
Abstract
Six genetic families of the enzyme carbonic anhydrase (CA, EC 4.2.1.1) were described to date. Inhibition of CAs has pharmacologic applications in the field of antiglaucoma, anticonvulsant, anticancer, and anti-infective agents. New classes of CA inhibitors (CAIs) were described in the last decade with enzyme inhibition mechanisms differing considerably from the classical inhibitors of the sulfonamide or anion type. Five different CA inhibition mechanisms are known: (i) the zinc binders coordinate to the catalytically crucial Zn(II) ion from the enzyme active site, with the metal in tetrahedral or trigonal bipyramidal geometries. Sulfonamides and their isosters, most anions, dithiocarbamates and their isosters, carboxylates, and hydroxamates bind in this way; (ii) inhibitors that anchor to the zinc-coordinated water molecule/hydroxide ion (phenols, carboxylates, polyamines, 2-thioxocoumarins, sulfocoumarins); (iii) inhibitors which occlude the entrance to the active site cavity (coumarins and their isosters), this binding site coinciding with that where CA activators bind; (iv) compounds which bind out of the active site cavity (a carboxylic acid derivative was seen to inhibit CA in this manner), and (v) compounds for which the inhibition mechanism is not known, among which the secondary/tertiary sulfonamides as well as imatinib/nilotinib are the most investigated examples. As CAIs are used clinically in many pathologies, with a sulfonamide inhibitor (SLC-0111) in Phase I clinical trials for the management of metastatic solid tumors, this review updates the recent findings in the field which may be useful for a structure-based drug design approach of more selective/potent modulators of the activity of these enzymes.
Introduction
Six distinct genetic families of carbonic anhydrases (CAs, EC 4.2.1.1), the superfamily of metalloenzymes which catalyze the interconversion between CO2 and bicarbonate are known to date, the α-, β-, γ-, δ-, ζ-, and η-CAsCitation1–11. The catalytic/inhibition mechanisms for the physiologic reaction were reviewed in earlier papersCitation12–21, but the discovery of new classes of CA inhibitors (CAIs) possessing different inhibition mechanisms compared to the classical inhibitors of the sulfonamide/anion type, prompted me to review again this field.
The inhibition and activation of CAs are well-understood processes: most types of classical inhibitors bind to the metal centerCitation12–21, whereas activators bind at the entrance of the active site cavity and participate in proton shuttling processes between the metal ion-bound water molecule and the environmentCitation22–24. This leads to enhanced formation of the metal hydroxide, catalytically active species of the enzymeCitation1,21–24.
CO2, bicarbonate, and protons are essential molecules/ions in many important physiologic processes in all life kingdoms (Bacteria, Archaea, and Eukarya), throughout the tree of life, and for this reason, relatively high amounts of CAs are present in different tissues/cell compartments of most investigated organismsCitation1–11. The α-CAs are present in vertebrates, protozoa, algae, and cytoplasm of green plants and in some BacteriaCitation1,Citation3; the β-CAs are predominantly found in Bacteria, algae and chloroplasts of both mono- as well as dicotyledons, but also in many fungi and some ArchaeaCitation1–9. The γ-CAs were found in Archaea, cyanobacteria, and most types of BacteriaCitation3,Citation25, the δ - and ζ-CAs seem to be present only in marine diatomsCitation11, whereas the η-CAs in protozoaCitation4. In many organisms these enzymes are involved in crucial physiological processes connected with respiration and transport of CO2/bicarbonate, pH, and CO2 homeostasis, electrolyte secretion in a variety of tissues/organs, biosynthetic reactions (e.g. gluconeogenesis, lipogenesis, and ureagenesis), bone resorption, calcification, tumorigenicity, and many other physiologic or pathologic processes (thoroughly studied in vertebrates)Citation1,4–7,9. In algae, plants, and some bacteria they play an important role in photosynthesis and other biosynthetic reactionsCitation3,Citation5. In diatoms δ- and ζ-CAs play a crucial role in carbon dioxide fixation, whereas in protozoa the role of the η-CAs is poorly understood for the momentCitation11. Many such enzymes from vertebrates, nematodes, fungi, protozoa and bacteria are well-known drug targetsCitation1,3,4,26–43.
The zinc binders as CAIs
Sulfonamides are the most important class of CAIsCitation1,2,5,6,8,16–19, with at least 20 such compounds in clinical use for decades or clinical development in the last period ()Citation44–79. They include acetazolamide 1, methazolamide 2, ethoxzolamide 3, sulthiame 4, dichlorophenamide 5, dorzolamide 6, brinzolamide 7, sulpiride 8, zonisamide 9, topiramate 10, saccharin 11, celecoxib 12, chlorothiazide/highceiling diuretics of types 13–19, including the widely prescribed hydrochlorothioazide 13a, furosemide 18, bumethanide 19, compounds in clinical use for many years, as diuretics, antiglaucoma agents, antiepilepticsCitation1,2,6–8,19,44–79. Compound 20, SLC-0111, is in Phase I clinical trials as an antitumor/antimetastatic agent, and was discovered in the author’s laboratoryCitation19,Citation54,Citation63 (). Most of the sulfonamides 1–19 act as potent CAIs and are in clinical use for decades, but their main problem is related to the fact that by inhibiting most of the catalytically active CA isoforms found in vertebrates (12 such isoforms are known to dateCitation1,Citation21), they show a wide range of side effectsCitation1,2,5,6,8,16–19. Newer generation inhibitors, among which 20 is a rather recent example, were designed in such a way as to act as isoform-selective inhibitors for the transmembrane, tumor-associated isoforms hCA IX and XII (h = human), and as thus, they probably will show less side effects compared to the classical CAIs of types 1–19 discussed aboveCitation19,Citation54,Citation63. Furthermore, sulfonamides and sulfamates strongly inhibit CAs belonging to most families, not only the α-class enzymes, and this may be exploited in the future for obtaining antiinfectives with a new mechanism of actionCitation1,3–6,9,11,13–18,21.
Figure 1. Clinically used sulfonamide and sulfamate CAIs 1–19 and compound 20 in Phase I clinical development.
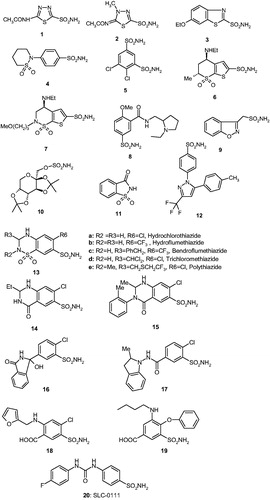
A general CA inhibition mechanism with zinc binders, including sulfonamides, sulfamates, and sulfamides (which are in fact sulfonamide isosters) is shown in . These CAIs bind in deprotonated form, as anions, to the Zn(II) ion from the enzyme active site, which is in a tetrahedral geometry, with the zinc-binding group (ZBG) (SO2NH− moieties for the three main classes of inhibitors mentioned above) directly interacting with the metal ion (). There is overwhelming X-ray crystallographic evidence showing this type of binding for sulfonamides, sulfamates, and sulfamides belonging to a variety of chemical classesCitation21,44–46,57–65. In addition, the ZBG also interact with two other conserved residues in all α-CAs, acting as “gate keepers”, i.e. Thr199 (hydrogen bonded through its OH group with the water molecule/hydroxide ion coordinated to the zinc in the uninhibited enzyme, and with the ZBG, as shown in , in the enzyme-inhibitor adducts) and Glu106, which is hydrogen bonded to Thr199 through its carboxylate moiety ()Citation21,44–46,57–65.
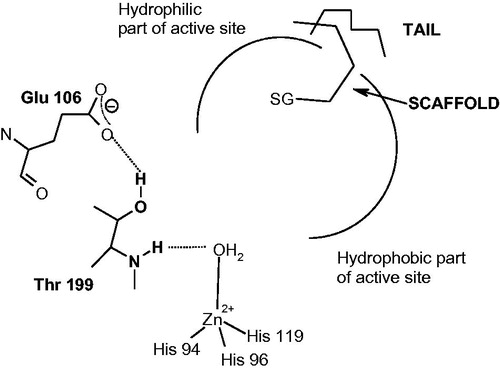
Figure 2. CAIs belonging to the zinc binders (sulfonamides, sulfamates, sulfonamides, carboxylates, hydroxamates, phosphonates). The ZBG is coordinated to the metal ion and participates in strong interactions with the gate keeper residues Thr199–Glu106. The scaffold may occupy either the hydrophylic or hydrophobic (or both) halves of the active site, whereas the tails generally are orientated toward the exit of the cavity where the most variable amino acid residues among the different mammalian isoforms are foundCitation1,Citation21,Citation80.
