solved in the binding site.
Molecular modeling
Docking studies were carried out using a conformational ensemble of hSR formed by five enzyme conformations. The closed conformation is represented by crystal structure 3L6B; three partially closed conformations, namely the half-closed, intermediate, and half-open, were extracted from a Targeted MD simulation previously performed by usCitation33. Finally, the open hSR conformation was built by comparative modeling using the rat SR crystal structure in its open conformation as a template (PDB code 3HMK, model previously reported by us)Citation33. Ligands were prepared using LigPrep at pH 7 ± 1Citation34. The Protein Preparation Workflow was used to add hydrogen atoms, assign bond orders, optimize the hydrogen-bonding network and, finally, to refine the protein structures with a maximum RMSD of tolerance of 0.3 ÅCitation35. At first, water molecules were removed from all the enzyme conformations. Then, 3L6B was also prepared for docking studies retaining structural water molecules W403, W372, and W373. All docking studies were performed with three different software: Glide SP 6.1 from Schrödinger suiteCitation36,Citation37, Autodock4.2Citation38, and PlantsCitation39. For all the three software and for all the enzyme conformations used, the binding site grid box was centred on the centroid of the following residues: PLP, Ser84, Ser85, Arg135, Asn154, Ser242, and Thr285 (3L6B numbering). For the docking studies using 3L6B retaining the conserved water molecules, we applied the same protocol reported by Vorlová et al., with the grid centred on malonateCitation25.
Results
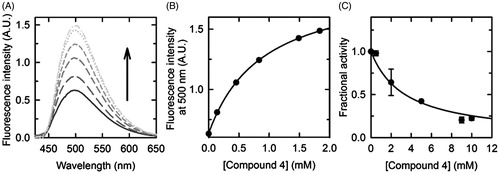
The main goal of our study was the identification of a new series of SR inhibitors based on the cyclopropane scaffold given its 3D similarity with malonate, the most well-known serine racemase inhibitor. We prepared and tested as potential SR inhibitors the cyclopropane derivatives reported in . Compound 4 was synthesized on the basis of the similarity with malonate and compound 5, trans 1,2-cyclopropanedicarboxylic acid, was synthesized to probe the effect of a trans-configuration of the two carboxylate moieties, also on the basis of the mild inhibition observed for fumaric acidCitation23. With compounds 6 and 7, we aimed at exploring the effect of the esterification of one of the two carboxylate moieties, while with 8 we explored the effect of a further functionalization of the cyclopropane ring. Finally, compound 9 was purchased from Sigma-Aldrich (St. Louis, MO) as a constrained analogue of cinnamic acid, previously reported to be a fairly good inhibitor of SRCitation23. The six compounds were assayed in vitro on recombinant human SR. To our surprise, all the cyclopropane derivatives resulted to be much less potent than malonate. The most active compound was compound 4 which showed a Kd of 900 μM and an IC50 value around 3 mM (), while all the other derivatives had a Kd higher than 2.5 mM.
Figure 3. Dissociation constant and half-maximal inhibitory concentration of compound 4. (A) Fluorescence emission spectra, upon excitation at 412 nm, of a solution containing 2.4 μM hSR, 50 mM TEA, 150 mM NaCl, 5 mM TCEP, 1 mM MgCl2, 10% DMSO (v/v), pH 8.0, and increasing concentrations of compound 4, at 20 °C. (B) Dependence on the concentration of compound 4 of the fluorescence emission intensity at 500 nm upon excitation at 412 nm. The experimental points were fitted by a binding isotherm, yielding a KD of 0.90 ± 0.03 mM. (C) Dependence on the concentration of compound 4 of hSR β-elimination activity, at 37 °C. Experiments were carried out in an assay solution containing 0.4 μM hSR, 50 mM TEA, 150 mM NaCl, 36 mM l-Ser, 5 mM DTT, 1 mM MgCl2, 50 μM PLP, 2 mM ATP, 10% DMSO (v/v), pH 8.0, and variable concentrations of compound 4. Experimental data were fitted to the Equation (1), yielding an IC50 value of 3.3 ± 0.4 mM.
