
Abstract
The carboxylic acid group (–COOH) present in classical NSAIDs is partly responsible for the gastric toxicity associated with the administration of these drugs. This concept has been extensively proven using NSAID prodrugs. However, the screening of NSAIDs with no carboxylic acid at all has been neglected. The goal of this work was to determine if new NSAID derivatives devoid of acidic moieties would retain the anti-inflammatory activity of the parent compound, without causing gastric toxicity. To test this concept, we replaced the carboxylic acid group in ibuprofen, flurbiprofen, and naproxen with three ammonium moieties. We tested the resulting water-soluble NSAID derivatives for anti-inflammatory and ulcerogenic activity in vitro and in vivo. In this regard, we observed that all non-acidic NSAIDs exerted a potent anti-inflammatory activity, suggesting that the acid group in commercial 2-phenylpropionic acid NSAIDs not be an essential requirement for anti-inflammatory activity. These data provide complementary evidence supporting the discontinuation of ulcerogenic acidic NSAIDs.
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are one of the most extensively used therapeutic agents worldwide. The use of NSAIDs accounts for nearly 5% of all prescribed medicationsCitation1 because of their proven efficacy in a wide variety of inflammatory disordersCitation2–8. The principal pharmacological mechanism of action exerted by NSAIDs is the inhibition of cyclooxygenase (COX)-1 and COX-2 enzymes, which leads to decreased levels of prostaglandins (PGs) and thromboxanes (TXs) systemically. The scientific literature describes COX-1 as a constitutive enzyme involved in the physiological production of PGs. In contrast, COX-2 has been regarded as an inducible enzyme expressed as a response to different stimuli. However, the “traditional” roles attributed to both enzymes are now under scrutiny and constitute a challenging research area. It is now clear that we need to reevaluate the “traditional” roles for each enzyme, not only in inflammation but also in the underlying mechanisms of NSAID-induced toxicityCitation9. Gastrointestinal (GI) erosions and bleeding are two of the most common toxic side-effects associated with the administration of NSAIDs. These effects have been observed even with small prophylactic doses of aspirin (81 mg/d)Citation10. Approximately 50% of patients taking NSAIDs on a long-term basis may develop mucosal damage in the small intestineCitation11.
It has been observed that the GI toxicity of NSAIDs is partly due the presence of a free carboxylic acid groupCitation12. Because of this, the scientific literature describes the design of a high number of NSAID prodrugs [mainly ester and amide derivativesCitation13–15]. However, scientists have neglected the study of drugs entirely devoid of the –COOH moiety. In this regard, we thought of the following two alternatives: (a) to reduce an acid/ester prodrug to an alcohol and (b) to reduce an amide prodrug to an amine. The presence of an amine was more attractive to us considering that we could also form the corresponding polar (and potentially water-soluble) ammonium salt.
Therefore, as part of an ongoing research work aimed to obtain effective and safe anti-inflammatory agents, we report the synthesis and the biological evaluation of nine different ammonium salts derived from naproxen (1), ibuprofen (2), and flurbiprofen (3, Scheme 1). We also conducted docking studies to evaluate and compare the binding interactions exerted by the test compounds on the active site of the COX enzyme.
Scheme 1. Chemical synthesis of naproxen (1), ibuprofen (2), and flurbiprofen (3) derivatives possessing a polar pyrrolidinium (13a, 14a, and 15a), 2 -(hydroxymethyl)pyrrolidinium (13 c, 14 c, and 15 c), or a hydroxyethylammonium chloride moiety (13 b, 14 b, and 15 b). Reagents and conditions: (a) oxalyl chloride, DCM, 25° C, 3–12 h; (b) pyrrolidine or l-proline, DCM, 25 °C, 3 h; (c) 2-hydroxyethylamine, DCM, 25 °C, 3 h; (d) LiAlH4, THF, 25 °C (compounds 10a, 10c, 11a, 11c, 12a, and 12c) or reflux (compounds 10b, 11b, and 12b) for 3 h; (e) dry HCl, ether, N2, 25 °C.
Materials and methods
Chemistry
We determined melting points for solid products using an Electrothermal Mel-Temp® melting point apparatus (Barnstead, Dubuque, IA) and are uncorrected. We recorded 1H- and 13C-NMR spectra on a Bruker AVANCE 600, or a Bruker AM-300 NMR spectrophotometer (Bruker BioSpin Corporation, Billerica, MA). In this paper, we reported all coupling constants (J) in Hertz (Hz), but the corresponding chemical shifts in relative units (δ units, or ppm) using TMS as an internal standard. We recorded mass spectra (ESI-MS) using a Water’s micro mass ZQ-4000 single quadruple mass spectrometer (Waters Corporation, Milford, MA). We determined elemental analyzes (C, H, N) for some products, accepting those samples that were within ± 0.4 of the theoretical values (Microanalytical Service Laboratory, Department of Chemistry, University of Alberta, Edmonton, Alberta, Canada). We observed that compounds 13–15 showed a single spot on RediSep® silica gel glass plates (Axel Semrau GmbH & Co, Sprockhövel, Germany) (UV254, 0.2 mm) when we used a high, medium, and low polarity solvent mixtures. No residue remained after combustion, indicating a purity greater than 95%. We performed column chromatography separations using a CombiFlash Retrieve, or a CombiFlashRf system using RediSepRf silica gel® (40–60 µm) cartridges (Axel Semrau GmbH & Co, Sprockhövel, Germany), or prepacked RediSep Gold® columns (Axel Semrau GmbH & Co, Sprockhövel, Germany). We synthesized the corresponding acid chlorides of naproxen (4), ibuprofen (5), and flurbiprofen (6), following reported literature proceduresCitation16. We purchased all other reagents from Aldrich Chemical Company (Milwaukee, WI) and were used without further purification.
General procedure for the synthesis of amides (7–9)
We added a solution of the corresponding amine (2 eq.) dissolved in dichloromethane (5 mL), to a solution of naproxen acid chloride (4, 1 eq.), ibuprofen acid chloride (5, 1 eq.), or flurbiprofen acid chloride (6, 1 eq.) in dichloromethane (10 mL) at 25 °C. In all cases, we monitored the reaction progress by TLC while stirring at room temperature (about 25 °C). We detected the formation of amides in all mixtures within 3 h. Then we filtered the corresponding reaction mixture and evaporated the solvent under vacuum. Finally, we purified the crude products by flash chromatography using a mixture of n-hexane/ethyl acetate (gradient).
2-(6-Methoxynaphthalen-2-yl)-1-(pyrrolidin-1-yl)propan-1-one (7a): 90% yield; m.p. 109–110 °C; 1H-NMR: (CDCl3) δ 7.76–7.65 (m, 3H, naphthyl), 7.43 (dd, J: 8.55 Hz, J: 1.18 Hz, 1H, naphthyl), 7.16–7.12 (m, 2H, naphthyl), 3.92 (s, 3H, CH3O), 3.86 (q, J: 7.32, 1H, CHCH3), 3.46 (broad singlet, 4H, pyrrolidinyl H-2, H-5), 1.73 (broad singlet, 4H, pyrrolidinyl H-2, H-4), 1.51 (d, J: 7.32 Hz, 3H, CH3CH).