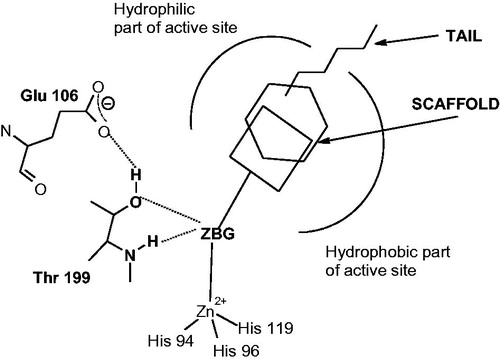
The main scope of the drug design campaigns in the last years was to obtain isoform-selective CAIs for the various isoforms involved specifically in different pathologies, as noted aboveCitation1,21,44–46,57–65. This is however not an easy task, considering that the 12 catalytically active isoforms present in primates, have an active site architecture quite similar with each otherCitation1,21–24,44–46. In fact, all human isoforms have the three conserved His residues (His94, 96, and 119) acting as zinc ligands, as well as half the active site mainly lined with hydrophobic residues and the opposite one with hydrophilic residuesCitation1,Citation21,Citation80. However, there are also important differences in amino acid residues mainly in the middle and toward the exit of the active site cavityCitation1,Citation21,Citation80. Most of the inhibitors mentioned above, of types 1–9, do not make extensive contacts with them as their scaffolds are rather compact and these sulfonamides bind deep within the active site cavity which is rather similar in all mammalian isoformsCitation1,Citation21,Citation80. Thus, it appeared of considerable interest to devise alternative approaches for obtaining isoform-selective CAIs. One of the most successful one was termed “the tail approach”Citation1,21,48,81,82. It consists in appending “tails” to the scaffolds of aromatic/heterocyclic sulfonamides possessing derivatizable moieties of the amino, imino, or hydroxy type, in such a way that an elongated molecule is obtained with its tail being able to interact with amino acid residues from the middle and edge of the active site cavity, as shown in for a general zinc-binder CAICitation1,21,48,81,82. By choosing tails with a diverse chemical nature, it was also possible to modulate the physico-chemical properties of such CAIs, which are crucial for their biological activityCitation1,21,48,81,82. For example, antiglaucoma agents should possess an appropriate hydrophilicity and water solubility in order to be formulated as eye drops but also a balanced lipophilicity for being able to penetrate through the plasma membranes and arrive at the ciliary processes where the enzymes responsible for aqueous humor secretion are found (i.e. CA II, IV, and XII)Citation48,Citation81,Citation82. This approach was subsequently expanded to all classes of CAIs, such as among others to the dithiocarbamates (DTCs)Citation28–30,83, xanthatesCitation84, carboxylatesCitation31,Citation41,Citation85,Citation86, phosphonatesCitation87–92, and hydroxamatesCitation93–100, etc. X-Ray crystal structures are available for adducts of various isoforms (mainly hCA I and II) with all these classes of CAIs except the xanthates for which computational methods were used to assess their inhibition mechanismCitation30,83,88,100,101. For example, DTCs 21–23 were crystallized bound to hCA IICitation83. As seen from , one of the sulfur atoms of the inhibitor is coordinated to the Zn(II) ion and makes the canonical interactions of all ZBGs () with the gate keeper residues Thr199-Glu106. For these DTCs, the scaffolds have very diverse orientation within the active site, which may explain their high inhibitory activity against many CA isoformsCitation28–30,83. Using these derivatives as leads, new DTCs were reported recently, which showed better selectivity profiles compared to the first class of DTCs, including 21–23Citation102.
Figure 3. (A) Structure of DTCs 21–23. (B) View of compounds 21 (cyan), 22 (magenta), 23 (green), superposed in the active site of hCA II. The zinc ion is shown as the central blue sphere and the amino acid residues involved in the binding are evidenced and numbered (hCA I numbering system)Citation83.
Many DTCs showed excellent intraocular pressure (IOP) lowering effects in animal models of glaucoma, being equivalent and superior to the sulfonamide CAIs, and due to their excellent water solubility (and high CA inhibitory action) this class of compounds might be exploited in the future for obtaining antiglaucoma agentsCitation30,Citation74,Citation102.
CAIs anchoring to the zinc-coordinated water/hydroxide ion
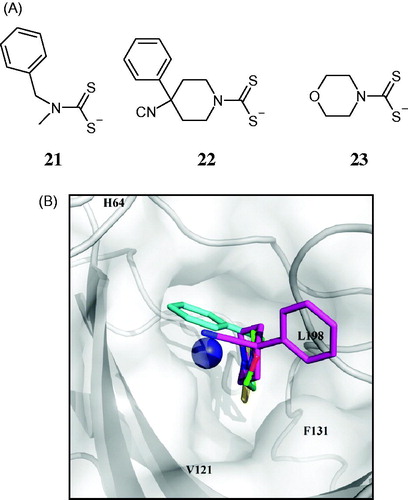
The first compound for which this new CA inhibition mechanism, i.e. anchoring to the zinc-coordinated water molecule/hydroxide ion, was reported, is phenol 24Citation103. The polyamine spermine 25 was the second oneCitation105. Compounds inhibiting CAs by this mechanism () are characterized by a presence of an anchoring group (AG) which is attached to a scaffold possibly incorporating tails which can interact with the two halves of the active site (as for the zinc binders). The main difference between the zinc binders and the inhibitors anchoring to the zinc nucleophile consists in the fact that the inhibitor is not in a direct contact with the metal ion. As reported in the last years by several groups, the AGs belong to a variety of chemical functionalities. They include phenolic OHCitation104,106–113, primary amineCitation105, COOHCitation113, COOMeCitation111, and SO3HCitation114 moieties. The scaffolds of these compounds can be of the aromatic, aliphatic, heterocyclic, or sugar typeCitation114–121, and many of them display efficient inhibitory activity against CAs belonging to the α- and β-CA classesCitation105–122.
Figure 4. Compounds which anchor to the Zn(II)-coordinated water molecule/hydroxide ion. The anchoring group (AG) may be of the OH (phenol), amino (polyamine), ester (COOR), sulfonate (sulfocoumarin) type or just a simple sulfur atom (thioxocoumarin)Citation103–114.
As shown in , phenol 24 is anchored to the non-protein zinc ligand (probably a hydroxide anion at the pH at which the experiments were carried outCitation103) by a hydrogen bond involving the H atom of the inhibitor and the donor OH from the zinc hydroxide. In addition, a second hydrogen bond involves the gate-keeping residue Thr199, whose NH group participates in a second hydrogen bond with the phenol (). Spermine 25 binds in a rather similar manner with phenol, although the hydrogen bonding network is slightly different (). Thus, one of the primary amine moieties of the inhibitor (probably as ammonium salt) anchors by means of a hydrogen bond to the zinc-coordinated water molecule/hydroxide ion, and also makes a second hydrogen bind with the OH (not NH as phenol) moiety of Thr199. The other terminal primary amine of spermine participates in hydrogen bonding with Thr200 and Pro201, whereas its main scaffold makes some clashes with a water molecule and Gln92, which explains why this is a rather weak hCA II inhibitor, but significantly inhibits other mammalian isoforms, such as for example hCA IV (KI of 10 nM)Citation104.
Figure 5. The first compounds which inhibit CAs by anchoring to the zinc-coordinated water molecule: (A) Phenol 24Citation103; (B) Spermine 25Citation104.
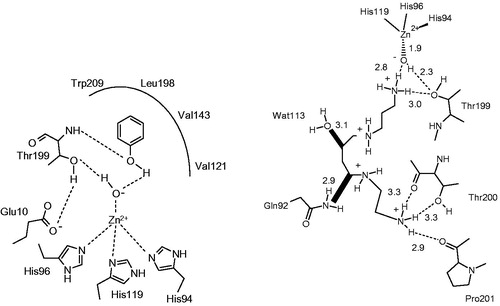
As shown in , X-ray crystallographic studies evidenced the same type of CA inhibition with resorcinol 26 (a diphenol)Citation113, 2,5-dihydroxybenzoic acid 27 (compound for which the anchoring is assured not by the phenolic OH moieties but by the COOH oneCitation113), xylariamide 28 (the anchoring is assured by the ester moiety, COOMe, although the CAs possess esterase activity)Citation111, as well as the hydrolyzed sulfocoumarin derivative 29 (see discussion latter in the text for sulfocoumarins).
Figure 6. Other compounds which inhibit CAs by anchoring to the non-protein zinc ligand: (A) resorcinol 26Citation113; (B) 2,5-dihydroxybenzoic acid 27Citation113; (C) spermine 25 (stick view, not the schematic one as in Figure 5B)Citation104; (D) xylariamide A 28Citation111; (E) hydrolyzed 6-bromo-sulfocoumarin 29Citation114. (F) Chemical structures of the discussed CAIs. All these adducts have been characterized by high resolution X-ray crystallography. The Zn(II) ion (gray sphere), its three His ligands and coordinated water molecule (red sphere) and amino acid residues involved in the binding of the inhibitors are shown.
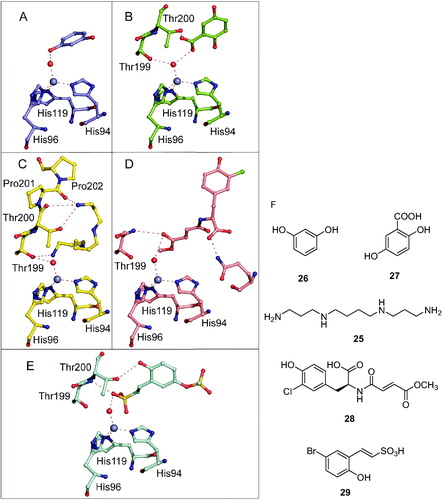
More recently a 2-thioxocoumarin derivative, 30, was also observed to bind by anchoring to the zinc-coordinated water moleculeCitation122 (), whereas iodide, an anion which was considered to be a zinc binder as all inorganic anions investigated so farCitation12, was also observed to anchor to the zinc-bound water/hydroxide ion, in the β-CA from the alga CoccomyxaCitation123. In the case of 30, the exocyclic sulfur atom of the inhibitor is hydrogen bonded to the zinc-coordinated water molecule/hydroxide ion, whereas the organic scaffold makes favorable interactions with water molecules and amino acid residues from the active siteCitation122 (). Thus, this CA inhibition mechanism seems to be much more general as initially considered when phenols were discovered as inhibitors. It should be also noted that CAs belonging to other genetic families than the α- and β-ones were not yet investigated for this type of alternative inhibition mechanism.
Figure 7. (A) Superposition of the adducts of hCA II with 6-hydroxy-2-thioxocoumarin 30 (sky blue, 4WL4) with the hCA II-hydrolyzed coumarin 31 adduct (5BNL) (silver). The zinc ion, its three His ligands and amino acid residues involved in the binding of inhibitors are shown in Ref. Citation122. (B) Chemical structures of compounds 30 and 31.
CA inhibition by occlusion of the active site entrance
shows schematically the third main CA inhibition mechanism, i.e. active site entrance occlusion. These inhibitors bind even further away from the metal ion compared to the zinc binders ( and ) or the compounds anchoring to the zinc-coordinated water molecule (), basically at the entrance of the active site cavity (which is the most variable region between various isoforms otherwise rather homologous with each other, as the 16 mammalian onesCitation1,5–8). Compounds acting by this mechanism of inhibition possess a sticky group (SG) attached to a scaffold which can be aromatic, heterocyclic, or aliphatic (). They may also incorporate a tail, which can extend away from the active site, since, as mentioned above, these compounds bind in a rather external region of the cavity. This inhibition mechanism has been evidenced for the first time for coumarinsCitation124,Citation125 but thereafter other compounds such as the antiepileptic drug lacosamideCitation126, substituted coumarinsCitation127–131, 5- and 6-membered lactones and thiolactonesCitation132 or quinolinonesCitation133 (many of them display some structural similarity to coumarin 31, the simplest such derivative) were observed to possess significant CA inhibitory properties and probably share a common mechanism of action.