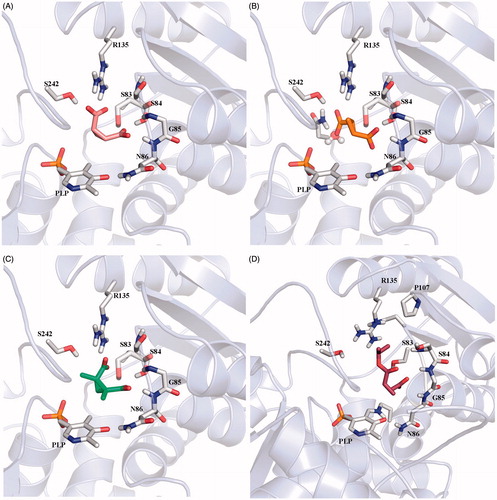
To understand the reason behind these poor results, we decided to investigate the interaction between the enzyme and the cyclopropane derivatives at molecular level by computational tools. Docking studies were carried out for the compounds 4–8 to verify whether they could be accommodated in the SR binding pocket. Crystal structures of SR have been recently solvedCitation20–22 and the comparison between the holo SR structures and those in the presence of malonate revealed that the binding of the inhibitor triggers a transition from an open to a closed conformation. For this reason, in a previous work by us, SR conformational flexibility was investigated, identifying a few intermediate conformations that were included in our docking studies together with the open and closed conformationsCitation33. As we expected from the structural similarity with malonate, compound 4 assumed a binding mode very similar to that of malonate in crystal structure 3L6B, in which the enzyme assumes a closed conformation, establishing the same hydrogen bonds with Ser83, Ser84, Asn86, Arg135, and Ser242 (, numbering from 3L6B crystal structure). Docking results for compound 5, which resulted completely inactive in the experimental assays, displayed a different orientation of the ligand in the SR-binding site. Due to the trans orientation of the carboxylic groups, this compound cannot form a bi-dentate interaction with Arg135 but seems able to establish a mono-dentate interaction with Arg135 and an H-bond with Asn154. Nevertheless, according to the docking results, it seemed it could be equally well accommodated in the binding pocket ( and ). Also compound 8, experimentally inactive, was predicted by docking to bind fairly well to SR, with an affinity only slightly lower than malonate ( and ). For compounds 6 and 7, the two monoester derivatives of 4, no docking poses were found when the closed enzyme conformation was used. These compounds were predicted to bind to the intermediate and open enzyme conformations, but with worse docking scores in comparison to malonate ().
Figure 4. (A) Docking pose of compound 4; (B) binding mode of compound 5: the carboxylic group is located in proximity of a water molecule, that was omitted during docking simulation; (C) pose of compound 8; (D) docking pose of compound 7.
This almost complete lack of agreement between docking and experimental results deserved more investigation for a better exploitation of the available crystal structures of SR on route towards new potent inhibitors. In this respect, we drew inspiration from the work by Vorlová et al.Citation25 and looked for the presence of water molecules in the crystal structures of SR which were not considered in the first docking experiments. In particular, in their work, they were quite successful in discerning qualitatively between good and weak binders. They undertook a thorough analysis of the binding site hydration using WaterMap, then performed docking studies with Glide and finally quantum mechanical refinement of the binding poses. The results of the WaterMap analysis are completely in agreement with what can be inferred just analyzing the B factors of the water molecules in the different crystal structures. Moreover, we decided to not perform any quantum mechanical refinement since it demonstrated to be little informative in their work. We identified a water molecule (W403, 3L6B crystal structure numbering for all water molecules) that was not taken into account in our initial docking study, which is located approximately where one of the carboxylic groups of compound 5, the one interacting with Asn154, is accommodated in its best docking pose. This water molecule should be considered as a structural water because it is conserved in almost all the available crystal structures stored in the PDB (also in the open enzyme form, establishing H bonds with Asn154 and the phosphate group of PLP, see ). We repeated our docking experiment in its presence, and no productive poses for compound 5 were observed. As also pointed out by Vorlová et al.Citation25, the active site of SR X-ray crystal structures is indeed filled with ordered water molecules, suggesting a possible structural role for at least some of them. For example, W355 (3L6B numbering scheme) is present in all the crystal structures (, W8) and it can be speculated having an important role in the stabilization of the phosphate group of PLP cofactor. Additional well-conserved water molecules, present in almost all the crystal structures so far available, are W372, W373, W403, and W436, that form a dense network of H-bonds with the surrounding residues and malonate (, 3L6B numbering scheme).
