
Abstract
Neuronal autophagy is increased in numerous excitotoxic conditions including neonatal cerebral hypoxia-ischemia (HI). However, the role of this HI-induced autophagy remains unclear. To clarify this role we established an in vitro model of excitotoxicity combining kainate treatment (Ka, 30 µM) with hypoxia (Hx, 6% oxygen) in primary neuron cultures. KaHx rapidly induced excitotoxic death that was completely prevented by MK801 or EGTA. KaHx also stimulated neuronal autophagic flux as shown by a rise in autophagosome number (increased levels of LC3-II and punctate LC3 labeling) accompanied by increases in lysosomal abundance and activity (increased SQSTM1/p62 degradation, and increased LC3-II levels in the presence of lysosomal inhibitors) and fusion (shown using an RFP-GFP-LC3 reporter). To determine the role of the enhanced autophagy we applied either pharmacological autophagy inhibitors (3-methyladenine or pepstatinA/E64) or lentiviral vectors delivering shRNAs targeting Becn1 or Atg7. Both strategies reduced KaHx-induced neuronal death. A prodeath role of autophagy was also confirmed by the enhanced toxicity of KaHx in cultures overexpressing BECN1 or ATG7. Finally, in vivo inhibition of autophagy by intrastriatal injection of a lentiviral vector expressing a Becn1-targeting shRNA increased the volume of intact striatum in a rat model of severe neonatal cerebral HI. These results clearly show a death-mediating role of autophagy in hypoxic-excitotoxic conditions and suggest that inhibition of autophagy should be considered as a neuroprotective strategy in HI brain injuries.
Introduction
Macroautophagy, the main type of autophagy (hereafter called autophagy), consists of the engulfment of part of the cytoplasm in a multimembrane compartment (autophagosome), which then fuses with a lysosome to produce a large secondary lysosome (autolysosome).Citation1 Autophagy is essential to the long-term health of cells (including neurons), and by preserving cellular homeostasis and supplying nutrients an increase in autophagy can contribute to cell survival in some stress conditions such as nutrient and growth factor deprivation or accumulation of toxic molecules (pathogens, misfolded proteins, damaged organelles). However, in other situations, autophagy can be deleterious and contribute to cell death either by triggering apoptosis or necrosis or as an independent cell death mechanism (“autophagic cell death” or type II programmed cell death).Citation2,Citation3 In neurons the question of whether enhanced autophagy promotes cell death or cell survival is a subject of considerable debateCitation3-Citation8 especially in excitotoxic conditions such as cerebral ischemia. Some studies have suggested a protective role of neuronal autophagy in cerebral hypoxia/ischemia,Citation9,Citation10 whereas others have demonstrated a death-mediating role.Citation11-Citation14 In the standard model of cerebral HI in rodent pups,Citation15 which is widely used for investigating the neuropathological processes of perinatal asphyxia, autophagy is greatly increased in dying cortical and hippocampal neurons,Citation11,Citation16 but the role of this autophagy in neonatal HI brain injury is unclear. Whereas experiments involving pharmacological modulation of autophagy have suggested a protective role,Citation9 neuron-specific deletion of the autophagy-related (Atg) gene Atg7 has provided evidence for a deleterious effect.Citation11
The present study aims to clarify the role of enhanced neuronal autophagy in HI-induced injury. Excitotoxicity, a pathological process involving the overstimulation of excitatory neurotransmitter receptors (mainly to glutamate), and hypoxia/reoxygenation-induced detrimental pathways are considered as central players in neuronal death after HI. Our aim was to develop an in vitro model involving doses of an excitotoxin and hypoxia that were toxic in combination but not when used separately so as to imitate as closely as possible the situation in hypoxic-ischemic brain damage. For the reasons explained above and in Results, we selected as excitotoxin, kainate (Ka), a potent glutamate receptor agonist that targets non-NMDA (N-methyl-D-aspartate) glutamate receptors. Furthermore, Ka induces both excitotoxicityCitation17,Citation18 and autophagy in vitroCitation19 and in vivo.Citation18,Citation20 Ka treatment was applied in a hypoxic environment since hypoxia can exacerbate the mitochondrial production of reactive oxygen species (ROS), especially H2O2 and O2-, which can regulate autophagy,Citation21-Citation25 or inversely autophagy can contribute to oxidative stress by its involvement in ROS accumulation.Citation26,Citation27 We here show that the combination of Ka and hypoxia (KaHx) activates autophagy and that this mediates neuronal death as demonstrated by the protective effects of downregulating 2 important ATG proteins, BECN1/Beclin 1 and ATG7, and by the sensitization to KaHx when both proteins are upregulated. Importantly, the downregulation of BECN1 in a rat model of neonatal HI affords neuroprotection. Taken together these results provide strong evidence for a death-mediating role of enhanced autophagy in HI neuronal injury.
Results
Kainate-hypoxia treatment induces excitotoxic neuronal death
In order to develop an in vitro model of cerebral asphyxia, we subjected cultured neurons to hypoxia and excitotoxicity since these are the main mediators of hypoxic-ischemic neuronal death. In initial experiments with NMDA as excitotoxin, we failed to find a dose that was toxic in combination with hypoxia but not in isolation (see Introduction), but further experiments with kainate (Ka) fulfilled this criterion. Thus, primary cortical neurons were subjected to both 30 µM Ka and hypoxia (Hx) at 6% oxygen for 30 min. Whereas the separate treatments with Ka or Hx were found not to be neurotoxic, their combination was highly effective in promoting neuronal cell death as shown by a strong increase in propidium iodide (PI)-positive nuclei at 3 h and 6 h after KaHx treatment ().