Figure 8. Compounds which inhibit the CAs by occluding the entrance to the active site. SG represents a sticky group, of the phenol, carboxylic acid or amide type, the scaffold binds at the entrance of the active site cavity, occluding it, and a tail may be also present, which will interact with residues on the surface of the proteinCitation115–117.
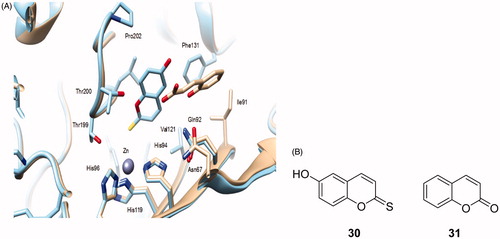
The first compound with this interesting CA inhibition mechanism was the natural product coumarin 32, isolated from the Australian plant Leionema ellipticumCitation124. When this natural product, 6-(1S-hydroxy-3-methylbutyl)-7-methoxy-2H-chromen-2-one 32 as well as the simple, unsubstituted coumarin 31 were cocrystallized with hCA II, the electron density data showed the presence of the hydrolyzed derivatives 32a and 31a, respectively, bound within the CA active site (). The formation of the 2-hydroxy-cinnamic acids 31a and 31b, which represent the de facto enzyme inhibitors is shown schematically in . The α-CAs possess esterase activity which is responsible for the hydrolysis of the lactone ring of coumarins or thiocoumarins (these last compounds were also shown to act as CAIsCitation125). However, the rather bulky nature of the obtained hydrolysis products interferes with their binding in the neighborhood of the metal ion. Thus, the obtained 2-hydroxy-cinnamic acids may bind as cis isomers (as for 32aCitation124) or as trans isomers (as for 31aCitation125) depending on how bulky the R and R′ moieties present on the scaffold are. Indeed, for less bulky groups (R = R′= H), the trans isomer was observedCitation125, whereas for bulkier such groups the isomerization did not occurCitation124. This difference is well observed in the superposition of the two hCA II – hydrolyzed coumarin adducts shown in . The most notable aspect of this inhibition mechanism is the fact that the inhibitors bind in an active site region which is the most variable between the various isoforms, i.e. the entrance to the cavityCitation1,5–8. This has important consequences for the drug design of CAIs, since compounds binding in that region should in principle interact differently with the various CAs and thus show isoform-selective inhibitory profiles. In fact, this was indeed the case when a larger series of diversely substituted coumarin/thiocoumarin derivatives of types 33–51 were investigatedCitation125 (), which showed a high level of isoform selective behavior against many isoforms such as CA IX, XII, XIII, XIV, etc.Citation125. Subsequent work from our group showed this situation to be the case for many classes of coumarins/thiocoumarins as well as structurally related derivatives which were investigated as CAIs against a large number of human isoformsCitation125–133.
Figure 9. (A) Binding of the coumarin 31 hydrolysis product (trans-2-hydroxy-cinnamic acid 31a, in yellow) and coumarin 32 hydrolysis product (cis-2-hydroxycinnamic acid 32a, magenta) to the hCA II active site. The protein backbone is shown as green ribbon, the catalytic Zn(II) ion as violet sphere, with its three protein ligands (His94, 96, and 119, CPK colors) also evidenced. The proton shuttle residue (His64) is shown in red. (B) Coumarins 31 and 32 and their CA-mediated hydrolysis to 31a and 32a, respectivelyCitation124,Citation125.
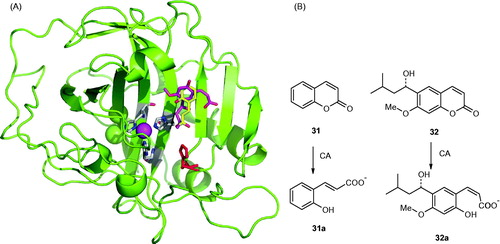
Figure 10. Proposed inhibition mechanism of CAs by coumarins/thiocoumarins, leading to cis- or trans-2-hydroxy/mercapto-cinnamic acids. (A) Hydrolysis of the lactone ring. (B) Movement of the hydrolysis product (as cis stereoisomer) toward the entrance of the active site cavity. (C) Cis-trans isomerization of the hydrolysis productCitation124,125.
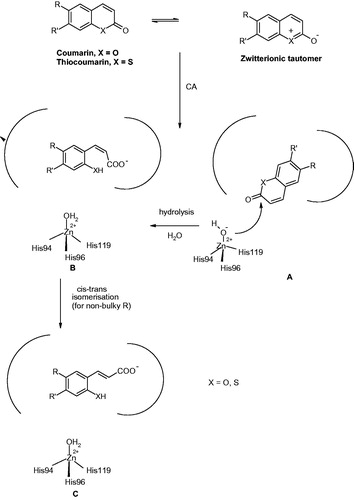
Figure 11. Some of the investigated coumarins 33–51 as CAIs which showed highly isoform-selective inhibition profile against various human CA isoformsCitation124,125.
It is also interesting to note that considering coumarins as leads, the sulfocoumarins were reported (in which the CO moiety of the lactone was replaced by a SO2 group)Citation114. Although the coumarins and the sulfocoumarins have a different inhibition mechanism (coumarins bind in hydrolyzed form at the entrance of the cavity, as discussed here, whereas sulfocoumarins also bind in hydrolyzed form but by anchoring to the zinc-coordinated water) many sulfocoumarins reported so far showed a very high degree of isoform selectivity toward the trans-membrane tumor-associated isoforms hCA IX and XII compared to the cytosolic ones hCA I and IICitation114,134–138.
Out of the active site binding as a CA inhibition mechanism
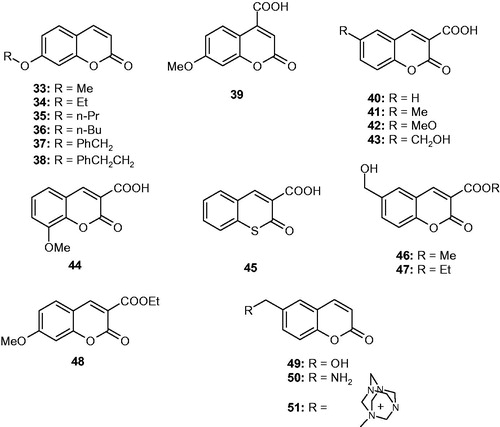
The CA inhibition mechanism shown in is the last one to be discovered, quite recentlyCitation139. When the 2-(benzylsulfonyl)-benzoic acid 52 was co-crystallized with hCA II, the electronic density of the inhibitor was not observed within the active site but in an adjacent binding pocket to the active site, where other inhibitors were never observed earlier (). Thus, this compound binds quite far away, outside the active site of the enzyme. But how does it inhibit the CA activity when the binding is in an outside region of the active site pocket? The same crystallographic studyCitation139 showed that 52 blocks the proton shuttle of the enzyme, His64, in its out conformation, interfering thus with the rate determining step of the entire catalytic cycle, which is indeed the transfer of a proton from the zinc-coordinated water molecule to the environment, with generation of the zinc hydroxide species of the enzymeCitation1,5–10,139. This amino acid residue (His64) has a high flexibility with two main conformations, the in one (closer to the metal ion) and the out one, toward the exit of the active site cavity. By accepting a proton from the zinc-coordinated water, the imidazole moiety of this residue becomes protonated (in its in conformation) whereas when it is in the out one, the same proton may be transferred to the environment. Interfering with this process may block the entire catalytic cycle, and this exactly how compound 52 inhibits the enzyme, blocking the proton shuttle through a network of hydrogen bonds showed in detail in Citation139.
Figure 12. Out of the active site CA inhibition mechanism. (A) Schematic representation of the inhibitor binding site. (B) hCA II complexed with the carboxylic acid 52. (C) Interactions between the inhibitor 52 and amino acid residues from the binding pocket. (D) Chemical structure of 52Citation139.
Compounds acting as CAIs with an unknown mechanism of action
The four inhibition mechanisms of the compounds discussed up until now are well understood as they were thoroughly documented by means of kinetic and more importantly, X-ray crystallographic studies. However in the last period relevant evidence accumulated on other classes of compounds, which show various levels of CA inhibitory properties, but for which the precise mechanism of action is not known due to the fact that they were not yet crystallized in adducts with the enzyme. This is most of the time due to solubility problems for some of these derivatives, but it should be also stressed that many classes of CAIs only recently started to be investigatedCitation140–146.
The secondary sulfonamide moiety is present in a large variety of clinically used drugs, among which the sulfa drugs are the best-known examplesCitation17,Citation140. Alp et al.Citation141 investigated a panel of such derivatives, of type 53–70 () – including sulfadiazine 69 and sulfapyridine 70 – for their interaction with the physiologically dominant isoforms hCA I and II, proving that many of them show sub-micromolar affinity for both enzymes. The investigated derivatives incorporated both secondary sulfonamide (53–62, 69, and 70) as well as tertiary sulfonamide (63–68) functionalities in their molecules, as well as quite bulky substituents at the sulfonamide group, and clearly are unable to bind to the metal ion from the CA active site due to steric hindrance. In fact the available space in the active site funnel, at its bottom, is rather restricted to allow such sterically impaired compoundsCitation1,5,140–146. It has been hypothesized that similar to lacosamideCitation126 and the hydrolyzed coumarinsCitation124,Citation125, these CAIs may bind at the entrance of the active site, in the coumarin-binding site (discussed in the section “CA inhibition by occlusion of the active site entrance” of this review), but this hypothesis has not yet been confirmed by X-ray crystallography. However many such secondary/tyertiary sulfonamides were thereafter investigated as CAIsCitation142–153 as well as structurally related sulfanates/sulfamidesCitation153, some of which are shown in . By using superacid chemistry Thibaudeau’s group reported a wide range of halogenated secondary/tertiary derivatives (e.g. 71–73), some of which showed selective inhibitory action against the tumor-associated isoforms hCA IX/XII, without significant inhibition of hCA I and IICitation145–148. By using saccharin 11 as leadCitation46, Carradori’s group reported a range of N-substituted saccharins, among which 74–77 (and some of their congeners) showed CA XII selective inhibitory actionCitation149. AcesulfameCitation150 or probenecid 78 derivatives of type 79, incorporating a variety of aliphatic, aromatic, or amino acid moieties derivatizing the carboxylic acid moiety of 78Citation151–153 were also prepared and investigated as inhibitors of at least four relevant isoforms, hCA I, II; IX and XII; by the same groups. Again notable CA selective inhibitory activity has been observed for many such tertiary sulfonamide derivatives, although their inhibition mechanism is unclear at this moment.