Figure 5. Conserved water molecules in the closed structure of SR. (A) Crystal structure of 3L6B; malonate is depicted in sticks and lemon green, water molecules are reported in ball and sticks. (B) Hbond network between malonate, water molecules and protein residues in the active site of 3L6B crystal structure.
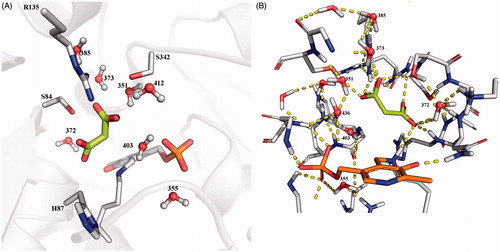
Therefore, docking experiments were repeated using 3L6B crystal structure with structural water molecules W372, W373, and W403 explicitly considered, applying the same docking settings as in the studies by Vorlová et al.Citation25. Unfortunately, the results did not help us to finally have a full understanding of the situation. In fact, given the applied van der Waals radius scaling factor of 0.5 for atoms with partial atomic charge less than 0.15 in their proposed experimental settings, reasonable poses for all the inactive derivatives were obtained. On the contrary, if the scaling factor is not applied, poses are retrieved only for compounds 4 and 5, with the last one adopting again an orientation slightly different from malonate and compound 4. Therefore, we can assume that the inactivity of compound 5 is possibly due to an inefficient binding mode. However, poses and docking scores comparable if not better than malonate or 2,2-dichloromalonate were still found for compounds 4 and 8, regardless of the application of the scaling factor (). To verify that the good docking poses and scores obtained were not just due to the algorithm and the scoring function of the selected docking software, we also performed our analysis using other two docking software, namely AutoDock and PLANTS, trying all the different conditions applied so far, however obtaining the same results ().
Table 3. Docking experiments performed using the same conditions reported in the paper by Vorlova et al. performed using Glide, PLANTS, and AutoDock.
Discussion
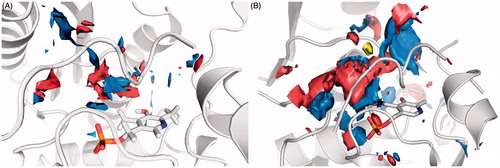
In this work, we presented our efforts for the development of novel SR inhibitors. Even if the results are apparently discouraging, they are in line with data published so far, confirming that targeting SR is a strenuous challenge. Besides malonate, the most effective known inhibitors presenting inhibition constants in the micromolar range are the unselective l-erithrohydroxyaspartate and 2,2-dichloromalonateCitation40,Citation41. Other research groups tried to develop SR inhibitors starting from substrate analogues, yet with results similar to oursCitation40,Citation42. A different approach was applied by Dixon et al.Citation43 who screened a small library of peptides for SR inhibitory activity. He found two short peptides behaving as slow-binding inhibitors of SR, with Ki values in the high micromolar range. One of these peptides, that reasonably target the open conformation of SR, has been used as a template for a ligand-based virtual screening to identify novel inhibitors by Mori et al.Citation24 but again the results are not particularly encouraging, also in the light of the unusually high IC50 value measured for malonate in their study. We decided to develop a novel series of potential SR inhibitors using a scaffold which resembles the 3D conformation of malonate. However, the results are substantially different from those reported for malonate. To understand the experimental behavior of the cyclopropane derivatives, we decided to analyze thoroughly the SR binding site. As recently pointed out by Vorlová et al.Citation25, the active site of SR is highly polar, as suggested by the presence of many polar residues and highly conserved water molecules. Indeed, both the closed and open conformations of SR lack appreciable hydrophobic area in the active site ().