N-(2-hydroxyethyl)-2–(6-methoxynaphthalen-2-yl)propanamide (7b): 48% yield; m.p. 81–83 °C; 1H-NMR (CDCl3) δ 7.75–7.68 (m, 3H, naphthyl), 7.37 (d, J: 7.92 Hz, 1H, naphthyl), 7.18–7.13 (m, 2H, naphthyl), 5.85 (broad singlet, 1H, OH), 4.33 (t, 1H, NH), 3.93 (s, 3H, CH3O), 3.74 (q, J: 6.72 Hz, 1H, CHCH3), 3.65 (t, J: 4.26 Hz, 2H, CH2OH), 3.36 (t, J: 4.26 Hz, 2H, CH2NH), 1.62 (d, J: 6.09 Hz, 3H, CHCH3).
1-[2-(6-Methoxynaphthalen-2-yl)-1-oxo-1-propyl]proline (7c): 81% yield; m.p. 156–157 °C; 1H-NMR (CDCl3) δ 7.75–7.61 (m, 3H, naphthyl), 7.35 (dd, J: 8.52 Hz, J: 1.83 Hz, 1H, naphthyl), 7.17–7.11 (m, 2H, naphthyl), 4.71 (m, 1H, CHCOOH), 3.92 (s, 3H, CH3O), 3.88 (q, J: 6.69 Hz, 1H, CHCH3), 3.53 (m, 1H, prolinyl), 3.30 (m, 1H, prolinyl), 2.47 (m, 1H, prolinyl), 1.97–1.84 (m, 3H, prolinyl), 1.54 (d, J: 7.32 Hz, 3H, CH3CH).
2-(4-Isobutylphenyl)-1-(pyrrolidin-1-yl)propan-1-one (8a): 87% yield; 1H-NMR (CDCl3) δ 7.18 (d, J: 7.95 Hz, 2H, phenyl H-2, H-6), 7.07 (d, J: 7.95 Hz, 2H, phenyl H-3, H-5), 3.71 (q, J: 6.72 Hz, 1H, CHCH3), 3.43 (br, 4H, pyrrolidinyl H-2, H-5), 2.43 (d, J: 7.32 Hz, 2H, CH2Ar), 1.86 (m, 5H, CH(CH3)2 and pyrrolidinyl H-3, H-4), 1.43 (d, J: 6.72 Hz, 3H, CH3CH), 0.89 (d, J: 6.72 Hz, 6H, (CH3)2CH).
N-(2-Hydroxyethyl)-2–(4-isobutylphenyl)propanamide (8b): 85% yield; 1H-NMR (CDCl3) δ 7.19 (d, J: 7.95 Hz, 2H, phenyl H-2, H-6), 7.11 (d, J: 7.95 Hz, 2H, phenyl H-3, H-5), 6.04 (br, 1H, NH), 3.64 (t, J: 5.49 Hz, 2H, CH2OH), 3.58 (q, J: 7.32 Hz, 1H, CHCH3), 3.34 (m, 2H, CH2N), 3.08 (s, 1H, OH), 2.44 (d, J: 7.32 Hz, 2H, CH2Ar), 1.84 (m, 1H, CH(CH3)2), 1.51 (d, J: 7.32 Hz, 3H, CH3CH), 0.90 (d, J: 6.69 Hz, 6H, (CH3)2CH).
1-[2-(4-Isobutylphenyl)-1-oxo-1-propyl]proline (8c): 91% yield; m.p. 113–114 °C; 1H-NMR (CDCl3) δ 7.24 (d, J: 7.95 Hz, 2H, phenyl H-2, H-6), 7.09 (d, J: 7.95 Hz, phenyl H-3, H-5), 4.72 (m, 1H, prolinyl H-2), 3.53 (q, J: 7.32 Hz, 1H, CHCH3), 3.41–3.16 (m, 2H, prolinyl), 2.45 (d, J: 7.32 Hz, 2H, CH2Ph), 1.94–1.69 (m, 5H, prolinyl and CH(CH3)2), 1.43 (d, J: 6.72 Hz, 3H, CH3CH), 0.88 (d, J: 6.72 Hz, 6H, (CH3)2CH).
2-[4-(Phenyl)-3-fluorophenyl)-1-(pyrrolidin-1-yl)propan-1one (9a): 98% yield; m.p. 86–87 °C; 1H-NMR (CDCl3) δ 7.55–7.33 (m, 6H, phenyl), 7.16–7.11 (m, 2H, phenyl), 3.79 (q, J: 6.72 Hz, 1H, CHCH3), 3.48 (br, 4H, pyrrolidinyl H-2, H-5), 1.86 (br, 4H, pyrrolidinyl H-3, H-4), 1.49 (d, J: 6.72 Hz, 3H, CH3 CH).
2 -(4-Phenyl-3-fluorophenyl)-N-(2-hydroxyethyl)propanamide (9b): 92% yield; m.p. 82–83 °C; 1H-NMR (CDCl3) δ 7.55–7.36 (m, 6H, phenyl), 7.17–7.11 (m, 2H, phenyl), 6.00 (br, 1H, NH), 3.69 (t, 2H, J: 4.8 Hz, CH2OH), 3.64 (q, J: 7.32 Hz, 1H, CHCH3), 3.39 (m, 2H, CH2N), 2.33 (br, 1H, OH), 1.55 (d, J: 7.32 Hz, 3H, CH3 CH).
1-[2-(4-Phenyl-3-fluorophenyl)-1-oxo-1-propyl]proline (9c). 66% yield; 1H-NMR (CDCl3) δ 7.55–7.34 (m, 5H, phenyl), 7.14–7.09 (m, 3H, phenyl), 4.61 (m, 1H, CH prolinyl, 3.84 (q, J: 7.32 Hz, CHAr), 3.69 (m, 1H, pyrrolidin-1-yl), 3.35 (m, 1H, prolinyl), 2.09–1.91 (m, 4H, prolinyl), 1.53 (d, J: 7.32 Hz, 3H, CH3 CH).
General procedure for the preparation of amines (10a–c to 12a–c)
We added (dropwise) a solution of the corresponding amide (7–9, 1 eq.) in dry THF (5 mL), to a suspension of lithium aluminum hydride (2–4 eq.) in dry THF (15 mL). Then we stirred the reaction mixture at about 25 °C for 3–4 h (except for compounds 10b, 11b, and 12b, in which case we stirred the mixture under reflux for 3 h). In all cases, we monitored the reaction progress by TLC. After completion, we quenched the reaction by adding water (dropwise), 1 N NaOH solution, and ethyl acetate. After stirring for about 2 min, we separated the organic layer and dried it with sodium sulfate. Finally, we evaporated the solvent under vacuum, and we purified the corresponding amines by flash chromatography. All products (10–12) were used immediately after purification to obtain the corresponding ammonium salts (13–15) described below.