Figure 1. Kainate-hypoxia treatment induces neuronal death. (A) KaHx induces neuronal death as shown morphologically in brightfield images or by combined propidium iodide (PI)-staining (red, dead cells) and MAP2-immunolabeling (green) 6 h after control and KaHx stimulations. (B) Representative images of PI-stained (red) and Hoechst-stained nuclei (blue, total cells) of cultured cortical neurons 6 h following 30 min of different stimulations: ct, control; KaHx, kainate-hypoxia; Ka, kainate; Hx, hypoxia. (C) Quantification of PI-positive nuclei as a percentage of all nuclei showing a rapid and strong cell death induced by the combination of kainate and hypoxia whereas the separate treatments were not toxic (ct: 5 ± 2%, KaHx 3 h: 44 ± 6%, KaHx 6 h: 59 ± 3%, Ka: 11 ± 2%, Hx 6 ± 2%). Values are mean ± SEM, *P < 0.05, ***P < 0.001, Tukey-Kramer test. n = 6 independent experiments. Scale bar: 20 µm.
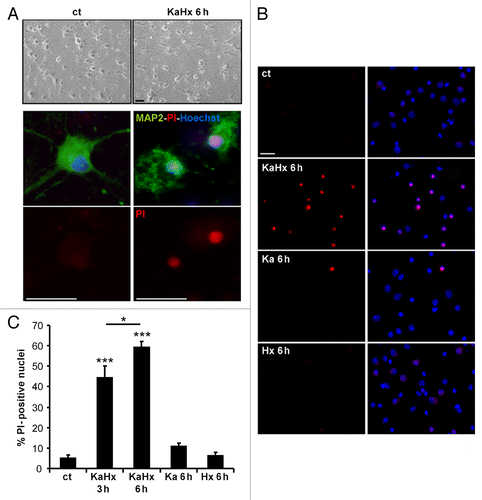
To characterize further this KaHx-induced neuronal death, we pretreated cortical neuronal cultures with either EGTA (a chelator of extracellular calcium) (5 mM) or the NMDA receptor antagonist MK801 (40 μM). Both pretreatments were strongly neuroprotective against KaHx treatment (Fig. S1A). These results demonstrate that the KaHx-induced neuronal death is calcium dependent and mediated by NMDA receptors, even though Ka does not directly activate NMDA receptors.
Inhibition of apoptosis did not prevent kainate-hypoxia-induced neuronal death
We investigated whether KaHx treatment induced apoptosis, also using the classical apoptotic stimulus, staurosporine (STS) (1 μM),Citation28 as a positive control. KaHx-induced neuronal death, unlike that induced by STS, appeared to be caspase-independent. This was suggested by a very weak activation of CASP3/caspase 3 (Fig. S2A) and was confirmed by the fact that apoptosis inhibition—either by overexpression of the anti-apoptotic protein BCL2 or by treatment with the pan-caspase inhibitor Q-VD-OPH—failed to protect neurons against KaHx. Overexpression of BCL2 (Fig. S2B), which was very efficient in preventing STS-induced CASP3 activation (Fig. S2C), and eliminated the weak KaHx-induced CASP3 activation that was induced by KaHx (Fig. S2D), was unable to protect cortical cultured neurons against KaHx-induced cell death (Fig. S2E). A similar lack of protection was observed when KaHx-treated neurons were pretreated with the pan-caspase inhibitor Q-VD-OPH (25 µM) (Fig. S2F).
Kainate-hypoxia treatment enhances neuronal autophagy
To investigate whether autophagy could be involved in KaHx-induced neuronal death, the effect of KaHx treatment on autophagosome formation was studied. As shown in , KaHx-treated neurons showed more autophagosomes compared with control conditions as demonstrated by an increase both in LC3-II expression by immunoblot () and in the number of LC3-positive dots per neuron revealed by immunocytochemistry and confocal microscopy () at 3 h and 6 h following KaHx treatment. Furthermore, we also showed that this KaHx-induced autophagosome accumulation is dependent on NMDA receptor stimulation and extracellular Ca2+ since pre-treatment with either MK801 or EGTA were able to prevent the LC3-II increase induced by KaHx treatment (Fig. S1B). Individual treatments with Ka or Hx did not result in any significant change in LC3-II expression (Fig. S3).