Figure 13. Secondary/tertiary sulfonamides 53–70 initially investigated as CAIsCitation141.
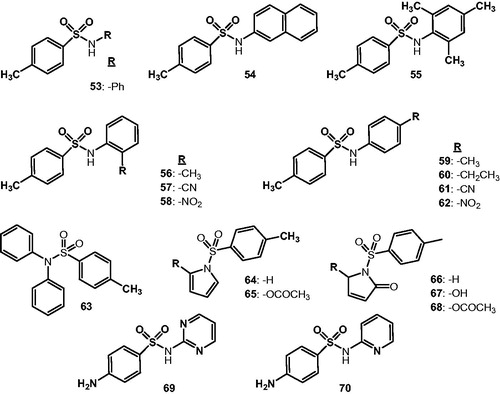
Figure 14. Secondary/tertiary sulfonamides, sulfamates, and sulfamides investigated as CAIsCitation145–154.
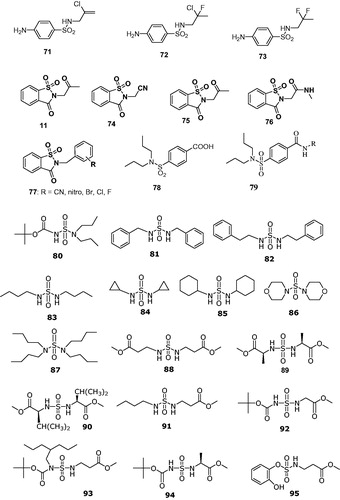
Gavernet et al.Citation154 explored N,N′-disubstituted sulfamides of types 80–94 or the N,O-disubstituted sulfamate 95, which showed a broad range of inhibitory activity and selectivity: some aromatic sulfamides were active against hCA I, hCA II and/or hCA VII, while they were less active against hCA XII and XIV. On the other hand, several bulky sulfamides were selective hCA VII inhibitorsCitation154. The inhibitory mechanism of these compounds is also unknown, but it is rather probable that their bulky substituents at the NHSO2X (X = NH or O) fragment of the molecule impairs their binding to the metal ion within the CA active site, as for derivatives 53–79 discussed earlier.
Effective, nanomolar inhibitory activity against several CA isoforms was also reported for the protein tyrosine kinase inhibitors and clinically used drugs imatinib and nilotinibCitation155, for some amino acid derivatized fullerenesCitation156 as well as for the murine CA inhibitor mICA, a protein belonging to the transferrin familyCitation157. Also in these cases the inhibition mechanism is unknown.
Fabricated data and scientific misconduct issues of the CA field
As with all successful fields there is however also the reverse side of the medal, as the CA inhibitors field showed ultimately an increase in the number of fabricated data and similar scientific misconduct issues, some of which I will dealt with in the last part of the review. I have already mentioned in an earlier reviewCitation6 that the “two-prong” approachCitation158 (based on our tail approach published several years earlierCitation81,Citation82) is probably fabricated. According to this approach, ideated by Srivastava’s group, iminodiacetic (IDA) moieties (which complexate Cu2+) were attached to benzenesulfonamides via different chain length spacers, leading to the so-called “two-prong” inhibitors. In these CAIs one prong (the SO2NH− moiety) would coordinate the zinc ion, whereas the second one (containing the copper ions) would bind to the various His residues from the histidine cluster comprising His3, 4, 10, 15, and 64 (at least in the hCA II active site)Citation23. The His cluster was reported by Briganti et al.Citation23 whereas Zn(II), Cu(II), Co(II) and many other metal complexes of aromatic/heterocyclic sulfonamides incorporating polyamino-polycarboxylic acids (including IDA) in 2002 by Scozzafava et al.Citation48 In all cases in which such metal-containing CAIs were investigated by our group, we observed only binding of the metal ion to His64 and not to the other His residues from the His cluster mentioned aboveCitation48. Compounds sent by Srivastava in our laboratory in 2005–2006, did not show the inhibition pattern published in their paperCitation158 and the X-ray crystallography performed with some of these compounds (unpublished data from this and De Simone laboratory) showed the normal binding of the sulfonamide to the zinc ion and the Cu(II) always bound to His64. A subsequent crystallographic work from the same groupCitation159 again evidenced binding of the Cu(II) only to His64, but the conclusion of the work was that the two-prong approach was working. Finally, we investigated some compounds prepared by Vomasta et al.Citation160 following exactly the two-prong approach, and their inhibition data against four CA isoforms with various His residues in their cluster, among which hCA I, II, IX, and XII. They did not show any two-prong effects, as both mono-and two-prong compounds showed rather similar inhibitory propertiesCitation160. We thus clearly demonstrated that this apparently interesting approach, unfortunately does not work, probably because His64, unlike the other His residues from the cluster, has a high mobility, with its imidazole ring not only participating in the proton transfer processes as mentioned earlier in this review, but also having higher affinity for complexating metal ions (e.g. Cu2+) compared to the other His residues from the cluster (which are less exposed to the solvent).
The second unfortunate example that I shall present here is from a research group in Shanghai, ChinaCitation161–164. In several papers this group presented interesting scaffolds of aromatic/heterocyclic sulfonamides and their inhibition data mainly against hCA II and IX (as this last isoform is an attractive target due to its involvement in tumorigenesisCitation1,Citation2,Citation19). Initially I was rather enthusiastic of these papersCitation161–164 as they were mainly based on the tail approach developed by my group in the late 90sCitation81,Citation82. However when I read their experimental parts I realized that the enzyme inhibition procedure was basically exactly the one from our papers. Furthermore, all (but really all) the instrumentation that this group claimed to possess was identical to the one from my laboratory in Florence (e.g. stopped-flow spectrophotometer, UV-VIS spectrophotometer, etc.) and the enzymes were claimed to have been cloned exactly by our methods, although these authors did not ask the plasmids or the cDNAs encoding those CAs from me or my other associates. I thus started to suspect misconduct and contacted the group, asking just one of their published inhibitors to be sent to my laboratory. Initially they told me that it impossible to send compounds from China to Europe (which is not true as we received samples from other Chinese laboratories), and afterwards that they need to re-prepare the compounds (there were many of them in the papers, and I asked whatever one to be sent). I concluded that this are all fabricated data, and informed the Editors of the journals in which these papers were published, whom however did not take any action. It is of course a pity that this type of scientific misconduct pollutes our wonderful field. What is even worse is that many of these dishonest authors act thereafter as reviewers of our or other groups’ papers (which indeed present high-quality science) and create problems for the publication of the genuine data (whereas the falsified ones are still there in the literature). This is also one of the reasons why I raised this problem in the present review. Unfortunately there are many other similar examples which I will not present here for lack of space.
Conclusions
By catalyzing the simple but highly important hydration of carbon dioxide to bicarbonate and protons, CAs are involved in critical steps of the life cycle of many organisms, including eukaryotes, Bacteria and Archaea. In fact a large number of new such enzymes are constantly being cloned, purified, and characterized in detail from many pathogenic as well as nonpathogenic organisms. Modulation of the activity of these enzymes, mainly by using selective inhibitors, has been used pharmacologically for decades for various applications (diuretics, antiglaucoma agents, antiepileptics, antiobesity drugs and more recently anti-tumor therapies/diagnostic agents as well as novel anti-infectivesCitation1–10,14–19,21 Citation165–167). One of the main problems with the CAIs was however the lack of selectivity of the sulfonamides for the inhibition of the many mammalian (human) CAs, which led to a range of side effects for the first and second generation inhibitors of types 1–19Citation1–10,14–19,21,165–167. However, this situation changed substantially in the last decade, when a large number of isoform-selective primary sulfonamidesCitation63,Citation64,Citation70,Citation71 were reported by using the tail approachCitation47–49,80–82 and with the discovery of new classes of CAIs, such as the coumarins and their isosters, the polyamines, DTCs, xanthates, etc., as shown in detail in this review. An overview on the history of CAI drug discovery is shown in .
Figure 15. Historic overview of the CAI drug design panorama.
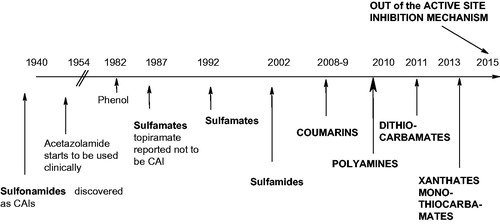
As mentioned throughout this review, the sulfonamides are the most important class of CAIs, discovered already in 1940, with the first clinically approved drug, acetazolamide 1, launched in 1954Citation1,5–9. For the next 5 decades, there were only variations on the sulfonamide theme with the sulfamates and sulfamides the most logical class of compounds to be considered as CAIsCitation161. Indeed, such an activity has been demonstrated for the simple, inorganic sulfamide and sulfamateCitation168, although Maryanoff et al.Citation169 reported that some sugar sulfamates (one of which later became the antiepileptic drug topiramate 10) were devoid of CA inhibitory properties. We later showed that topiramate is a low nanomolar CAI against many isoformsCitation170. Indeed, a wide range of primary sulfamates and sulfamides were shown to possess potent such properties, in all aspects similar to the isosteric sulfonamides, considering the binding to the enzyme, mechanism of action, pharmacology, etc.Citation1,Citation5,Citation6,Citation167.