General procedure for the preparation of ammonium salts (13–15)
We passed dry HCl gas through a solution of the corresponding amine (10–12; 2 mmol) dissolved in diethyl ether (20 mL), under magnetic stirring until we did not observe any more precipitate coming out of solution. We filtered the resulting insoluble ammonium salt, washed it twice with ether, and dried it under vacuum in a desiccator for 18 h. To generate HCl gas, we produced it in situ by adding (dropwise) conc. H2SO4 to a commercial 37% solution of HCl under vigorous magnetic stirring. Then we transferred the HCl gas produced in this reaction using Teflon tubing connected to a CaCl2 trap. While passing the HCl gas, we isolated the system (three-necked round bottom flask) by using a septum inlet adapter with stopcock, to which we had attached a balloon to store any excess of HCl gas.
1-(2-(6-Methoxynaphthalen-2-yl)propyl)pyrrolidinium chloride (13a): 89.36% yield; m.p. 197–198 °C; 1H-NMR (D2O) δ 7.88 (m, 3H, naphthyl), 7.52 (dd, J: 8.55 Hz, 1.83 Hz, 1H, naphthyl), 7.38 (d, J: 2.43 Hz, 1H, naphthyl), 7.25 (dd, J: 8.55 Hz, 2.43 Hz, 1H, naphthyl), 3.95 (s, 3H, CH3O), 3.62 (m, 2H, CH2N), 3.4 (m, 1H, CHCH3), 3.28 (br, 4H, pyrrolidinyl), 1.95 (br, 4H, pyrrolidinyl), 1.38 (d, J: 6.72 Hz, 3H, CH3CH). 13C-NMR (D2O): 156.4, 136.4, 132.9, 128.6, 128.1, 127.2, 125.4, 124.9, 118.1, 105.5, 60.5, 54.7, 53.9, 36.1, 21.8, 18.3. ESI-MS (m/z); 292 [M + Na]+(100%), 269 [M + H]+.
N-(2-Hydroxyethyl)-2–(6-methoxynaphthalen-2-yl)propan-1-ammonium chloride (13b): 85.0% yield; m.p. 230–231 °C; 1-NMR (D2O): δ 7.91–7.81 (m, 3H, naphthyl), 7.51 (dd, J: 8.55 Hz, J: 1.23 Hz, 1H, naphthyl), 7.38 (d, J: 2.43 Hz, 1H, naphthyl), 7.23 (dd, J: 9.15 Hz, J: 2.43 Hz, 1H, naphthyl), 3.95 (s, 3H, CH3O), 3.77 (t, J: 5.49 Hz, 2H, CH2OH), 3.39 (m, 1H, CHCH3), 3.35 (m, 2H, CH2NH), 3.15 (q, J: 5.49 Hz, CH2NH), 1.41 (d, J: 6.72 Hz, 3H, CH3CH). 13C-NMR (D2O): 156.3, 136.4, 132.8, 128.6, 128.1, 127.1, 125.2, 125.0, 118.1, 105.5, 55.5, 54.6, 52.5, 48.5, 36.1, 18.3. ESI-MS (m/z); 296 [M + Na]+(100%), 273 [M + H]+.
2-(Hydroxyethyl)-1-[2-(6-methoxynaphtalen-2-yl)-1-propyl]pyrrolidinium chloride (13c): 85.0% yield, m.p. 153–154 °C; 1H-NMR (D2O) δ 7.91–7.83 (m, 3H, naphthyl), 7.51 (dd, J: 8.55 Hz, J: 1.23 Hz, 1H, naphthyl), 7.37 (d, J: 2.44 Hz, 1H, naphthyl), 7.24 (dd, J: 8.55 Hz, J: 2.44 Hz, 1H, naphthyl), 3.94 (s, 3H, CH3O), 3.87 (m, 2H, CH2OH), 3.66 (m, 1H, CHCH3), 3.59 (m, 2H, CH2N), 3.42 (m, 2H, pyrrolidinyl), 3.28 (m, 1H, pyrrolidinyl), 2.15 (m, 2H pyrrolidinyl), 1.90–1.67 (two m, 2H, pyrrolidinyl), 1.36 (d, J: 6.81 Hz, 3H, CH3CH). 13C-NMR (D2O): 156.4, 136.0, 132.9, 128.6, 128.1, 127.3, 125.4, 124.8, 118.1, 105.5, 69.2, 58.3, 54.6, 53.6, 53.5, 35.9, 24.7, 21.1, 19.5. ESI-MS (m/z); 322 [M + Na]+ (100%), 299 [M + H]+.
1-(2-(4-Isobutylphenyl)propyl)pyrrolidinium chloride (14a): 90.9% yield; m.p. 187–188 °C; 1H-NMR (D2O) δ 7.33–7.27 (m, 4H, phenyl), 3.59 (m, 4H, pyrrolidinyl), 3.15 (m, 2H, CH2N), 2.98 (m, 1H, CHAr), 2.49 (d, J: 6.69 Hz, 2H, CH2Ar), 2.07–1.80 (two m, 4H, pyrrolidinyl), 1.81 (m, 1H, CH(CH3)3, 1.30 (d, J: 6.72 Hz, 3H, CH3CH), 0.87 (d, J: 6.09 Hz, 6H, (CH3)2CH). 13C-NMR (D2O): 141.1, 138.3, 129.3, 126.3, 60.6, 54.5, 53.2, 43.4, 35.7, 28.9, 21.8, 20.8, 18.9. ESI-MS (m/z); 268 [M + Na]+(100%), 245 [M + H]+.
N-(2-Hydroxyethyl)-2-(4-isobutylphenyl)propan-1-ammonium chloride (14b): 89.6% yield; m.p. 137–138 °C; 1H-NMR (D2O) δ 7.32–7.26 (m, 4H, phenyl), 3.78 (t, J: 4.89 Hz, 2H, CH2OH), 3.32 (d, J: 7.32 Hz, 2H, CH2N), 3.22–3.09 (m, 3H, CH2N and CHAr), 2.49 (d, J: 7.32 Hz, 2H, CH2Ar), 1.85 (m, 1H, CH(CH3)2), 1.32 (d, J: 7.32 Hz, 3H, CH3), 0.87 (d, J: 6.72 Hz, 6H, (CH3)2CH). 13C-NMR (D2O): 141.1, 138.2, 129.3, 126.3, 55.5, 52.7, 48.5, 43.4, 35.7, 28.9, 20.8, 18.3. ESI-MS (m/z); 258 [M + Na]+(100%), 235 [M + H]+.