Figure 6. Surface representation of the binding site features map: in blue are reported the Hbond donor areas, in red the Hbond acceptor areas and in yellow the hydrophobic areas. (A) 3L6B crystal structure (closed enzyme conformation). (B) Human model built on the rat 3HMK crystal structure (open enzyme conformation).
Scheme 1. Reagents and conditions A: (a) for 4, H2O, 2 h at reflux; for 6, EtOH, 2 h at reflux; for 7, iPrOH, 2 h at reflux; (b) 1.KOH, reflux, 2 h; 2.HCl 1N; (c) NaH, toluene, 0 °C to RT, 48–72 h; (d) KOH, THF/water (1:2) from 0 °C to 50 °C, 4 d. Yields and purification methods are reported in the Supplementary information.
As analyzed before, structural water molecules conserved among all the crystal structures solved until now, regardless of the species and of the form of the enzyme (apo or inhibitor bound), are W355, W372, W373, W403, and W436, accordingly with the 3L6B crystal structure numbering, and they contribute to establish a dense network of H-bonds all around the catalytic site (). Their effective role and the importance they could assume for enzyme function have not been investigated yet and it is beyond the scope of this paper. However, it can be speculated that they could have a key role in the stabilization of the carbanion intermediate, cooperating with the polar active site residues, as was demonstrated in alanine racemaseCitation44,Citation45. It can be postulated that this well-ordered network of water molecules together with the polar features of the residues defining the binding pocket creates a completely hostile environment for the presence of even small hydrophobic moiety in the ligand. In this scenario, even the additional methylene groups of the cyclopropane moiety with respect to malonate are sufficient to prevent an effective pairing of hydrophobic/hydrophilic surfaces and, hence, a productive binding. Retrospectively, this is confirmed by the lack of activity of other malonate derivatives. For instance, the addition of a single methyl group to malonate has a detrimental effect on the activity, giving a compound with 30-fold decreased activityCitation25, despite an almost perfect docking into the binding site. To compare our results with those previously published and to prove the detrimental effects of hydrophobic contributions for SR inhibitors, we purchased and tested cinnamic acid, that was reported to act as a moderate SR inhibitorCitation23, with an affinity for the enzyme higher than that of l-serine. Not surprisingly, it turned out to be completely inactive under our test conditions.
Conclusions
With this work, in the attempt to discover new SR inhibitors, we tested and docked several cyclopropane derivatives. The analysis of the results and the lack of agreement between computational and experimental data provide valuable hints for future studies. In fact, to design SR inhibitors, we highlighted the need of taking into account the extremely high polarity of the binding site and not to neglect the role of the structural water molecules, probably key players in the catalytic reactions and thus not displaceable by inhibitors. The reason why all the docking software employed in this study failed in recognizing this strong requirement for polar substituents and all yielded excellent docking scores for inactive compounds is not apparent, but may constitute a serious drawback for the further identification of SR inhibitors by virtual screening and molecular docking.
Supplementary material available online
IENZ_1057720_Supp.pdf
Download PDF (52.3 KB)Declaration of interest
The authors report no declarations of interest. G. C. acknowledges the Italian Ministry of University and Research (MIUR) for financial support (PRIN2010-11).