However the breakthrough in the field was represented by the discovery of coumarins as a totally new chemotype with a particular inhibition mechanism, which has been discussed in detail in this review articleCitation124,Citation125. As shown here, coumarins were also used as lead compounds, leading to the discovery of a large number of structurally related chemotypes possessing prodrug-like CA inhibitory properties, among which the thiocoumarins, thioxocoumarins, coumarin oximes, 5-/6-membered ring lactones and thiolactones, sulfocoumarins, etc.Citation114,127–138. Polyamines were then discovered as CAIs in 2010Citation104 (), followed shortly thereafter by DTCs and xanthatesCitation28–30,83,84. Finally the out of the active site CA inhibition mechanism was reported in 2015Citation139. A particular feature of the new classes of CAIs discovered in the last years is that they show a much higher isoform-selectivity profile compared to most sulfonamides and sulfamates (of which 1–19 are the typical representatives, see ). Thus, there is enthusiasm and optimism that the new generations of CAIs will show less side effects and more efficacy for the treatment of the classical pathologies associated with these enzymes but also to newer ones such as neuropathic painCitation171, cerebral ischemiaCitation172,Citation173, and obviously tumors (with SLC-0111 20 in clinical trials, as mentioned earlierCitation2,Citation19,Citation54).
Despite the problem of the fabricated data and similar scientific misconduct issues described in paragraph 7, I will conclude this article in an optimistic mannerCitation174. The review presented here is a continuation of the highly cited ones from 2008Citation1 and 2012Citation5. Moreover, the field of the CAs and their inhibitors lived a great period in the last decade, with a lot of important discoveries being made. Several important mammalian isoforms (e.g. CA VA in obesity; CA VII in neuropathic pain; CA IX and XII in tumors) have been validated as drug targets and a large number of highly potent and selective inhibitors, belonging to a large number of diverse classes, reported and characterized in detail. The classical sulfonamide inhibitors are still massively used clinically but the hope is that in the nearby future novel CAIs belonging to the newly identified classes will reach the clinics and the patients in need for them.
Declaration of interest
The author declares conflict of interest, being author of several patents in the field of CA inhibitors. Research from my laboratory was financed by several EU projects (Euroxy, Metoxia, DeZnIt, and Dynano).
References
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77
- Capasso C, Supuran CT. An overview of the alpha-, beta- and gamma-carbonic anhydrases from Bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 2015;30:325–32
- Supuran CT, Capasso C. The η-class carbonic anhydrases as drug targets for antimalarial agents. Expert Opin Ther Targets 2015;19:551–63
- Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72
- Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;20:3467–74
- Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enzyme Inhib Med Chem 2004;19:199–229
- Supuran CT. Carbonic anhydrase inhibitors and activators for novel therapeutic applications. Future Med Chem 2011;3:1165–80
- Supuran CT. Bacterial carbonic anhydrases as drug targets: toward novel antibiotics? Front Pharmacol 2011;2:34
- Supuran CT, Scozzafava A, Casini A. Carbonic anhydrase inhibitors. Med Res Rev 2003;23:146–89
- Del Prete S, Vullo D, De Luca V, et al. Biochemical characterization of the δ-carbonic anhydrase from the marine diatom Thalassiosira weissflogii, TweCA. J Enzyme Inhib Med Chem 2014;29:906–11
- De Simone G, Supuran CT. (In)organic anions as carbonic anhydrase inhibitors. J Inorg Biochem 2012;111:117–29
- Del Prete S, Vullo D, De Luca V, et al. Biochemical characterization of recombinant β-carbonic anhydrase (PgiCAb) identified in the genome of the oral pathogenic bacterium Porphyromonas gingivalis. J Enzyme Inhib Med Chem 2015;30:366–70
- Supuran CT. Carbonic anhydrase inhibitors: an editorial. Expert Opin Ther Pat 2013;23:677–9
- Supuran CT. Carbonic anhydrases: from biomedical applications of the inhibitors and activators to biotechnological use for CO(2) capture. J Enzyme Inhib Med Chem 2013;28:229–30
- Capasso C, Supuran CT. Anti-infective carbonic anhydrase inhibitors: a patent and literature review. Expert Opin Ther Pat 2013;23:693–704
- Capasso C, Supuran CT. Sulfa and trimethoprim-like drugs – antimetabolites acting as carbonic anhydrase, dihydropteroate synthase and dihydrofolate reductase inhibitors. J Enzyme Inhib Med Chem 2014;29:379–87
- Supuran CT, Scozzafava A. Carbonic anhydrases as targets for medicinal chemistry. Bioorg Med Chem 2007;15:4336–50
- Lou Y, McDonald PC, Oloumi A, et al. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res 2011;71:3364–76
- Zimmerman S, Innocenti A, Casini A, et al. Carbonic anhydrase inhibitors. Inhibition of the prokaryotic beta and gamma-class enzymes from Archaea with sulfonamides. Bioorg Med Chem Lett 2004;14:6001–6
- Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68
- Temperini C, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. X-Ray crystal studies of the carbonic anhydrase II-trithiocarbonate adduct-an inhibitor mimicking the sulfonamide and urea binding to the enzyme. Bioorg Med Chem Lett 2010;20:474–8
- Briganti F, Mangani S, Orioli P, et al. Carbonic anhydrase activators: X-ray crystallographic and spectroscopic investigations for the interaction of isozymes I and II with histamine. Biochemistry 1997;36:10384–92
- Temperini C, Scozzafava A, Vullo D, Supuran CT. Carbonic anhydrase activators. Activation of isozymes I, II, IV, VA, VII and XIV with L- and D-histidine and crystallographic analysis of their adducts with isoform II: engineering proton transfer processes within the active site of an enzyme. Chemistry 2006;12:7057–66
- Vullo D, De Luca V, Del Prete S, et al. Sulfonamide inhibition studies of the γ-carbonic anhydrase from the Antarctic cyanobacterium Nostoc commune. Bioorg Med Chem 2015;23:1728–34
- Schlicker C, Hall RA, Vullo D, et al. Structure and inhibition of the CO2-sensing carbonic anhydrase Can2 from the pathogenic fungus Cryptococcus neoformans. J Mol Biol 2009;385:1207–20
- Pacchiano F, Carta F, Vullo D, et al. Inhibition of β-carbonic anhydrases with ureido-substituted benzenesulfonamides. Bioorg Med Chem Lett 2010;20:102–5
- Carta F, Aggarwal M, Maresca A, et al. Dithiocarbamates strongly inhibit carbonic anhydrases and show antiglaucoma action in vivo. J Med Chem 2012;55:1721–30
- Monti SM, Maresca A, Viparelli F, et al. Dithiocarbamates strongly inhibit the beta-class fungal carbonic anhydrases from Cryptococcus neoformans, Candida albicans and Candida glabrata. Bioorg Med Chem Lett 2012;22:859–62
- Maresca A, Carta F, Vullo D, Supuran CT. Dithiocarbamates strongly inhibit the β-class carbonic anhydrases from Mycobacterium tuberculosis. J Enzyme Inhib Med Chem 2013;28:407–11
- Carta F, Innocenti A, Hall RA, et al. Carbonic anhydrase inhibitors. Inhibition of the β-class enzymes from the fungal pathogens Candida albicans and Cryptococcus neoformans with branched aliphatic-/aromatic carboxylates and their derivatives. Bioorg Med Chem Lett 2011;21:2521–6
- Nishimori I, Onishi S, Takeuchi H, Supuran CT. The alpha and beta classes carbonic anhydrases from Helicobacter pylori as novel drug targets. Curr Pharm Des 2008;14:622–30
- Minakuchi T, Nishimori I, Vullo D, et al. Molecular cloning, characterization, and inhibition studies of the Rv1284 beta-carbonic anhydrase from Mycobacterium tuberculosis with sulfonamides and a sulfamate. J Med Chem 2009;52:2226–32
- Nishimori I, Minakuchi T, Vullo D, et al. Carbonic anhydrase inhibitors. Cloning, characterization, and inhibition studies of a new β-carbonic anhydrase from Mycobacterium tuberculosis. J Med Chem 2009;52:3116–20
- Güzel Ö, Maresca A, Scozzafava A, et al. Discovery of low nanomolar and subnanomolar inhibitors of the mycobacterial beta-carbonic anhydrases Rv1284 and Rv3273. J Med Chem 2009;52:4063–7
- Carta F, Maresca A, Suarez Covarrubias A, et al. Carbonic anhydrase inhibitors. Characterization and inhibition studies of the most active beta-carbonic anhydrase from Mycobacterium tuberculosis, Rv3588c. Bioorg Med Chem Lett 2009;19:6649–54
- Nishimori I, Minakuchi T, Maresca A, et al. The β-carbonic anhydrases from Mycobacterium tuberculosis as drug targets. Curr Pharm Des 2010;16:3300–9
- Winum JY, Kohler S, Supuran CT. Brucella carbonic anhydrases: new targets for designing anti-infective agents. Curr Pharm Des 2010;16:3310–16
- Vullo D, Nishimori I, Minakuchi T, et al. Inhibition studies with anions and small molecules of two novel β-carbonic anhydrases from the bacterial pathogen Salmonella enterica serovar Typhimurium. Bioorg Med Chem Lett 2011;21:3591–5
- Maresca A, Vullo D, Scozzafava A, Supuran CT. Inhibition of the alpha- and beta-carbonic anhydrases from the gastric pathogen Helycobacter pylori with anions. J Enzyme Inhib Med Chem 2013;28:388–91