2-(Hydroxymethyl)-1-[2-(4-isobutylphenyl)-1-propyl]pyrrolidinium chloride (14c): 67.5% yield; m.p. 145–146 °C; 1H-NMR (D2O) δ 7.35–7.25 (m, 4H, phenyl), 3.88 (m, 2H, CH2OH), 3.77 (m, 2H, CH2N) 3.42–3.21 (m, 3H, pyrrolidinyl), 2.99 (m, 1H, CHAr), 2.49 (d, J: 6.72 Hz, 2H, CH2Ar), 2.22–2.11 (m, 4H, pyrrolidinyl), 1.83 (m, 1H, CHCH2), 1.31 (d, J: 6.69 Hz, 3H, CH3CH), 0.87 (d, J: 6.72 Hz, 6H, (CH3)2CH). 13C-NMR (D2O): 140.8, 139.1, 129.5, 129.2, 126.4, 126.2, 69.3, 60.8, 60.3, 58.1, 43.4, 35.7, 28.9, 24.4, 21.2, 20.7, 19.5, 19.3. ESI-MS (m/z); 298 [M + Na]+ (100%), 275 [M + H]+.
1-[2-(4-phenyl-3-fluorophenyl)-1-propyl]pyrrolidinium chloride (15a): 70% yield; m.p. 197–198 °C; 1H-NMR (D2O) δ 7.60–7.43 (m, 6H, phenyl), 7.27–7.22 (m, 2H, phenyl), 3.56–3.39 (m, 6H, 4H pyrrolidinyl, and CH2N), 3.24 (m, 1H, CHCH3), 2.00 (br, 4H, pyrrolidinyl), 1.33 (d, J: 6.72 Hz, 3H, CH3 CH). 13C-NMR (D2O): δ 160.4, 146.0, 137.4, 133.8, 131.2, 130.5, 130.0, 126.0, 117.4, 117.1, 63.4, 57.7, 57.0, 38.9, 24.9, 21.8. ESI-MS (m/z); 306 [M + Na]+(100%), 283 [M + H]+.
N-(2-Hydroxyethyl)-2-(4-phenyl-3-fluorophenyl)propan-1-ammonium chloride (15b): 65% yield; m.p. 157–158 °C; 1H-NMR (D2O) δ 7.63–7.44 (m, 6H, phenyl), 7.26 (m, 2H, phenyl), 3.83 (t, J: 4.89 Hz, 2H, CH2OH), 3.37 (d, J: 7.95 Hz, 2H, CH2N), 3.31 (m, 1H, CHCH3), 3.19 (m, 2H, CH2N), 1.37 (d, J: 6.72 Hz, 3H, CH3 CH). 13C-NMR (D2O): 160.4, 146.1, 146.0, 137.5, 133.8, 131.6, 130.6, 129.3, 117.4, 117.1, 58.7, 55.5, 51.8, 38.9, 21.4. ESI-MS (m/z); 296 [M + Na]+(100%), 273 [M + H]+.
1-[2-(4-Phenyl-3-fluorophenyl)propan-1-yl]-2-hydroxymethylpyrrolidinium chloride (15c): 70% yield; m.p. 54 °C; 1H-NMR (D2O) δ 7.76–7.47 (m, 8H, phenyl), 3.90 (m, 2H, CH2OH), 3.69 (m, 3H, CH2CH and CH pyrrolidinyl), 3.47 (m, 2H, CH2 pyrrolidinyl), 3.32 (m, 1H, CHAr), 2.15–1.87 (m, 4H, pyrrolidinyl), 1.38 (d, J: 6.72 Hz, 3H, CH3 CH). 13C-NMR (D2O): δ 160.1, 146.5, 137.4, 133.6, 131.4, 130.6, 130.1, 127.1, 117.3, 117.1, 70.0, 60.9, 59.0, 54.4, 36.3, 25.1, 21.8, 20.1. ESI-MS (m/z); 336 [M + Na]+ (100%), 313 [M + H]+.
Cyclooxygenase inhibition assay
We determined the ability of the test compounds (listed in ) to inhibit ovine COX-1 and human recombinant COX-2 by using a fluorescent inhibitor screening assay kit (Catalog no. 700100, Cayman Chemicals Inc., Ann Arbor, MI). We followed the procedure suggested by the manufacturer (the excitation wavelength = 530–540 nm, the emission wavelength of 585–595 nm).
Table 1. Cyclooxygenase (COX)-1 and COX-2 inhibition by water soluble NSAID derivatives lacking a carboxylic acid group (13a–13c, 14a–14c, and 15a–15c).
Carrageenan-induced paw edema anti-inflammatory assay
We evaluated the test compounds, as well as the reference drugs naproxen, ibuprofen, and flurbiprofen using the in vivo carrageenan-induced rat foot paw edema model reported previouslyCitation17. We administered all compounds orally, suspended in methylcellulose (1% aqueous solution) as the vehicle. We carried out this assay using protocols approved by the Health Sciences Animal Welfare Committee at the University of Alberta.
Docking studies
We performed the molecular docking experiments using crystal coordinates from the X-ray crystal structure of COX-2 (murine, 6COX, SC558 bound in the active site), obtained from the protein data bank. All the test compounds were built using the builder toolkit of the software package ArgusLab 4.0.1 (Mark, A. ArgusLab, Version 4.0.1; Thompson Planaria Software LLC, Seattle, WA). We carried out an energy minimization using the semi-empirical quantum mechanical method p.m.3. We chose the monomeric structure of the enzyme, and we defined the active site around the ligand. Then we inserted the molecule to be docked in the active site of the enzyme in the workspace, carrying the structure of the protein. The docking program implements an efficient grid-based docking algorithm that approximates an exhaustive search within the free volume of the binding site cavity. We surveyed the conformational space by geometry optimization of the flexible ligand (where we considered the rings as rigid), in combination with the incremental construction of the ligand torsions. Thus, the docking occurred between the flexible ligand parts of the compound and enzyme. We determined the ligand orientation by a shape scoring function based on A score, and we ranked the final positions by the lowest interaction energy values. The Einteraction is the sum of the energies involved in H-bond interactions, hydrophobic interactions, and van der Waal’s interactions. Finally, we determined the H-bond and hydrophobic interactions between the test compounds and the enzyme by distance measurements.
Ulcerogenic assay
We evaluated the ulcerogenic activity of naproxen (40 mg/kg) and the test compounds 13a, 13b, and 13c (equimolar dose to 40 mg/kg of naproxen), after oral administration of a single dose of the drug, suspended in 1.0 mL of a 1% methylcellulose solution. Control rats received only the vehicle (1.0 mL of 1% methylcellulose solution). We removed food 24 h before the administration of test compounds, but we allowed the animals to have a regular water intake. We euthanized the animals (CO2 chamber) 6 h after oral administration of the drug; then we removed the stomachs, we cut them along their greater curvature, we gently rinsed them with water, and we finally placed on ice. We determined the number, and the length of ulcers observed in each stomach using a magnifier lens. We assessed the severity of each gastric lesion along its greatest length (1 mm = rating of 1, 1–2 mm = score of 2, >2 mm = score according to their length in mm). We calculated the “ulcer index” (UI) for each test compound by adding the total length (L, in mm) of individual ulcers in each stomach, and averaging over the number of animals in each group (n = 4): UI = (L1 + L2 + L3 + L4)/4.