Figure 2. Kainate-hypoxia increases autophagosome formation and lysosomal markers in cultured cortical neurons. (A) Representative immunoblot of LC3 (upper part) and the corresponding quantification (lower part) showing that the LC3-II expression level is upregulated at 3 h and 6 h after kainate-hypoxia (KaHx) stimulation. (ct: 100 ± 0%, KaHx 3 h: 160 ± 8%, KaHx 6 h: 185 ± 19%). Values are mean ± SEM, ***P < 0.001, Steel-Dwass test. (B) Confocal images of LC3 immunolabeling (in red) of representative cultured cortical neurons at 6 h after control (ct) or KaHx stimulation (upper part), and quantification of LC3-positive dots per neuron per µm2 (lower part) demonstrating a strong increase in the number of autophagosomes after KaHx. (ct: 0.028 ± 0.004, KaHx 3 h: 0.189 ± 0.022, KaHx 6 h: 0.224 ± 0.024). Values are mean ± SEM, Tukey-Kramer test. Scale bar: 20 µm. (C) Immunolabeling for the lysosomal marker LAMP1 (green) shows an increase not only in the number of positive dots (upper graph; ct: 0.099 ± 0.006, KaHx 3 h: 0.196 ± 0.009, KaHx 6 h: 0.210 ± 0.017; Tukey-Kramer test) but also in the percentage of large positive dots (> 0.5 μm2) (lower graph; ct: 3.3 ± 1.5%, KaHx 3 h: 23.9 ± 3%, KaHx 6 h: 20 ± 4.8%; Steel-Dwass test) after KaHx. Values are mean ± SEM (D) Similar results are obtained with immunolabeling for the lysosomal enzyme CTSB (red). This phenomenon occurs in neurons as shown by double immunolabeling for CTSB and RBFOX3 (green). (Upper graph: ct: 0.06 ± 0.01, 3 h KaHx: 0.176 ± 0.02, 6 h KaHx: 0.19 ± 0.02; lower graph: ct: 3.4 ± 1.2%, 3 h KaHx: 16.1 ± 1.2%, 6 h KaHx: 16.9 ± 1.7.) Values are mean ± SEM, Steel-Dwass test. n ≥ 25. Scale bar: 20 µm. *P < 0.05, **P < 0.01, ***P < 0.001. n = 4 independent experiments.
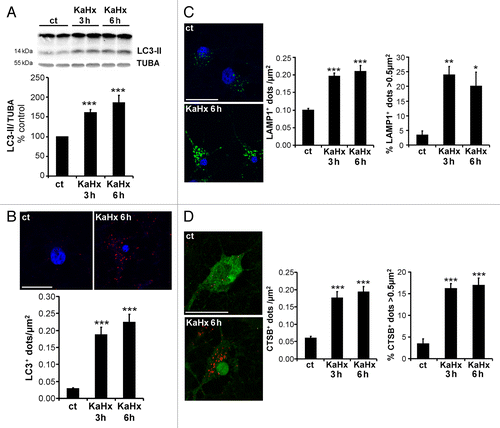
The effect of KaHx on lysosomes was then evaluated using immunocytochemistry against LAMP1 (lysosomal-associated membrane protein 1) () and CTSB (cathepsin B) (). KaHx induced not only an increase in the number of both LAMP1- (, left histogram) and CTSB-positive (, left histogram) dots, but also an increase in the size of both LAMP1- (, right histogram) and CTSB-positive dots (, right histogram) as shown by a rise in the number and the percentage of the largest dots (> 0.5 µm2) which are presumably autolysosomes produced by fusion between autophagosomes and lysosomes.
Finally, to demonstrate definitively that KaHx induced an increase in the autophagic flux, additional experiments were conducted. First, decreased expression of SQSTM1/p62, a long-lived protein preferentially degraded by autophagy, was shown at 3 h and 6 h after KaHx treatment based on immunoblot (). Moreover, treatments with Ka or Hx alone did not result in any significant change in SQSTM1/p62 expression (Fig. S3A), which is as expected since these separate treatments did not enhance autophagy. To check that the decrease in SQSTM1 expression after KaHx was not due to an effect on its production, we did additional experiments using a dose of 1 µg/µl cycloheximide (CHX), which prevents more than 95% of the protein synthesis. CHX treatment for 6 h significantly decreased SQSTM1 expression as expected; however, importantly, in the presence of CHX the stimulus KaHx (6 h) still induced a significant decrease in SQSTM1 expression, supporting the idea of KaHx-induced enhancement of autophagic flux (Fig. S3B). Then, cortical neurons were pretreated with a combination of the lysosomal enzyme inhibitors E64 (10 μg/ml) and pepstatin A1 (PepA) (10 μg/ml). The KaHx-induced accumulation of LC3-II was greater in E64/PepA-treated cultures than in untreated cultures (), showing that KaHx treatment induced the neoformation of autophagosomes. To further confirm an increase in the autophagic flux induced by KaHx and to demonstrate that KaHx did not impair the autophagosome-lysosome fusion process, autophagic flux was monitored with the tandem mRFP-GFP-LC3–expressing plasmid ptfLC3 ().Citation29 KaHx induced an increase in both the number of early autophagosomes (yellow dots, before fusion) and autolysosomes (red dot, after fusion with the lysosome) per neuron after both 3 h and 6 h of KaHx treatment.