- Maresca A, Vullo D, Scozzafava A, et al. Inhibition of the beta-class carbonic anhydrases from Mycobacterium tuberculosis with carboxylic acids. J Enzyme Inhib Med Chem 2013;28:392–6
- Vullo D, Nishimori I, Scozzafava A, et al. Inhibition studies of a beta-carbonic anhydrase from Brucella suis with a series of water soluble glycosyl sulfanilamides. Bioorg Med Chem Lett 2010;20:2178–82
- Nishimori I, Minakuchi T, Kohsaki T, et al. Carbonic anhydrase inhibitors: the beta-carbonic anhydrase from Helicobacter pylori is a new target for sulfonamide and sulfamate inhibitors. Bioorg Med Chem Lett 2007;17:3585–94
- Weber A, Casini A, Heine A, et al. Unexpected nanomolar inhibition of carbonic anhydrase by COX-2-selective celecoxib: new pharmacological opportunities due to related binding site recognition. J Med Chem 2004;47:550–7
- Di Fiore A, Pedone C, D’Ambrosio K, et al. Carbonic anhydrase inhibitors: valdecoxib binds to a different active site region of the human isoform II as compared to the structurally related, cyclooxygenase II “selective” inhibitor celecoxib. Bioorg Med Chem Lett 2006;16:437–42
- Köhler K, Hillebrecht A, Schulze Wischeler J, et al. Saccharin inhibits carbonic anhydrases: possible explanation for its unpleasant metallic aftertaste. Angew Chem Int Ed Engl 2007;46:7697–9
- Casini A, Scozzafava A, Mincione F, et al. Carbonic anhydrase inhibitors: water-soluble 4-sulfamoylphenylthioureas as topical intraocular pressure-lowering agents with long-lasting effects. J Med Chem 2000;43:4884–92
- Scozzafava A, Menabuoni L, Mincione F, Supuran CT. Carbonic anhydrase inhibitors. A general approach for the preparation of water-soluble sulfonamides incorporating polyamino-polycarboxylate tails and of their metal complexes possessing long-lasting, topical intraocular pressure-lowering properties. J MedChem 2002;45:1466–76
- Fabrizi F, Mincione F, Somma T, et al. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J Enzyme Inhib Med Chem 2012;27:138–47
- Ebbesen P, Pettersen EO, Gorr TA, et al. Taking advantage of tumor cell adaptations to hypoxia for developing new tumor markers and treatment strategies. J Enzyme Inhib Med Chem 2009;24:1–39
- Švastová E, Hulíková A, Rafajová M, et al. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett 2004;577:439–45
- Dubois L, Lieuwes NG, Maresca A, et al. Imaging of CA IX with fluorescent labelled sulfonamides distinguishes hypoxic and (re)-oxygenated cells in a xenograft tumour model. Radiother Oncol 2009;92:423–8
- Ahlskog JKJ, Dumelin CE, Trüssel S, et al. In vivo targeting of tumor-associated carbonic anhydrases using acetazolamide derivatives. Bioorg Med Chem Lett 2009;19:4851–6
- Pacchiano F, Carta F, McDonald PC, et al. Ureido-substituted benzenesulfonamides potently inhibit carbonic anhydrase IX and show antimetastatic activity in a model of breast cancer metastasis. J Med Chem 2011;54:1896–902
- Mincione F, Scozzafava A, Supuran CT. The development of topically acting carbonic anhydrase inhibitors as anti-glaucoma agents. Curr Top Med Chem 2007;7:849–54
- Carta F, Supuran CT, Scozzafava A. Novel therapies for glaucoma: a patent review 2007–2011. Expert Opin Ther Pat 2012;22:79–88
- Steele RM, Batugo MR, Benedini F, et al. Nitric oxide-donating carbonic anhydrase inhibitors for the treatment of open-angle glaucoma. Bioorg Med Chem Lett 2009;19:6565–70
- Mincione F, Benedini F, Biondi S, et al. Synthesis and crystallographic analysis of new sulfonamides incorporating NO-donating moieties with potent antiglaucoma action. Bioorg Med Chem Lett 2011;21:3216–21
- Alterio V, Vitale RM, Monti SM, et al. Carbonic anhydrase inhibitors: X-ray and molecular modeling study for the interaction of a fluorescent antitumor sulfonamide with isozyme II and IX. J Am Chem Soc 2006;128:8329–35
- Alterio V, De Simone G, Monti SM, et al. Carbonic anhydrase inhibitors: inhibition of human, bacterial, and archaeal isozymes with benzene-1,3-disulfonamides-Solution and crystallographic studies. Bioorg Med Chem Lett 2007;17:4201–7
- Wagner J, Avvaru BS, Robbins AH, et al. Coumarinyl-substituted sulfonamides strongly inhibit several human carbonic anhydrase isoforms: solution and crystallographic investigations. Bioorg Med Chem 2010;18:4873–8
- Biswas S, Aggarwal M, Guzel O, et al. Conformational variability of different sulfonamide inhibitors with thienyl-acetamido moieties attributes to differential binding in the active site of cytosolic human carbonic anhydrase isoforms. Bioorg Med Chem 2011;19:3732–8
- Pacchiano F, Aggarwal M, Avvaru BS, et al. Selective hydrophobic pocket binding observed within the carbonic anhydrase II active site accommodate different 4-substituted-ureido-benzenesulfonamides and correlate to inhibitor potency. Chem Commun (Camb) 2010;46:8371–3
- Carta F, Garaj V, Maresca A, et al. Sulfonamides incorporating 1,3,5-triazine moieties selectively and potently inhibit carbonic anhydrase transmembrane isoforms IX, XII and XIV over cytosolic isoforms I and II: solution and X-ray crystallographic studies. Bioorg Med Chem 2011;19:3105–19
- Hen N, Bialer M, Yagen B, et al. Anticonvulsant 4-aminobenzenesulfonamide derivatives with branched-alkylamide moieties: X-ray crystallography and inhibition studies of human carbonic anhydrase isoforms I, II, VII and XIV. J Med Chem 2011;54:3977–81
- Kolayli S, Karahalil F, Sahin H, et al. Characterization and inhibition studies of an α-carbonic anhydrase from the endangered sturgeon species Acipenser gueldenstaedti. J Enzyme Inhib Med Chem 2011;26:895–900
- Güzel Ö, Innocenti A, Scozzafava A, et al. Carbonic anhydrase inhibitors. Phenacetyl-, pyridylacetyl- and thienylacetyl-substituted aromatic sulfonamides act as potent and selective isoform VII inhibitors. Bioorg Med Chem Lett 2009;19:3170–3
- Güzel Ö, Innocenti A, Scozzafava A, et al. Carbonic anhydrase inhibitors. Aromatic/heterocyclic sulfonamides incorporating phenacetyl-, pyridylacetyl- and thienylacetyl-tails act as potent inhibitors of human mitochondrial isoforms VA and VB. Bioorg Med Chem 2009;17:4894–9
- Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Arylsulfonylureido and arylureido-substituted aromatic and heterocyclic sulfonamides: towards selective inhibitors of carbonic anhydrase isozyme I. J Enzyme Inhib 1999;14:343–63
- Garaj V, Puccetti L, Fasolis G, et al. Carbonic anhydrase inhibitors: synthesis and inhibition of cytosolic/tumor-associated carbonic anhydrase isozymes I, II and IX with sulfonamides incorporating 1,2,4-triazine moieties. Bioorg Med Chem Lett 2004;14:5427–33
- Garaj V, Puccetti L, Fasolis G, et al. Carbonic anhydrase inhibitors. Novel sulfonamides incorporating 1,3,5-triazine moieties as inhibitors of the cytosolic and tumor-associated carbonic anhydrase isozymes I, II and IX. Bioorg Med Chem Lett 2005;15:3102–8
- McDonald PC, Winum JY, Supuran CT, Dedhar S. Recent developments in targeting carbonic anhydrase IX for cancer therapeutics. Oncotarget 2012;3:84–97
- Carta F, Scozzafava A, Supuran CT. Sulfonamides (RSO2NH2): a patent review 2008-2012. Expert Opin Ther Pat 2012;22:747–58
- Masini E, Carta F, Scozzafava A, Supuran CT. Antiglaucoma carbonic anhydrase inhibitors: a patent review. Expert Opin Ther Pat 2013;23:705–16
- Monti SM, Supuran CT, De Simone G. Anticancer carbonic anhydrase inhibitors: a patent review (2008--2013). Expert Opin Ther Pat 2013;23:737–49
- Carta F, Supuran CT. Diuretics with carbonic anhydrase inhibitory action: a patent and literature review (2005--2013). Expert Opin Ther Pat 2013;23:681–91
- Supuran CT. Carbonic anhydrase inhibitors as emerging drugs for the treatment of obesity. Expert Opin Emerg Drugs 2012;17:11–15
- Scozzafava A, Supuran CT, Carta F. Antiobesity carbonic anhydrase inhibitors: a literature and patent review. Expert Opin Ther Pat 2013;23:725–35
- Supuran CT. The safety and clinical efficacy of acetazolamide for the treatment of idiopathic intracranial hypertension. Expert Rev Neurother 2015;15:851–6
- De Simone G, Alterio V, Supuran CT. Exploiting the hydrophobic and hydrophilic binding sites for designing carbonic anhydrase inhibitors. Expert Opin Drug Discov 2013;8:793–810
- Scozzafava A, Menabuoni L, Mincione F, et al. Carbonic anhydrase inhibitors. Synthesis of water-soluble, topically effective, intraocular pressure-lowering aromatic/heterocyclic sulfonamides containing cationic or anionic moieties: is the tail more important than the ring? J Med Chem 1999;42:2641–50
- Borras J, Scozzafava A, Menabuoni L, et al. Carbonic anhydrase inhibitors. Part 73. Synthesis of water-soluble, topically effective intraocular pressure lowering aromatic/heterocyclic sulfonamides containing 8-quinoline-sulfonyl moieties: is the tail more important than the ring? Bioorg Med Chem 1999;7:2397–406
- Carta F, Aggarwal M, Maresca A, et al. Dithiocarbamates: a new class of carbonic anhydrase inhibitors. Crystallographic and kinetic investigations. Chem Commun (Camb) 2012;48:1868–70
- Carta F, Akdemir A, Scozzafava A, et al. Xanthates and trithiocarbonates strongly inhibit carbonic anhydrases and show antiglaucoma effects in vivo. J Med Chem 2013;56:4691–700