Arthritic index assay
We randomly divided the rats into different groups (n = 4/group). On day one, we injected all rats with 0.2 mL of Mycobacterium butyricum in squalene (50 mg/mL). We injected the control animals 0.2 mL of normal saline solution. We started the drug administration (1 mg/kg) on the first day of the experiment as a preventive measure. During the experiment, we measured daily the body weight gain and paw thickness using a caliper (sensitivity = 25 μm; Mitutoyo Canada Inc., Toronto, ON). On day 18 after injection of the adjuvant, we calculated the arthritis index according to a previously described methodCitation18. Briefly, (a) for hinds (0–4 score): 0 = no virtual sign of arthritis; 1 = involved a single joint; 2 = more than one joint and/or ankle with swelling; 3 = several joints and ankle with moderate swelling; 4 = several joints and ankle with severe swelling. (b) for paws (0–3 score): 0 = no inflammation in any joint, 1 = involved inflammation a single joint, 2 = more than one joint and/or wrist with moderate inflammation, and 3 = wrist and joints with moderate-to-severe swelling.
Statistics
We presented the results as the mean values ± SEM. We determined statistical differences between treatment groups using one-way ANOVA, followed by the Student–Newman–Keuls post hoc test using SigmaStat 3.5 program for Windows, Systat Software Inc. (San Jose, CA).
Results and discussion
Chemistry
We describe the synthesis of polar NSAID derivatives possessing a pyrrolidinium, 2-(hydroxymethyl)pyrrolidinium, or a 2-hydroxyethylammonium chloride in Scheme 1. In this regard, we reacted the corresponding NSAID with oxalyl chloride in dichloromethane to form acid chlorides (4–6), which we then stirred with pyrrolidine, l-proline, or 2-hydroxylamine to afford the amide intermediates 7a–7c, 8a–8c, and 9a–9c. The subsequent reduction of the carbonyl group using lithium aluminum hydride in dry THF afforded the corresponding amines 10a–10c, 11a–11c, and 12a–12c. The final step was the formation of ammonium chloride salts by reacting solutions of the corresponding amine in ether with dry hydrochloric acid. Following this procedure, we obtained the final products (13a–13c, 14a–14c, and 15a–15c) in moderate to good yields.
In vitro COX inhibition
The mechanism of action of classical NSAIDs is one of the most well-established drug targets in Medicinal Chemistry. Therefore, the obligated step toward the biological evaluation of any investigational drug with potential anti-inflammatory activity involves an in vitro screening for COX inhibition. In this regard, the ammonium salts obtained from naproxen (13a–13c) showed a significant inhibitory profile when we incubated them with human recombinant COX-2, exhibiting IC50 values in the 1.6–8.1 µM range (). We observed the same inhibitory profile for the ibuprofen derivatives 14a–14c (COX-2 IC50’s in the 20.7–28.1 µM range), and the flurbiprofen derivatives 15a-15c (COX-2 IC50’s in the 19.2–60.4 µM range). Under the experimental conditions used in our laboratory, we could not detect a significant inhibition of the COX-1 (bovine) isoform by any of the test compounds, at the maximum compound concentration (100 µM). The relative potency of all ammonium salts tested in vitro was 13 (naproxen derivatives) >14 (ibuprofen derivatives) >15 (flurbiprofen derivatives). Moreover, we observed that all compounds were relatively weaker COX inhibitors compared to the reference drug celecoxib (COX-2 IC50 value = 0.02 µM; COX-1 IC50 value >100 µM).
These results suggest a selective and relatively potent inhibitory activity of the COX-2 enzyme, while sparing the enzymatic activity of the COX-1 isoform (the extent at which the test compounds inhibited COX-1 activity at the highest test compound concentration of 100 µM was in the 17–44% range).
In vivo anti-inflammatory activity (carrageenan-induced inflammatory model)
To evaluate the potential in vivo anti-inflammatory profile exerted by the ammonium salts derived from naproxen, ibuprofen, and flurbiprofen, we carried out the conventional carrageenan-induced rat paw edema assayCitation17 using male Sprague–Dawley rats.
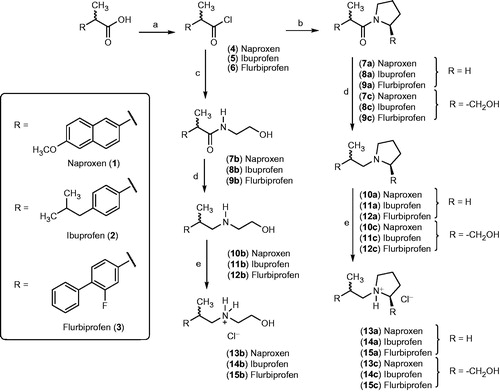
When we administered (orally) the ammonium salts derived from naproxen (13a–13c), we observed a significant and time-dependent anti-inflammatory effect exerted by the test drugs (at a dose equivalent to 5 mg/kg of naproxen). In this regard, derivative 13a showed a statistically significant (p < 0.05) anti-inflammatory effect 1 h after the injection of the carrageenan reagent, but unlike naproxen, this compound was inactive from the second time point (2 h) onward; this effect can be observed in the corresponding dose–response curve (). However, compound 13b significantly decreased inflammation at all time-points (1–4 h, ), but derivative 13c showed a significant anti-inflammatory effect (compared with the control group), only during the first 2 h ().
Figure 1. Time-dependent anti-inflammatory effect exerted by naproxen derivatives. (A) Compound 13a, (B) compound 13b, (C) compound 13c; four rats were used in each group (n = 4); individual values represent the average of 40 measurements (10 measurements per animal) ± SEM; (*) = p < 0.05 versus the control (vehicle) group for the time points marked.
Importantly, when we compared the anti-inflammatory profile of derivatives 13a–13c, to that obtained with the parent drug naproxen, at the same molar dose, it is evident that the carboxylic acid group present in the NSAID is not an essential requirement for the therapeutic effect. At least one derivative (compound 13b), maintained the desired in vivo anti-inflammatory activity ().