Figure 3. Kainate-hypoxia enhances autophagic flux in cultured cortical neurons. (A) Representative immunoblots for SQSTM1 (upper part) and the corresponding quantification (lower part) indicate that kainate-hypoxia (KaHx) significantly decreases SQSTM1 expression. (ct: 100 ± 0%, 3 h KaHx: 76 ± 4%, 6 h KaHx: 73 ± 5%). n = 13 independent experiments. (B) Representative immunoblots against LC3 and SQSTM1 and the corresponding quantifications demonstrate that exposure to pepstatin A (PepA) and E64 in cultured neurons increases the level of LC3-II (Pep/E64: 174 ± 13%) and SQSTM1 (Pep/E64: 144 ± 14%) relative to control (ct) stimulation, indicating that autophagic degradation is prevented. Addition of Pep/E64 6 h before, during or after KaHx treatment (6 h) results in a greater increase in LC3-II than treatment with KaHx or E64/PepA alone (Ka+Hx: 169 ± 5%, E64/PepA + KaHx: 277 ± 27%) and diminishes the KaHx-induced degradation of SQSTM1 (Ka+Hx: 78 ± 3%, E64/PepA + KaHx: 118 ± 9%). Values are mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001, Tukey-Kramer test. n = 4 independent experiments. (C) Confocal images of representative cultured cortical neurons transfected with the tandem mRFP-GFP-LC3–expressing plasmid at 3 h and 6 h after Ka/Hx. (D) Quantification of the number of LC3-positive dots per neuron per µm2 shows an active autophagic flux (TOTAL = GFP+ RFP+ and GFP- RFP+: ct: 0.03 ± 0%, 3 h: 0.17 ± 0.02%, 6 h: 0.23 ± 0.01%; early autophagosomes = GFP+ RFP+: ct: 0.01 ± 0%, 3 h: 0.10 ± 0.02%, 6 h: 0.13 ± 0.01%; autolysosomes = GFP- RFP+: ct: 0.02 ± 0%, 3 h: 0.07 ± 0.01%, 6 h: 0.10 ± 0.01%). Values are mean ± SEM,. **P < 0.01, ***P < 0.001. Tukey-Kramer test. n = 4 independent experiments. Scale bar: 20 µm.
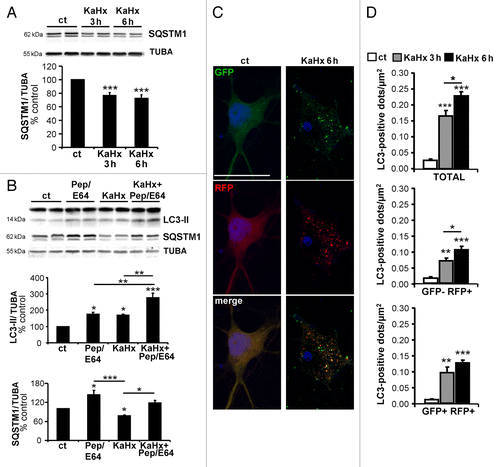
Taken together, these results clearly demonstrate that KaHx treatment induced an enhancement of the autophagic flux.
Autophagy contributes to kainate-hypoxia-induced neuronal death
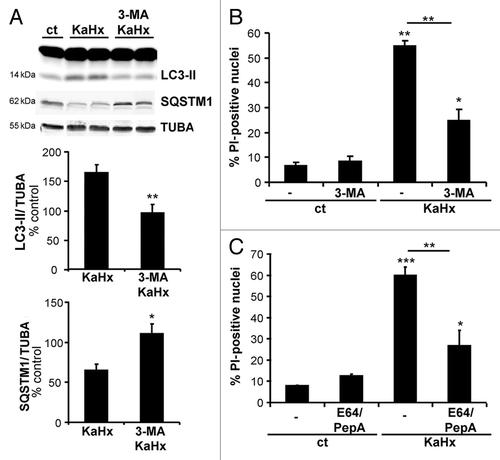
Having demonstrated that autophagy is induced by KaHx treatment, the next step was to evaluate the functional role of this enhanced autophagy in KaHx-induced cell death. First, the effect of pharmacological inhibition of autophagy with 3-methyladenine (3-MA) was tested. As expected, pretreatment with 3-MA (10 mM) prevented the increases in both LC3-II expression and SQSTM1 degradation (). Then 3-MA treatment displayed a significant neuroprotective effect as demonstrated by a decrease in PI-positive nuclei 6 h after KaHx treatment (). We also evaluated the effect of lysosomal enzyme inhibition on KaHx-induced neuronal death by using combined treatment with the above-mentioned cocktail of E64 and PepA. Pretreatment with E64/PepA, that efficiently blocked autophagic degradation (), significantly reduced neuronal death induced by KaHx ().
Figure 4. Pharmacological inhibition of the autophagic process with 3-methyladenine or E64/pepstatinA protects cultured cortical neurons against kainate-hypoxia treatment. (A) Representative immunoblots against LC3 and SQSTM1 and the corresponding quantifications confirm that 3-methyladenine (3-MA) prevents accumulation of LC3-II (KaHx: 164 ± 14%, 3-MA KaHx: 95 ± 14; Welch ANOVA, n = 5 independent experiments) and reduction of SQSTM1 (KaHx: 65 ± 8%, 3-MA KaHx: 110 ± 12%; Welch ANOVA, n = 5 independent experiments) following exposure to KaHx. (B and C) Quantification of PI-positive nuclei as a percentage of all nuclei (Hoechst-stained) demonstrates that inhibition of the autophagic process with (B) 3-MA (ct: 6.6 ± 1.4, 3-MA: 8.4 ± 1.9, KaHx: 54.8 ± 2, 3-MA KaHx: 24.6 ± 4.8, Steel-Dwass test, n = 3 independent experiments) or (C) E64/PepstatinA (PepA) (ct: 7.9 ± 0.1, E64/PepA: 12.5 ± 1, KaHx: 60 ± 3.8, E64/PepA KaHx: 26.7 ± 7.4, Tukey-Kramer test, n = 4 independent experiments) is neuroprotective. Values are mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001.