References
- Hashimoto A, Nishikawa T, Hayashi T, et al. The presence of free d-serine in rat brain. FEBS Lett 1992;296:33–6
- Hashimoto A, Oka T. Free d-aspartate and d-serine in the mammalian brain and periphery. Prog Neurobiol 1997;52:325–53
- Soglia JR, Contillo LG, Kalgutkar AS, et al. A semiquantitative method for the determination of reactive metabolite conjugate levels in vitro utilizing liquid chromatography – tandem mass spectrometry and novel quaternary ammonium glutathione analogues. Chem Res Toxicol 2006;19:480–90
- Conti P, Tamborini L, Pinto A, et al. Drug discovery targeting amino acid racemases. Chem Rev 2011;111:6919–46
- Matsui TA, Sekiguchi M, Hashimoto A, et al. Functional comparison of d-serine and glycine in rodents: the effect on cloned NMDA receptors and the extracellular concentration. J Neurochem 1995;65:454–8
- Mothet JP, Parent AT, Wolosker H, et al. d-serine is an endogenous ligand for the glycine site of the N-methyl-d-aspartate receptor. Proc Natl Acad Sci 2000;97:4926–31
- Wolosker H, Blackshaw S, Snyder SH. Serine racemase: a glial enzyme synthesizing d-serine to regulate glutamate-N-methyl-d-aspartate neurotransmission. Proc Natl Acad Sci 1999;96:13409–14
- Santangelo RM, Acker TM, Zimmerman SS, et al. Novel NMDA receptor modulators: an update. Expert Opin Thera Pat 2012;22:1337–52
- Van-Horn MR, Sild M, Ruthazer ES. d-Serine as a gliotransmitter and its roles in brain development and disease. Front Cell Neurosci 2013;7:39. doi: 10.3389/fncel.2013.00039
- Amadasi A, Bertoldi M, Contestabile R, et al. Pyridoxal 5′-phosphate enzymes as targets for therapeutic agents. Curr Med Chem 2007;14:1291–324
- Marchetti M, Bruno S, Campanini B, et al. Regulation of human serine racemase activity and dynamics by halides, ATP and malonate. Amino Acids 2015;47:163–73
- Wolosker H. NMDA receptor regulation by d-serine: new findings and perspectives. Mol Neurobiol 2007;36:152–64
- Campanini B, Spyrakis F, Peracchi A, Mozzarelli A. Serine racemase: a key player in neuron activity and in neuropathologies. Front Biosci 2013;18:1112–28
- Campanini B, Pieroni M, Raboni S, et al. Inhibitors of the sulfur assimilation pathway in bacterial pathogens as enhancers of antibiotic therapy. Curr Med Chem 2015;22:187–213
- De Miranda J, Panizzutti R, Foltyn VN, Wolosker H. Cofactors of serine racemase that physiologically stimulate the synthesis of the N-methyl-d-aspartate (NMDA) receptor coagonist d-serine. Proc Natl Acad Sci 2002;99:14542–7
- Verrall L, Walker M, Rawlings N, et al. d-Amino acid oxidase and serine racemase in human brain: normal distribution and altered expression in schizophrenia. Eur J Neurosci 2007;26:1657–69
- Xia M, Liu Y, Figueroa DJ, et al. Characterization and localization of a human serine racemase. Mol Brain Res 2004;125:96–104
- Wolosker H, Mori H. Serine racemase: an unconventional enzyme for an unconventional transmitter. Amino Acids 2012;43:1895–904
- Stříšovský K, Jirásková J, Bařinka C, et al. Mouse brain serine racemase catalyzes specific elimination of l-serine to pyruvate. FEBS Lett 2003;535:44–8
- Smith MA, Mack V, Ebneth A, et al. The structure of mammalian serine racemase: evidence for conformational changes upon inhibitor binding. J Biol Chem 2010;285:12873–81
- Goto M, Yamaouchi T, Kamiya N, et al. Crystal structure of a homolog of mammalian serine racemase from Schizosaccharomyces pombe. J Biol Chem 2009;284:25944–52
- Yamauchi T, Goto M, Wu HY, et al. Serine racemase with catalytically active lysinoalanyl residue. J Biochem 2009;145:421–4