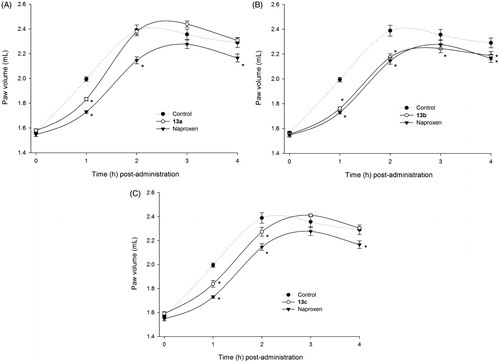
The results obtained with the ammonium salts derived from ibuprofen (14a–14c), provided additional evidence to support the non-essential role of the carboxylic acid group. Even though compounds 14a and 14b were not as potent as ibuprofen, they exerted a statistically significant (p < 0.05) anti-inflammatory effect compared with the control group (). Nevertheless, compound 14c (oral dose equivalent to 60 mg/kg of ibuprofen) was not active at any time point.
Figure 2. Time-dependent anti-inflammatory effect exerted by ibuprofen derivatives. (A) Compound 14a, (B) compound 14b, (C) compound 14c; four rats were used in each group (n = 4); individual values represent the average of 40 measurements (10 measurements per animal) ± SEM; (*) = p < 0.05 versus the control (vehicle) group for the time points marked.
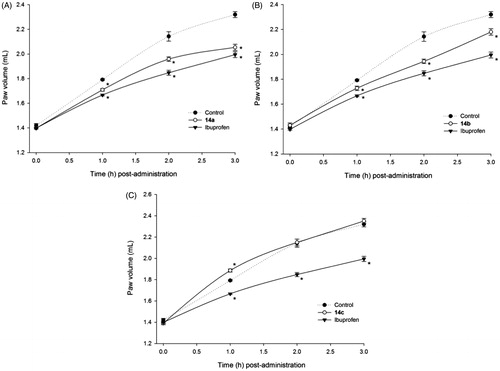
Finally, when we determined the anti-inflammatory activity exerted by the flurbiprofen ammonium salts 15a–15c (oral dose equivalent to 0.5 mg/kg of flurbiprofen), we observed that these drugs exerted a significant anti-inflammatory effect (p < 0.05) compared to control animals. This difference was bigger at 2 h and 3 h post-injection of the carrageenan reagent (), suggesting that the ammonium salts 15a–15c are absorbed at low rates from the gastrointestinal tract of rats (“slow-acting” drugs). In this regard, derivative 15c showed a very similar anti-inflammatory profile compared with the parent drug flurbiprofen ().
Figure 3. Time-dependent anti-inflammatory effect exerted by flurbiprofen derivatives. (A) Compound 15a, (B) compound 15b, (C) compound 15c; four rats were used in each group (n = 4); individual values represent the average of 40 measurements (10 measurements per animal) ± SEM; (*) = p < 0.05 versus the control (vehicle) group for the time points marked.
Docking studies
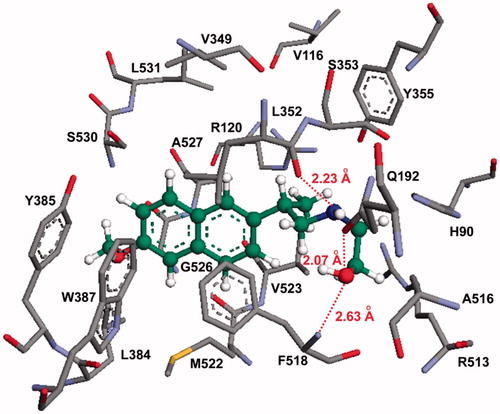
To find out if the NSAID derivatives 13–15 can bind to the active site of the COX enzyme, despite not having the carboxylic acid group present in their parent counterparts, we conducted a molecular modeling (docking) study using the active naproxen derivative 13b. According to our docking studies, compound 13b oriented favorably within the COX-2 active site, in such a way that the terminal hydroxyl group (OH) is close to Gln192, Phe518, Arg513, and Val523 (active site residues; ). The hydroxyl group of compound 13b formed hydrogen bonding interactions with a nitrogen atom in Phe518 (OH–N distance = 2.63 Å). At the same binding pose, the hydroxyl group also formed a hydrogen bond with the carbonyl oxygen of Gln192 (OH–O distance = 2.07 Å; COX-2 secondary pocket residue). Additionally, we observed that the nitrogen atom in compound 13b formed an additional hydrogen bond with Leu352 (COOH–N distance = 2.23 Å), and it is close to Gln192 (the distance between the carbonyl oxygen and the OH = 3.01 Å). Interestingly, the two conjugated aromatic rings of the naproxen derivative 13b were located near the hydrophobic region of the COX-2 active site formed by Trp387, Phe518, and Tyr385.
Figure 4. Molecular modeling (docking) of compound 13b (carbon atoms in green color) in the binding site of COX-2 (PDB ID: 6COX; Eintermolecular = −12.23 kcal/mol). Hydrogen atoms of amino acid residues have been omitted for clarity.
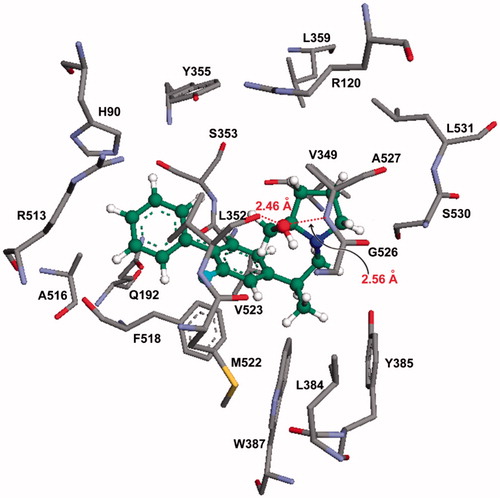
An interesting case is that observed with compound 15c, which is a flurbiprofen derivative that also showed significant efficacy in vivo. The best binding mode obtained from the molecular docking study, showed that compound 15c resided at the center of the COX-2 active site (), where the fluorine atom present at one of the phenyl rings is close to both the Phe518 (1.98–2.89 Å range), and the Val523 (2.01–3.02 Å range) residues. Notably, the hydroxyl group (–OH) of the prolinol ring is located in the vicinity of Leu352, Ala527, and Gly526 residues. Also, the hydroxyl group showed hydrogen bonding interactions with the carbonyl oxygen of Val523 (2.46 Å) and the NH group of Ala527 (2.56 Å). In the best binding mode calculated for compound 15c, the unsubstituted phenyl ring is located in the vicinity of Gln192, Phe518, Ala516, Arg513, and Val523 amino acid residues ().
Figure 5. Molecular modeling (docking) of compound 15c (carbon atoms in green color) in the binding site of COX-2 (PDB ID: 6COX; Eintermolecular = −12.16 kcal/mol). Hydrogen atoms of amino acid residues have been omitted for clarity.
Previous reports describe a particular role for the carboxylic acid group in the overall binding modes of classical NSAIDs within the COX active site. For example, Ermondi et al.Citation19 described the binding interaction between the carboxylate group present in flurbiprofen and a cavity in the enzyme formed by Leu531, Val116, Val349, Leu359, and Tyr355. Authors also suggest that the –COOH group in NSAIDs limits their selectivity toward COX-1 or COX-2, due to decreased “freedom of movement” created by a binding interaction with Arg120. Additional binding interactions with Ser530 and Tyr385 were also considered significant, accounting for some enzyme–ligand interactions that are also relevant to the inhibition of COX-2Citation19.