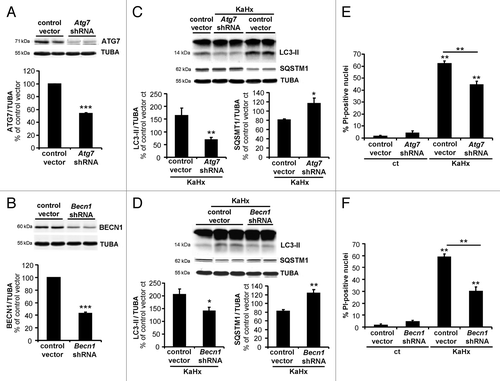
To confirm this death-promoting role of enhanced autophagy in KaHx-induced neuronal death, we performed knockdown of 2 different autophagy genes (Atg7 and Becn1) using lentiviral vectors encoding short hairpin RNAs (shRNAs). Transduction of cultured cortical neurons with Atg7- and Becn1-targeting shRNA vectors resulted in large reductions in the levels of the corresponding proteins (). As expected, downregulation of both ATG7 and BECN1 decreased KaHx-induced autophagy as shown by both a decrease in autophagosome formation (58% and 31% reduction in LC3-II for shRNAs targeting Atg7 and Becn1, respectively) and the inhibition of SQSTM1 degradation (). Quantification of PI-positive nuclei or of LDH (lactate dehydrogenase) release 6 h after KaHx treatment demonstrated a significantly reduced toxicity in Atg7 shRNA- and Becn1 shRNA-transduced neurons, confirming a prodeath role of autophagy following KaHx treatment (; Fig. S4A). This neuroprotective effect was still observed at 24 h after KaHx treatment (Fig. S4A and S4B).
Figure 5. Inhibition of autophagy by downregulation of ATG7 or BECN1 protects cultured cortical neurons against kainate-hypoxia treatment. (A and B) Representative immunoblots and corresponding quantifications showing that lentiviral transduction of shRNAs against (A) Atg7 or (B) Becn1 reduces the expression of ATG7 (53 ± 2%) or BECN1 (43 ± 2%), respectively, compared with control vector-transduced neurons. Values are mean ± SEM, ***P < 0.001, Wilcoxon test. n = 4 independent experiments. (C and D) Downregulation of ATG7 or BECN1 inhibits autophagy induced 6 h after KaHx. (C) ATG7 downregulation significantly reduces LC3-II (control vector KaHx: 163 ± 30%, Atg7 shRNA KaHx: 68 ± 10%, Kruskal-Wallis test, n = 6 independent experiments) as occurs to a lesser extent with (D) BECN1 downregulation (control vector KaHx: 205 ± 22%, Becn1 shRNA KaHx: 140 ± 15%, ANOVA, n = 5 independent experiments). Knockdown of either protein is however enough to completely block SQSTM1 degradation following KaHx (for ATG7: control vector KaHx: 81 ± 2%, Atg7 shRNA KaHx: 116 ± 12%, n = 5 independent experiments; for BECN1: control vector KaHx: 82 ± 4%, Becn1 shRNA KaHx: 123 ± 7%, Welch ANOVA, n = 7 independent experiments). Values are mean ± SEM expressed as a percentage of control (ct)-stimulated neurons transduced with control vector, **P < 0.01, ***P < 0.001. (E and F) Transduced neurons with Atg7 and Becn1 shRNAs are less sensitive to KaHx than control vector-transduced neurons as demonstrated by a significant decreased number of PI-positive nuclei 6 h after the stimulation (for ATG7: control vector: 2 ± 0.7%, Atg7 shRNA: 5 ± 2%, control vector KaHx: 62 ± 2%, Atg7 shRNA KaHx: 46 ± 3%; for BECN1: control vector: 2 ± 0.8%, Becn1 shRNA: 5 ± 1%, control vector KaHx: 59 ± 2%, Becn1 shRNA KaHx: 30 ± 4%). Values are mean ± SEM expressed as a percentage of all nuclei (Hoechst-stained). **P < 0.01, Steel-Dwass test, n = 8 independent experiments.
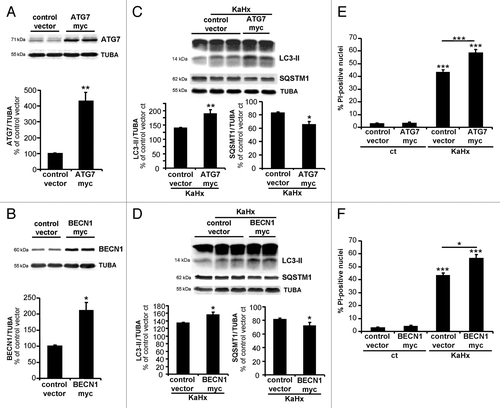
To corroborate these results, we overexpressed BECN1 and ATG7 in cortical neuronal cultures using lentiviral vectors encoding ATG7-myc and BECN1-myc. Expecting that KaHx-induced neuronal death would occur more rapidly when ATG7 and BECN1 were overexpressed, we chose an earlier time-point (3 h) point for the overexpression experiments. Overexpression of either BECN1 or ATG7 () resulted in an increased autophagic response to KaHx as shown by significantly higher expression of LC3-II and reduced expression of SQSTM1 (). Furthermore, overexpression of ATG7 or BECN1 exacerbated KaHx-induced neuronal death, as shown by increased numbers of PI-positive neurons 3 h after KaHx treatment in both cases ().