- Mori M, Cau Y, Vignaroli G, et al. Hit recycling: discovery of a potent carbonic anhydrase inhibitor by in silico target fishing. ACS Chem Biol 2015;10:1964–9
- Innocenti A, Vullo D, Scozzafava A, et al. Carbonic anhydrase inhibitors. Interaction of isozymes I, II, IV, V and IX with carboxylates. Bioorg Med Chem Lett 2005;15:573–8
- Rusconi S, Innocenti A, Vullo D, et al. Carbonic anhydrase inhibitors. Interaction of isozymes I, II, IV, V and IX with phosphates, carbamoyl phosphate and the phosphonate antiviral drug foscarnet. Bioorg Med Chem Lett 2004;14:5763–7
- Temperini C, Innocenti A, Guerri A, et al. Phosph(on)ate as a zinc binding group in metalloenzyme inhibitors: X-ray crystal structure of the antiviral drug foscarnet complexed to human carbonic anhydrase I. Bioorg Med Chem Lett 2007;17:2210–15
- Tauro M, Loiodice F, Ceruso M, et al. Dual carbonic anhydrase/matrix metalloproteinase inhibitors incorporating bisphosphonic acid moieties targeting bone tumors. Bioorg Med Chem Lett 2014;24:2617–20
- Tauro M, Loiodice F, Ceruso M, et al. Arylamino bisphosphonates: potent and selective inhibitors of the tumor-associated carbonic anhydrase XII. Bioorg Med Chem Lett 2014;24:1941–3
- Reich R, Hoffman A, Veerendhar A, et al. Carbamoylphosphonates control tumor cell proliferation and dissemination by simultaneously inhibiting carbonic anhydrase IX and matrix metalloproteinase-2. Toward nontoxic chemotherapy targeting tumor microenvironment. J Med Chem 2012;55:7875–82
- Reich R, Hoffman A, Suresh RR, et al. Carbamoylphosphonates inhibit autotaxin and metastasis formation in vivo. J Enzyme Inhib Med Chem 2015;30:767–72
- Scozzafava A, Supuran CT. Carbonic anhydrase and matrix metalloproteinase inhibitors: sulfonylated amino acid hydroxamates with MMP inhibitory properties act as efficient inhibitors of CA isozymes I, II, and IV, and N-hydroxysulfonamides inhibit both these zinc enzymes. J Med Chem 2000;43:3677–87
- Santos MA, Marques S, Gil M, et al. Protease inhibitors: synthesis of bacterial collagenase and matrix metalloproteinase inhibitors incorporating succinylhydroxamate and iminodiacetic acid hydroxamate moieties. J Enzyme Inhib Med Chem 2003;18:233–42
- Scozzafava A, Supuran CT. Hydroxyurea is a carbonic anhydrase inhibitor. Bioorg Med Chem 2003;11:2241–6
- Nuti E, Orlandini E, Nencetti S, et al. Carbonic anhydrase and matrix metalloproteinase inhibitors. Inhibition of human tumor-associated isozymes IX and cytosolic isozyme I and II with sulfonylated hydroxamates. Bioorg Med Chem 2007;15:2298–311
- Santos MA, Marques S, Vullo D, et al. Carbonic anhydrase inhibitors. Inhibition of cytosolic/tumor-associated isoforms I, II and IX with iminodiacetic carboxylates/hydroxamates also incorporating benzenesulfonamide moieties. Bioorg Med Chem Lett 2007;17:1538–43
- Marques SM, Nuti E, Rossello A, et al. Dual inhibitors of matrix metalloproteinases and carbonic anhydrases: iminodiacetyl-based hydroxamate-benzenesulfonamide conjugates. J Med Chem 2008;51:7968–79
- Di Fiore A, Maresca A, Supuran CT, De Simone G. Hydroxamate represents a versatile zinc binding group for the development of new carbonic anhydrase inhibitors. Chem Commun (Camb) 2012;48:8838–40
- Supuran CT. Hydroxamates as carbonic anhydrase inhibitors. In: Gupta SP, ed. Hydroxamic acids: a unique family of chemicals with multiple biological activities. Heidelberg: Springer Verlag; 2013: 55–69
- Langella E, D’Ambrosio K, D’Ascenzio M, et al. A combined crystallographic and theoretical study explains the capability of carboxylic acids to adopt multiple binding modes within carbonic anhydrase active site. Chemistry 2015; in press. doi: 10.1002/chem.201503748
- Bozdag M, Carta F, Vullo D, et al. Synthesis of a new series of dithiocarbamates with effective human carbonic anhydrase inhibitory activity and antiglaucoma action. Bioorg Med Chem 2015;23:2368–76
- Nair SK, Ludwig PA, Christianson DW. Two-site binding of phenol in the active site of human carbonic anhydrase II: structural implications for substrate association. J Am Chem Soc 1994;116:3659–60
- Carta F, Temperini C, Innocenti A, et al. Polyamines inhibit carbonic anhydrases by anchoring to the zinc-coordinated water molecule. J Med Chem 2010;53:5511–22
- Innocenti A, Vullo D, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Interactions of phenols with the 12 catalytically active mammalian isoforms (CA I–XIV). Bioorg Med Chem Lett 2008;18:1583–7
- Innocenti A, Vullo D, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of mammalian isoforms I–XIV with a series of substituted phenols including paracetamol and salicylic acid. Bioorg Med Chem 2008;16:7424–8
- Bayram E, Senturk M, Kufrevioglu OI, Supuran CT. In vitro inhibition of salicylic acid derivatives on human cytosolic carbonic anhydrase isozymes I and II. Bioorg Med Chem 2008;16:9101–5
- Şentürk M, Gülçin I, Daştan A, et al. Carbonic anhydrase inhibitors. Inhibition of human erythrocyte isozymes I and II with a series of antioxidant phenols. Bioorg Med Chem 2009;17:3207–11
- Innocenti A, Öztürk Sarıkaya SB, Gülçin I, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of mammalian isoforms I-XIV with a series of natural product polyphenols and phenolic acids. Bioorg Med Chem 2010;18:2159–64
- Davis RA, Innocenti A, Poulsen SA, Supuran CT. Carbonic anhydrase inhibitors. Identification of selective inhibitors of the human mitochondrial isozymes VA and VB over the cytosolic isozymes I and II from a natural product-based phenolic library. Bioorg Med Chem 2010;18:14–18
- Davis RA, Hofmann A, Osman A, et al. Natural product-based phenols as novel probes for mycobacterial and fungal carbonic anhydrases. J Med Chem 2011;54:1682–92
- Carta F, Vullo D, Maresca A, et al. Mono-/dihydroxybenzoic acid esters and phenol pyridinium derivatives as inhibitors of the mammalian carbonic anhydrase isoforms I, II, VII, IX, XII and XIV. Bioorg Med Chem 2013;21:1564–9
- Martin DP, Cohen SM. Nucleophile recognition as an alternative inhibition mode for benzoic acid based carbonic anhydrase inhibitors. Chem Commun (Camb) 2012;48:5259–61
- Tars K, Vullo D, Kazaks A, et al. Sulfocoumarins (1,2-benzoxathiine-2,2-dioxides): a class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J Med Chem 2013;56:293–300
- Buchieri MV, Riafrecha LE, Rodríguez OM, et al. Inhibition of the β-carbonic anhydrases from Mycobacterium tuberculosis with C-cinnamoyl glycosides: identification of the first inhibitor with anti-mycobacterial activity. Bioorg Med Chem Lett 2013;23:740–3
- Riafrecha LE, Rodríguez OM, Vullo D, et al. Synthesis of C-cinnamoyl glycosides and their inhibitory activity against mammalian carbonic anhydrases. Bioorg Med Chem 2013;21:1489–94
- Ekinci D, Karagoz L, Ekinci D, et al. Carbonic anhydrase inhibitors: in vitro inhibition of α isoforms (hCA I, hCA II, bCA III, hCA IV) by flavonoids. J Enzyme Inhib Med Chem 2013;28:283–8
- Sahin H, Can Z, Yildiz O, et al. Inhibition of carbonic anhydrase isozymes I and II with natural products extracted from plants, mushrooms and honey. J Enzyme Inhib Med Chem 2012;27:395–402
- Supuran CT. Carbonic anhydrase inhibition with natural products: novel chemotypes and inhibition mechanisms. Mol Divers 2011;15:305–16
- Karioti A, Ceruso M, Carta F, et al. New natural product carbonic anhydrase inhibitors incorporating phenol moieties. Bioorg Med Chem 2015;23:7219–25
- Maresca A, Akyuz G, Osman SM, et al. Inhibition of mammalian carbonic anhydrase isoforms I-XIV with a series of phenolic acid esters. Bioorg Med Chem 2015;23:7181–8
- Ferraroni M, Carta F, Scozzafava A, Supuran CT. Thioxocoumarins inhibit carbonic anhydrases through a different mechanism compared to coumarins. J Med Chem 2015; submitted
- Huang S, Hainzl T, Grundström C, et al. Structural studies of β-carbonic anhydrase from the green alga Coccomyxa: inhibitor complexes with anions and acetazolamide. PLoS One 2011;6:e28458
- Maresca A, Temperini C, Vu H, et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–62
- Maresca A, Temperini C, Pochet L, et al. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem 2010;53:335–44
- Temperini C, Innocenti A, Scozzafava A, et al. The coumarin-binding site in carbonic anhydrase accommodates structurally diverse inhibitors: the antiepileptic lacosamide as an example and lead molecule for novel classes of carbonic anhydrase inhibitors. J Med Chem 2010;53:850–4
- Touisni N, Maresca A, McDonald PC, et al. Glycosyl coumarin carbonic anhydrase IX and XII inhibitors strongly attenuate the growth of primary breast tumors. J Med Chem 2011;54:8271–7
- Bonneau A, Maresca A, Winum JY, Supuran CT. Metronidazole-coumarin conjugates and 3-cyano-7-hydroxy-coumarin act as isoform-selective carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2013;28:397–401
- Sharma A, Tiwari M, Supuran CT. Novel coumarins and benzocoumarins acting as isoform-selective inhibitors against the tumor-associated carbonic anhydrase IX. J Enzyme Inhib Med Chem 2014;29:292–6
- Maresca A, Supuran CT. Coumarins incorporating hydroxy- and chloro- moieties selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II. Bioorg Med Chem Lett 2010;20:4511–4