Therefore, considering that derivatives 13a–13c, 14a–14c, and 15a–15c do not have a carboxylic acid functional group, and still exert in vitro inhibition of the COX-2 isoform, while producing significant in vivo anti-inflammatory activity, provides evidence to argue against the importance of the COOH group in the chemical structure of NSAIDs. Furthermore, the complete entry of compound 13b and 15c into the COX-2 active site, and the significant electronic interactions observed with these compounds within the COX-2 active site, is consistent with the significant anti-inflammatory activity determined experimentally.
In vivo anti-inflammatory activity (arthritic index)
We decided to challenge one of the new NSAID derivatives (15a) using a longer-term in vivo assay, in which Sprague–Dawley rats are injected with M. butyricum, inducing an arthritis-like state. As shown in , the effects produced by compound 15a were modest; we evaluated the increase in both right and left paw volume in animals treated with the test drug (1 mg/kg p.o.), and compared them to those obtained with vehicle-treated (control). We also tested the standard (flurbiprofen, equimolar dose to 15a). In this regard, we observed that only the animal group receiving flurbiprofen showed a statistically significant difference (p < 0.05) compared with control animals. The change in volume recorded for the right paw in animals dosed with 15a (), also experienced a significant decrease. However, due to the high variability associated with this experiment, we could not establish a potential anti-inflammatory associated to drug 15a. We observed a similar case when we compared the left and right joints of animals dosed with drug 15a; the decrease in joint volume suggested a potential anti-inflammatory effect exerted by both flurbiprofen (), and 15a (), but this effect was not statistically significant when we compared it with that obtained with the control group.
Figure 6. 15-Day anti-inflammatory study comparing flurbiprofen with compound 15a. (A) Percent increase in body weight; (B) percent increase of right paw volumes; (C) percent increase of left paw volumes; (D) percent increase of right joint volumes; (E) percent increase in left joint volume; (F) the arthritic index calculated for compound 15a and flurbiprofen. All values are expressed as percent increase at day 15 compared to day 1; N = 4; doses: flurbiprofen (1 mg/kg orally), 15a (equimolar dose) treated groups. (*) = p < 0.05 versus the control (vehicle) group.
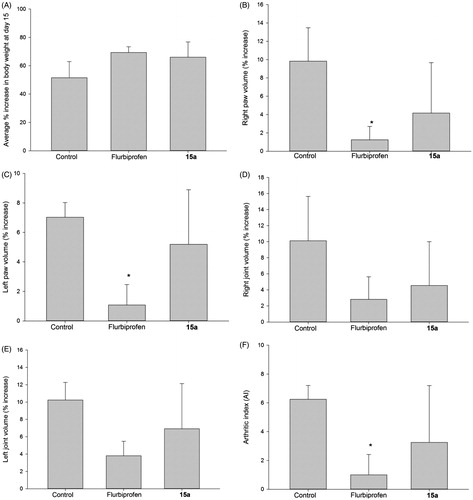
We observed an encouraging result when we recorded the change (increase) in body weight in animals dosed with flurbiprofen and compound 15a. This observation suggests that arthritic animals dosed with drug 15a and flurbiprofen, may have experienced some relief from pain and inflammation, which presumably, allowed these animals to feed better (hence the increase in body weight) than those in the control group ().
These results seem to suggest that the removal of the carboxylic acid group present in naproxen, ibuprofen, and flurbiprofen does not abolish their desirable anti-inflammatory activity, and therefore, the acid group may not be an essential requirement for these drugs. Moreover, these results showed that in vitro screening should not be the only parameter by which a new anti-inflammatory drug is are evaluated experimentally. For example, despite the weak in vitro COX-2 enzyme inhibition exerted by compound 15a (only 34% inhibition at 100 µM), this compound significantly decreased inflammation when tested in vivo (it displayed 50% of the anti-inflammatory effect exerted by flurbiprofen at 2 h, and almost the same effect at 3 h; see ).
One of the potential repercussions of this observation is the fact that NSAIDs are regarded as potential cancer chemopreventive drugsCitation20,Citation21 and consequently, in addition to complementary experiments aimed at validating the anti-inflammatory activity of these ammonium salts, we are also interested in exploring the effects of these compounds on cancer-related targets commonly associated with classical NSAIDs (future work).
Ulcer index assay
To complement our study, we needed to evaluate the potential gastric toxic effects exerted by the test compounds using an acute NSAID-induced ulcerogenic assayCitation22. In this regard, we dosed animals with oral naproxen (40 mg/kg), or an equimolar amount of compounds 13a, 13b, or 13c (single oral dose). After 6 h, we euthanized all animals, and we analyzed their stomachs looking for ulcers. We observed that only the rats dosed with the parent naproxen developed gastric lesions (ulcer index [UI] = 48), whereas the naproxen derivatives 13a–13c did not exert visually detectable gastric toxicity (UI = 0; see and ). This provides strong (but, we know, preliminary) evidence supporting the use of carboxyl-free molecules as effective and safe NSAIDs. Furthermore, it also provides evidence suggesting that inhibition of prostaglandin synthesis does not necessarily correlates with gastric damage, an issue that has been described previously in the literatureCitation23–25.
Figure 7. Ulcerogenicity assay data illustrating the extent of NSAID-induced gastric ulcers exerted by a single oral dose of naproxen (40 mg/kg), compared with that observed with derivatives 13a (“A”), and 13c (“B”). Experimental drugs were administered using equimolar doses to that of naproxen (“C”). Control animals (“D”) received methylcellulose as vehicle. Only one (representative) example from each group is presented. White arrows point to areas where we observed significant gastric bleeding.
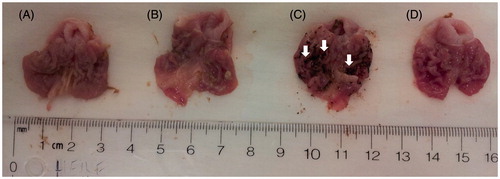
Table 2. Ulcer index (UI) determined for NSAID derivatives 13a–13c.
We realize that to provide conclusive evidence demonstrating the gastrointestinal safety of compounds 13–15, we will need to use complementary approaches outlined in the literatureCitation23. Some of these complementary assays include (a) the use of sub-chronic administrations of the drug (i.e. 25 mg/kg for 3 d); (b) an animal model of impaired mucosal defense; (c) detect hemoglobin in feces; (d) collect blood from animals to measure hematocrit; and (e) assess the effects of these new drugs on myeloperoxidase activityCitation26,Citation27. All these assays extend beyond the scope of this report and constitute the object of at least one research project being carried out in our group.