Figure 6. Upregulation of autophagy by overexpression of ATG7 and BECN1 sensitizes cultured cortical neurons against kainate-hypoxia treatment. (A and B) Representative immunoblots and corresponding quantifications showing that lentiviral transduction of (A) ATG7-myc and (B) BECN1-myc strongly increase expression of ATG7 (430 ± 32%) and BECN1 (220 ± 20%) compared with control vector transduced neurons. Values are mean ± SEM, Wilcoxon test. n = 4 independent experiments. (C) The very strong overexpression of ATG7 enhances the autophagic process induced 3 h after KaHx as demonstrated by an increase in LC3-II (control vector KaHx: 139 ± 3%, ATG7-myc KaHx: 189 ± 15%, Kruskal-Wallis test, n = 6 independent experiments) and by a reduction of SQSTM1 (control vector KaHx: 83 ± 1%, ATG7-myc KaHx: 65 ± 5%, Welch ANOVA, n = 5 independent experiments). (D) The 2-fold increase in BECN1 expression significantly increases the level of LC3-II (control vector KaHx: 134 ± 2%, BECN1-myc KaHx: 156 ± 7%, Welch ANOVA, n = 4 independent experiments) and decreases the level of SQSTM1 (control vector KaHx: 81 ± 2%, BECN1-myc KaHx: 72 ± 5%, Kruskal-Wallis test, n = 6 independent experiments) 3 h after KaHx. Values for (C and D) are mean ± SEM expressed as a percentage of control (ct)-stimulated neurons transduced with control vector. (E and F) Neurons transduced with ATG7-myc and BECN1-myc expression vectors are more sensitive to KaHx than control vector-transduced neurons as is demonstrated by a significantly increased number of PI-positive nuclei 3 h after the treatment (for ATG7: control vector: 2.6 ± 0.6%, ATG7-myc: 2.5 ± 1.1%, control vector KaHx: 43.3 ± 2.8%, ATG7-myc KaHx: 59.1 ± 3.3%, Steel-Dwass test; for BECN1: control vector: 2.6 ± 0.6%, BECN1-myc: 3.2 ± 1.2%, control vector KaHx: 43.3 ± 2.8%, BECN1-myc KaHx: 56.8 ± 5.4%, Tukey-Kramer test). Values are mean ± SEM expressed as a percentage of all nuclei (Hoechst-stained). n = 4 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
Since apoptosis and autophagy share common regulators we investigated the effect of apoptosis inhibition on KaHx-induced autophagy. Neither BCL2 overexpression using lentiviral vectors (Fig. S2) nor caspase inhibition using Q-VD-OPH (data not shown) had any effect on KaHx-induced autophagy.
Autophagy is increased and involved in neonatal cerebral hypoxic-ischemic brain injury
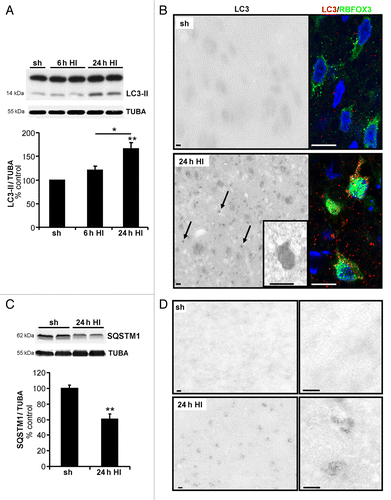
The functional role of autophagy in in vivo pathological situations involving hypoxic-excitotoxic brain injuries was then investigated. We had previously shown that neonatal cerebral hypoxia-ischemia (HI) enhanced neuronal autophagy in the cortex and the CA3 region of the hippocampus,Citation16 and we here show that a similar enhancement of autophagy occurred in the striatum in the same HI model. We selected the striatum rather than the cortex for the in vivo experiments because the striatum’s spheroidal form and limited extent permitted us to knock down genes throughout the entire structure using lentiviral vectors (Fig. S5). Autophagy was enhanced at 24 h after the insult based on an increase in the LC3-II level (), and by an increase in the number of autophagosomes in striatal neurons double-immunolabeled with the neuronal marker RBFOX3/NeuN and LC3 (). Moreover, immunoblots revealed that neonatal HI decreased the striatal levels of SQSTM1 expression, also at 24 h after HI (). Histochemistry for β-hexasosaminidase (HEXA-HEXB), a lysosomal enzyme, revealed a strong increase of β-hexasosaminidase-positive dots detected at 24 h after HI compared with sham animals (). Taken together, these results showed that autophagy is strongly increased in the striatum after neonatal cerebral HI.
Figure 7. Neuronal autophagy is enhanced in the striatum after neonatal cerebral hypoxia-ischemia. Cerebral hypoxia-ischemia (HI) increased the presence of neuronal autophagosomes in the striatum of 7-d old rats 24 h after HI as shown by (A) a higher level of LC3-II in immunoblots (sh: 100 ± 0%, 6 h: 121 ± 8%, n = 7 rats, 24 h: 166 ± 13%, n = 12 rats, Tukey-Kramer test) and (B) by strong punctate LC3-immunolabeling (arrows) in RBFOX3-positive neurons compared with sham-operated animals (sh). Scale bar: 20 µm. (C) Quantification of SQSTM1 expression level by immunoblot shows that neonatal HI induces SQSTM1 degradation (sh: 100 ± 4%, 24 h: 60 ± 7%, n = 8 rats, Welch ANOVA). Values are expressed as a percentage of sham-operated controls. (D) Neonatal cerebral HI increases β-hexosaminidase staining at 24 h compared with sham operated animals. Scale bar: 10 µm.