- Maresca A, Scozzafava A, Supuran CT. 78-Disubstituted- but not 6,7-disubstituted coumarins selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II in the low nanomolar/subnanomolar range. Bioorg Med Chem Lett 2010;20:7255–8
- Carta F, Maresca A, Scozzafava A, et al. 5- and 6-membered (thio)lactones are prodrug type carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2012;22:267–70
- Isik S, Vullo D, Bozdag M, et al. 7-Amino-3,4-dihydro-1H-quinoline-2-one, a compound similar to the substituted coumarins, inhibits α-carbonic anhydrases without hydrolysis of the lactam ring. J Enzyme Inhib Med Chem 2015;30:773–7
- Tanc M, Carta F, Bozdag M, et al. 7-Substituted-sulfocoumarins are isoform-selective, potent carbonic anhydrase II inhibitors. Bioorg Med Chem 2013;15:4502–10
- Grandane A, Tanc M, Zalubovskis R, et al. 6-Triazolylsubstituted sulfocoumarins are potent, selective inhibitors of the tumor-associated carbonic anhydrases IX and XII. Bioorg Med Chem Lett 2014;24:1256–60
- Grandane A, Tanc M, Zalubovskis R, et al. Synthesis of 6-tetrazolyl-substituted sulfocoumarins acting as highly potent and selective inhibitors of the tumor-associated carbonic anhydrase isoforms IX and XII. Bioorg Med Chem 2014;22:1522–8
- Grandane A, Tanc M, Di Cesare Mannelli L, et al. 6-Substituted sulfocoumarins are selective carbonic anhdydrase IX and XII inhibitors with significant cytotoxicity against colorectal cancer cells. J Med Chem 2015;58:3975–83
- Tanc M, Carta F, Scozzafava A, et al. 6-Substituted 1,2-benzoxathiine-2,2-dioxides are isoform-selective inhibitors of human carbonic anhydrases IX, XII and VA. Org Biomol Chem 2015;13:77–80
- D’Ambrosio K, Carradori S, Monti SM, et al. Out of the active site binding pocket for carbonic anhydrase inhibitors. Chem Commun 2015;51:302–5
- Scozzafava A, Carta F, Supuran CT. Secondary and tertiary sulfonamides: a patent review (2008–2012). Expert Opin Ther Pat 2013;23:203–13
- Alp C, Ozsoy S, Alp NA, et al. Sulfapyridine-like benzenesulfonamide derivatives as inhibitors of carbonic anhydrase isoenzymes I, II and VI. J Enzyme Inhib Med Chem 2012;27:818–24
- Ekinci D, Fidan I, Durdagi S, et al. Kinetic and in silico analysis of thiazolidin-based inhibitors of α-carbonic anhydrase isoenzymes. J Enzyme Inhib Med Chem 2013;28:370–4
- Abdel-Aziz AA, El-Azab AS, Ekinci D, et al. Investigation of arenesulfonyl-2-imidazolidinones as potent carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2015;30:81–4
- Arslan M, Şentürk M, Fidan I, et al. Synthesis of 3,4-dihydroxypyrrolidine-2,5-dione and 3,5-dihydroxybenzoic acid derivatives and evaluation of the carbonic anhydrase I and II inhibition. J Enzyme Inhib Med Chem 2015;30:896–900
- Liu F, Martin-Mingot A, Lecornué F, et al. Carbonic anhydrases inhibitory effects of new benzenesulfonamides synthesized by using superacid chemistry. J Enzyme Inhib Med Chem 2012;27:886–91
- Compain G, Martin-Mingot A, Maresca A, et al. Superacid synthesis of halogen containing N-substituted-4-aminobenzene sulfonamides: new selective tumor-associated carbonic anhydrase inhibitors. Bioorg Med Chem 2013;21:1555–63
- Métayer B, Mingot A, Vullo D, et al. New superacid synthesized (fluorinated) tertiary benzenesulfonamides acting as selective hCA IX inhibitors: toward a new mode of carbonic anhydrase inhibition by sulfonamides. Chem Commun (Camb) 2013;49:6015–17
- Le Darz A, Mingot A, Bouazza F, et al. Fluorinated pyrrolidines and piperidines containing tertiary benzenesulfonamides: selective carbonic anhydrase II inhibitors. J Enzyme Inhib Med Chem 2015;30:737–45
- D’Ascenzio M, Carradori SD, Monte C, et al. Design, synthesis and evaluation of N-substituted saccharin derivatives as selective inhibitors of tumor-associated carbonic anhydrase XII. Bioorg Med Chem 2014;22:1821–31
- De Monte C, Carradori S, Secci D, et al. Cyclic tertiary sulfamates: selective inhibition of the tumor-associated carbonic anhydrases IX and XII by N- and O-substituted acesulfame derivatives. Eur J Med Chem 2014;84:240–6
- D’Ascenzio M, Carradori S, Secci D, et al. Selective inhibition of human carbonic anhydrases by novel amide derivatives of probenecid: synthesis, biological evaluation and molecular modelling studies. Bioorg Med Chem 2014;22:3982–8
- Carradori S, Mollica A, Ceruso M, et al. New amide derivatives of probenecid as selective inhibitors of carbonic anhydrase IX and XII: biological evaluation and molecular modelling studies. Bioorg Med Chem 2015;23:2975–81
- Mollica A, Costante R, Akdemir A, et al. Exploring new probenecid-based carbonic anhydrase inhibitors: synthesis, biological evaluation and docking studies. Bioorg Med Chem 2015;23:5311–18
- Gavernet L, Gonzalez Funes JL, Palestro PH, et al. Inhibition pattern of sulfamide-related compounds in binding to carbonic anhydrase isoforms I, II, VII, XII and XIV. Bioorg Med Chem 2013;21:1410–18
- Parkkila S, Innocenti A, Kallio H, et al. The protein tyrosine kinase inhibitors imatinib and nilotinib strongly inhibit several mammalian α-carbonic anhydrase isoforms. Bioorg Med Chem Lett 2009;19:4102–6
- Innocenti A, Durdagi S, Doostdar N, et al. Nanoscale enzyme inhibitors: fullerenes inhibit carbonic anhydrase by occluding the active site entrance. Bioorg Med Chem 2010;28:2822
- Durdagi S, Vullo D, Pan P, et al. Protein-protein interactions: inhibition of mammalian carbonic anhydrases I-XV by the murine inhibitor of carbonic anhydrase and other members of the transferrin family. J Med Chem 2012;55:5529–35
- Banerjee AL, Eiler D, Roy BC, et al. Spacer-based selectivity in the binding of “carbonic anhydrase I” ligands to recombinant human carbonic anhydrase I. Biochemistry 2005;44:3211–24
- Jude KM, Banerjee AL, Haldar MK, et al. Ultrahigh resolution crystal structures of human carbonic anhydrases I and II complexed with “sis of high affinity” inhibitors reveal the molecular basis of high affinity. J Am Chem Soc 2006;128:3011–18
- Vomasta D, Innocenti A, König B, Supuran CT. Carbonic anhydrase inhibitors: two-prong versus mono-prong inhibitors of isoforms I, II, IX, and XII exemplified by photochromic cis-1,2-alpha-dithienylethene derivatives. Bioorg Med Chem Lett 2009;19:1283–6
- Wang ZC, Qin YJ, Wang PF, et al. Sulfonamides containing coumarin moieties selectively and potently inhibit carbonic anhydrases II and IX: design, synthesis, inhibitory activity and 3D-QSAR analysis. Eur J Med Chem 2013;66:1–11
- Wang L, Yang C, Lu W, et al. Discovery of new potent inhibitors for carbonic anhydrase IX by structure-based virtual screening. Bioorg Med Chem Lett 2013;23:3496–9
- Gao R, Liao S, Zhang C, et al. Optimization of heterocyclic substituted benzenesulfonamides as novel carbonic anhydrase IX inhibitors and their structure activity relationship. Eur J Med Chem 2013;62:597–604
- Zhang S, Yang C, Lu W, et al. A highly selective space-folded photo-induced electron transfer fluorescent probe for carbonic anhydrase isozymes IX and its applications for biological imaging. Chem Commun (Camb) 2011;47:8301–3
- Pettersen EO, Ebbesen P, Gieling RG, et al. Targeting tumour hypoxia to prevent cancer metastasis. from biology, biosensing and technology to drug development: the METOXIA consortium. J Enzyme Inhib Med Chem 2015;30:689–721
- Alafeefy AM, Abdel-Aziz HA, Vullo D, et al. Inhibition of human carbonic anhydrase isozymes I, II, IX and XII with a new series of sulfonamides incorporating aroylhydrazone-, [1,2,4]triazolo[3,4-b][1,3,4]thiadiazinyl- or 2-(cyanophenylmethylene)-1,3,4-thiadiazol-3(2H)-yl moieties. J Enzyme Inhib Med Chem 2015;30:52–6
- Winum JY, Supuran CT. Recent advances in the discovery of zinc binding motifs for the development of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2015;30:321–4
- Briganti F, Pierattelli R, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Part 37. Novel classes of carbonic anhydrase inhibitors and their interaction with the native and cobalt-substituted enzyme: kinetic and spectroscopic investigations. Eur J Med Chem 1996;31:1001–10
- Maryanoff BE, Nortey SO, Gardocki JF, et al. Anticonvulsant O-alkyl sulfamates. 2,3:4,5-Bis-O-(1-methylethylidene)-beta-D-fructopyranose sulfamate and related compounds. J Med Chem 1987;30:880–7
- Casini A, Antel J, Abbate F, et al. Carbonic anhydrase inhibitors: SAR and X-ray crystallographic study for the interaction of sugar sulfamates/sulfamides with isozymes I, II and IV. Bioorg Med Chem Lett 2003;13:841–5
- Carta F, Di Cesare Mannelli L, Pinard M, et al. A class of sulfonamide carbonic anhydrase inhibitors with neuropathic pain modulating effects. Bioorg Med Chem 2015;23:1828–40
- Bejaoui M, Panatzi E, De Luca V, et al. Acetazolamide protects steatotic liver grafts against cold ischemia reperfusion injury. J Pharmacol Exp Ther 2015;355:191–8
- Di Cesare Mannelli L, Micheli L, Carta F, et al. Carbonic anhydrase inhibition for the management of cerebral ischemia: in vivo evaluation of sulfonamide and coumarin inhibitors. J Enzyme Inhib Med Chem 2016;31:in press. doi: 10.3109/14756366.2015.1113407
- Palmer C, Supuran CT. A key opinion leader interview: insight into the research and career of Prof. Claudiu T Supuran. Expert Opin Ther Pat 2015;25:501–5