Conclusion
NSAID-induced gastropathy is still a significant clinical concern, and this work presents data to propose a new framework to reconsider the chemical features that are essential/required in classical non-steroidal anti-inflammatory drugs (NSAIDS). As it has been demonstrated previously with the vast number of publications describing ester and amide NSAID derivatives, we have shown that the carboxylic acid group present in these drugs may not be needed at all, and it can be replaced by an ammonium salt, which maintains the in vitro COX-2 selectivity with relatively high potency, and considerable in vivo efficacy (carrageenan assay).
Declaration of interest
The authors report that they have no conflict of interest. The authors alone are responsible for the content and writing of this article. The authors gratefully acknowledge the financial support provided by the Faculty of Pharmacy and Pharmaceutical Sciences at the University of Alberta (startup funds), the Higher Education Commission (HEC) of Pakistan for a graduate scholarship to N. U., the Saudi Cultural Bureau in Canada for a graduate scholarship to F. S. A., and the National Council of Science and Technology (CONACYT, Mexico) for a graduate scholarship to A. R. D.
References
- Smalley WE, Ray WA, Daugherty JR, Griffin MR. Nonsteroidal anti-inflammatory drugs and the incidence of hospitalizations for peptic ulcer disease in elderly persons. Am J Epidemiol 1995;141:539–45
- Simon LS. Biologic effects of nonsteroidal anti-inflammatory drugs. Curr Opin Rheumatol 1997;9:178–82
- Zochling J, van der Heijde D, Dougados M, Braun J. Current evidence for the management of ankylosing spondylitis: a systematic literature review for the ASAS/EULAR management recommendations in ankylosing spondylitis. Ann Rheum Dis 2006;65:423–32
- Kean WF, Buchanan WW. The use of NSAIDs in rheumatic disorders 2005: a global perspective. Inflammopharmacology 2005;13:343–70
- Schnitzer TJ. Update of ACR guidelines for osteoarthritis: role of the coxibs. J Pain Symptom Manage 2002;23:S24S30
- Connolly TP. Cyclooxygenase-2 inhibitors in gynecologic practice. Clin Med Res 2003;1:105–10
- Ong KS, Seymour RA. Maximizing the safety of nonsteroidal anti-inflammatory drug use for postoperative dental pain: an evidence-based approach. Anesth Prog 2003;50:62–74
- Lipton RB, Stewart WF, Ryan RE, et al. Efficacy and safety of acetaminophen, aspirin, and caffeine in alleviating migraine headache pain: three double-blind, randomized, placebo-controlled trials. Arch Neurol 1998;55:210–17
- Wallace JL. Prostaglandins, NSAIDs, and gastric mucosal protection: why doesn't the stomach digest itself? Physiol Rev 2008;88:1547–65
- Yeomans ND, Hawkey CJ, Brailsford W, Naesdal J. Gastroduodenal toxicity of low-dose acetylsalicylic acid: a comparison with non-steroidal anti-inflammatory drugs. Curr Med Res Opin 2009;25:2785–93
- Higuchi K, Umegaki E, Watanabe T, et al. Present status and strategy of NSAIDs-induced small bowel injury. J Gastroenterol 2009;44:879–88
- Bhandari SV, Kailash GB, Mayuresh KR, et al. Design, synthesis and evaluation of antiinflammatory, analgesic and ulcerogenicity studies of novel S-substituted phenacyl-1,3,4-oxadiazole-2-thiol and Schiff bases of diclofenac acid as nonulcerogenic derivatives. Bioorg Med Chem 2008;16:1822–31
- Kalgutkar AS, Crews BC, Saleh S, et al. Indolyl esters and amides related to indomethacin are selective COX-2 inhibitors. Bioorg Med Chem 2005;13:6810–22
- Harman CA, Turman MV, Kozak KR, et al. Sturctural basis of enantioselective inhibition of cyclooxygenase-1 S-alpha-substituted indomethacin ethanolamides. J Biol Chem 2007;282:28096–105
- Kalgutkar AS, Marnett AB, Crews BC, et al. Ester and amide derivatives of the nonsteroidal antiinflammatory drug, indomethacin, as selective cyclooxygenase-2 inhibitors. J Med Chem 2000;43:2860–70
- Abdellatif KRA, Chowdhury MA, Dong Y, et al. Dinitroglyceryl and diazen-1-ium-1,2-diolated nitric oxide donor ester prodrugs of aspirin, indomethacin and ibuprofen: synthesis, biological evaluation and nitric oxide release studies. Bioorg Med Chem Lett 2009;19:3014–8
- Winter CA, Risley EA, Nuss GW. Carrageenin-induced edema in hind paw of the rat as an assay for antiiflammatory drugs. Proc Soc Exp Biol Med 1962;111:544–7
- Piquettemiller M, Jamali F. Influence of severity of inflammation on the disposition kinetics of propranolol enantiomers in ketoprofen-treated and untreated adjuvant arthritis. Drug Metab Dispos 1995;23:240–5
- Ermondi G, Caron G, Lawrence R, Longo D. Docking studies on NSAID/COX-2 isozyme complexes using contact statistics analysis. J Comput Aided Mol Des 2004;18:683–96
- Steele VE, Rao CV, Zhang Y, et al. Chemopreventive efficacy of naproxen and nitric oxide-naproxen in rodent models of colon, urinary bladder, and mammary cancers. Cancer Prev Res 2009;2:951–6
- Tinsley HN, Grizzle WE, Abadi A, et al. New NSAID targets and derivatives for colorectal cancer chemoprevention. Recent Results Cancer Res 2013;191:105–20
- Cocco MT, Congiu C, Onnis V, et al. Synthesis of new 2-arylamino-6-trifluoromethylpyridine-3-carboxylic acid derivatives and investigation of their analgesic activity. Bioorg Med Chem 2004;12:4169–77
- Wallace JL, Caliendo G, Santagada V, Cirino G. Markedly reduced toxicity of a hydrogen sulphide-releasing derivative of naproxen (ATB-346). Br J Pharmacol 2010;159:1236–46
- Lichtenberger LM, Romero JJ, Dial EJ. Surface phospholipids in gastric injury and protection when a selective cyclooxygenase-2 inhibitor (Coxib) is used in combination with aspirin. Br J Pharmacol 2007;150:913–19
- Wallace JL, McKnight W, Reuter BK, Vergnolle N. NSAID-induced gastric damage in rats: requirement for inhibition of both cyclooxygenase 1 and 2. Gastroenterology 2000;119:706–14
- Li L, Rossoni G, Sparatore A, et al. Anti-inflammatory and gastrointestinal effects of a novel diclofenac derivative. Free Radic Biol Med 2007;42:706–19
- Chen K, Nezu R, Inouea M, et al. Beneficial effects of growth hormone combined with parenteral nutrition in the management of inflammatory bowel disease: an experimental study. Surgery 1997;121:212–18