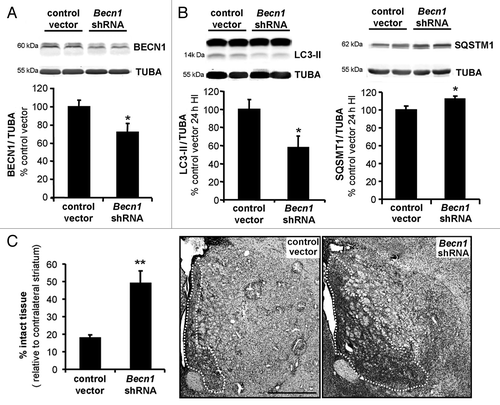
The role of this enhanced autophagy in striatum after neonatal cerebral HI was investigated by downregulation of BECN1 by intra-striatal injection of lentiviral vectors expressing Becn1 shRNA, which decreased BECN1 expression by 30% (). At 24 h after HI, BECN1 knockdown reduced the HI-induced LC3-II increase and SQSTM1 degradation (), and increased the percentage of striatal intact tissue (). Thus, our in vivo results provide evidence for a death-mediating role of autophagy in neonatal cerebral HI.
Figure 8. Inhibition of autophagy by lentiviral vectors expressing shRNA targeting Becn1 reduces brain damage following neonatal cerebral hypoxia-ischemia. (A) Representative immunoblot and corresponding quantification showing that lentiviral injections mediating Becn1 downregulation decreased BECN1 expression levels by 30% compared with control lentiviral vector injections (control vector: 100 ± 7%, Becn1 shRNA: 72 ± 10%), Welch ANOVA, n = 4 rats). When subjected to HI, animals injected with shRNA against Becn1 present (B) a reduced induction of LC3-II expression 24 h after the insult (control vector: 100 ± 10, Becn1 shRNA: 56 ± 12%, Welch ANOVA, n = 6 rats) and a decreased SQSTM1 degradation (control vector: 100 ± 4, Becn1 shRNA: 112 ± 3%, Welch ANOVA, n = 8 rats). (C) Rat pups with striatal injection of Becn1 shRNA show a higher percentage of “intact striatum,” meaning surviving or noninfarcted striatum, than do those with control vector-injection (control vector: 18 ± 2%, n = 15; Becn1 shRNA: 49 ± 7%, n = 12, Welch ANOVA). Values are mean ± SEM *P < 0.05, **P < 0.01. Scale bar: 1 mm.
Discussion
It is now generally recognized that autophagic processes are activated in excitotoxic conditions, and notably cerebral ischemia. Increased numbers of autophagosomes have been described in different in vitro situations of excitotoxicity such as long-term treatment and/or high doses of NMDACitation30,Citation31 or Ka,Citation19 or exposure to a specific glutamate uptake inhibitor.Citation32 The injection of toxic doses of Ka into the adult mice hippocampusCitation20,Citation33 or striatumCitation34,Citation35 also induces autophagy. Numerous in vivo studies of cerebral ischemia or HI over the past 20 y have likewise shown enhanced neuronal autophagy (autophagosome formation and lysosomal activity).Citation13,Citation14,Citation36-Citation42 Controversies remain, however, concerning the role of the autophagic activation in excitotoxic conditions both in vitro and in vivo. Even though most of the papers demonstrate a prodeath function of autophagy,Citation11-Citation14,Citation27,Citation31,Citation33,Citation34,Citation37,Citation43,Citation44 some studies suggest the opposite.Citation9,Citation10,Citation45-Citation49 These discrepancies could be due to differences in the excitotoxic conditions, to the use of pharmacological inhibitors with limited specificityCitation50,Citation51 or to the lack of complete analysis on autophagy flux, i.e., demonstration that autophagosome increase is not due to lysosomal dysfunction in excitotoxic situations. We here resolve these problems and provide clear evidence that neuronal autophagy in hypoxic-excitotoxic conditions is deleterious.
The combination of 2 stimuli, a low dose of Ka and moderate hypoxia (6% O2), each of which are nontoxic when applied for a very short period (30 min), produces a powerful and synergetic excitotoxic stimulus. By comparison with the in vivo situation, 6% O2 is in fact normoxic, not hypoxic, but when neurons cultured in standard conditions (at 21% O2) are exposed to 6% O2 this constitutes a hypoxic stress, probably because they have reduced mitochondrial function compared with ones cultured at 5% or 2% O2.Citation52 Ka is more potent than NMDA in release of endogenous glutamate.Citation53 Moreover different studies support the essential role of KA receptors (GRIK2/Gluk2, GRIK4/Gluk4) in excitotoxicity both in vitro and in vivo, partially involving MAPK activation.Citation54,Citation55 Interestingly, in the rat model of neonatal HI used in this study, neither the systemic hypoxia nor the common carotid occlusion alone produces brain damage, whereas the combination of both causes a very severe lesion.Citation15 Ka and hypoxia can mutually exacerbate their negative effects.Citation56,Citation57 A study in a guinea pig model of perinatal hypoxia has shown that hypoxia can decrease the affinity and change the kinetic of desensitization of Ka receptors suggesting a potential role of Ka receptors in hypoxia-induced excitotoxicity.Citation58 Interestingly, in humans a transient developmental elevation in KA binding in early life could correlate with the period of vulnerability of this cerebral region to hypoxia-ischemia.Citation59
We clearly demonstrated in this study that KaHx enhanced autophagic flux. The number of autophagosomes (LC3-positive dots, LC3-II expression level) was increased along with an activation of lysosomal activity (shown by degradation of SQSTM1, increase in the number and size of lysosomes labeled with LAMP1 or CTSB antibodies) and an increase in autophagosome-lysosome fusion (shown with an mRFP-GFP-LC3-expressing plasmid). Prevention of this KaHx-induced autophagy, not only pharmacologically with 3-MA but also more specifically by downregulating ATG7 or BECN1, significantly decreased neuronal death. Moreover, upregulation of either protein, which activated autophagy, aggravated neuronal death. These results strongly suggest that enhanced autophagy is involved in KaHx-induced cell death.