- Jirásková-Vaníčková J, Ettrich R, Vorlová B, et al. Inhibition of human serine racemase, an emerging target for medicinal chemistry. Curr Drug Targets 2011;12:1037–55
- Mori H, Wada R, Li J, et al. In silico and pharmacological screenings identify novel serine racemase inhibitors. Bioorg Med Chem Lett 2014;24:3732–5
- Vorlová B, Nachtigallová D, Jirásková-Vaníčková J, et al. Malonate-based inhibitors of mammalian serine racemase: kinetic characterization and structure-based computational study. Eur J Med Chem 2015;89:189–97
- Harty M, Nagar M, Atkinson L, et al. Inhibition of serine and proline racemases by substrate-product analogues. Bioorg Med Chem Lett 2014;24:390–3
- Oprea TI, Davis AM, Teague SJ, Leeson PD. Is there a difference between leads and drugs? A historical perspective. J Chem Inf Comput Sci 2001;41:1308–15
- Amori L, Katkevica S, Bruno A, et al. Design and synthesis of trans-2-substituted-cyclopropane-1-carboxylic acids as the first non-natural small molecule inhibitors of O-acetylserine sulfhydrylase. MedChemCommun 2012;3:1111–16
- Pieroni M, Annunziato G, Azzali E, et al. Further insights on the SAR of 1-substituted cyclopropylamine derivatives as inhibitors of histone demethylase KDM1A. Eur J Med Chem 2015;92:377–86
- McCoy LL. Three-membered rings. The preparation of some 1,2-cyclopropanedicarboxylic acids. J Am Chem Soc 1959;80:6568–72
- Zhao Q, Wong HNC. Synthetic studies toward plakortide E: application of the Feldman oxygenation to synthesis of highly substituted 1,2-dioxolanes. Tetrahedron 2007;63:6296–305
- Marchetti M, Bruno S, Campanini B, et al. ATP binding to human serine racemase is cooperative and modulated by glycine. FEBS J 2013;280:5853–63
- Bruno A, Amori L, Costantino G. Addressing the conformational flexibility of serine racemase by combining targeted molecular dynamics, conformational sampling and docking studies. Mol Inf 2011;30:317–28
- LigPrep, version 2.8. New York, NY: Schrödinger, LLC; 2013
- Cleland WW, Frey PA, Gerlt JA. The low barrier hydrogen bond in enzymatic catalysis. J Biol Chem 1998;273:25529–32
- Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 2004;47:1739–49
- Halgren TA, Murphy RB, Friesner RA, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 2004;47:1750–9
- Morris GM, Ruth H, Lindstrom W, et al. Software news and updates AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem 2009;30:2785–91
- Korb O, Stützle T, Exner TE. PLANTS: application of ant colony optimization to structure-based drug design. Lecture Notes Comput Sci. 2006;4150:247–58
- Strisovsky K, Jiráskova J, Mikulová A, et al. Dual substrate and reaction specificity in mouse serine racemase: identification of high-affinity dicarboxylate substrate and inhibitors and analysis of the β-eliminase activity. Biochemistry 2005;44:13091–100
- Hoffman HE, Jirásková J, Cígler P, et al. Hydroxamic acids as a novel family of serine racemase inhibitors: mechanistic analysis reveals different modes of interaction with the pyridoxal-5′-phosphate cofactor. J Med Chem 2009;52:6032–41
- Cook SP, Galve-Roperh I, Del Pozo AM, Rodríguez-Crespo I. Direct calcium binding results in activation of brain serine racemase. J Biol Chem 2002;277:27782–92
- Dixon SM, Li P, Liu R, et al. Slow-binding human serine racemase inhibitors from high-throughput screening of combinatorial libraries. J Med Chem 2006;49:2388–97
- Major DT, Nam K, Gao J. Transition state stabilization and α-amino carbon acidity in alanine racemase. J Am Chem Soc 2006;128:8114–15
- Major DT, Gao J. A combined quantum mechanical and molecular mechanical study of the reaction mechanism and α-amino acidity in alanine racemase. J Am Chem Soc 2006;128:16345–57