In vivo experiments have shown that intrastriatal injection of Ka in adult rats induced an excitotoxic injury that was decreased by both pre- and post-treatments with 3-MA.Citation34 More recently, both 3-MA injection and ATG7 downregulation have been shown to reduce the loss of hippocampal neurons provoked by intraperitoneal administration of Ka.Citation33 Both studies imply that autophagy can contribute to Ka-induced apoptosis. However in the present in vitro model, KaHx triggers a very rapid and strong neuronal death, with no apparent involvement of apoptosis since neither the pan-caspase inhibitor Q-VD-OPH nor BCL2 overexpression has a protective effect. The involvement of caspases is known to vary between excitotoxicity models.Citation60,Citation61 It has also been shown that 3-MA can suppress the mitochondrial ROS production associated with Ka-induced excitotoxicity in cultured striatal neurons.Citation19 Enhanced autophagy may therefore contribute to oxidative stress induced by KaHx. We showed also a protective effect of inhibiting lysosomal activity with E64/PepA suggesting that autophagic degradation is involved as well in neuronal death. Removal of some protective proteins such as antioxidantsCitation26 or generation of toxic molecules like ferric ironCitation62 may be implicated. Autophagosomes and (auto)lysosomes are also potential ROS sources.Citation27
There is controversy about the role of the autophagy in neonatal HI. Induction of autophagy with the pharmacological agent rapamycin provides some protective effects against cerebral HI in rat pups when injected before hypoxia.Citation9 Conversely, the work of Koike et al. (2008)Citation11 demonstrates that ATG7 deficiency specifically in neurons affords strong neuroprotection against HI-injury in the hippocampus of neonatal mice. Our results support the latter study by showing both in vitro, and for the first time in vivo, that autophagy inhibition by knockdown of autophagy genes is neuroprotective. This was the first time that downregulation of BECN1 was achieved in vivo requiring the use of lentiviral vectors. This downregulation decreased the stress-induced autophagy and reduced the striatal damage induced by HI in rat pups. We also previously demonstrated that post-treatment with 3-MA during the 4.5 h following the onset of ischemia decreased by half the volume of the cortical lesion in a model of middle cerebral artery occlusion in rat pups.Citation12,Citation43 Most, though not all,Citation47,Citation48 studies of adult cerebral ischemia likewise suggest a death-mediating role of autophagy when it is inhibited with 3-MA,Citation13,Citation27,Citation37 with lysosomal inhibitorsCitation13,Citation27,Citation63 or with BECN1 knockdown.Citation14
These results help open the way to possible therapy against neonatal HI by inhibition of autophagy, but this would need to be by transient inhibition, since constitutive autophagy is required for neuronal health and even survival.Citation64,Citation65
In conclusion, our study gives a clear demonstration of the pro-death role played by induced autophagy in excitotoxic-hypoxic conditions in cultured neurons and neonatal rats, and it consolidates the idea that strategies targeting excessively activated autophagy, to inhibit it transiently, should be considered in the development of new therapeutic approaches against hypoxic-ischemic brain injuries.
Materials and Methods
Primary cortical neuronal culture
Sprague-Dawley rat pups (from Janvier, France) were euthanized in accordance with the Swiss Laws for the protection of animals, and the procedures were approved by the Vaud Cantonal Veterinary Office. Primary neuronal cultures from cortices of 2-d-old rats were prepared and maintained at 37 °C with a 5% CO2-containing atmosphere in neurobasal medium (Life Technologies, 21103-049) supplemented with 2% B27 (Invitrogen, 17504044), 0.5 mM glutamine (Sigma, G7513) and 100 μg/ml penicillin-streptomycin (Invitrogen, 15140122) as described previously.Citation66 Western blot analyses were done on neurons plated at a density of ~7 × 105 cells/dish (35-mm poly-d-lysine precoated dishes, BD Biosciences, 356467) and immunocytochemistry on neurons plated at a density of ~3 × 105 cells on 12-mm glass coverslips coated with 0.01% poly-l-lysine (Sigma, P4832). Experiments were performed at 12–13 d in vitro (DIV). These cultures contain around 10–15% of non-neuronal cells (MAP2 or RBFOX3-negative). The n values in the different results refer to the number of independent experiments, each involving 2 or 3 culture dishes or coverslips. Thus, the mean value of 2–3 dishes or coverslips was counted as a single observation.
Rat model of perinatal asphyxia
All experiments were performed in accordance with Swiss laws for the protection of animals and were approved by the Vaud Cantonal Veterinary Office. Seven-day-old male rats (Sprague Dawley, from Janvier, France) underwent hypoxia-ischemia (HI) according to the Rice-Vannucci modelCitation15 adapted from the Levine procedureCitation67 as described previously.Citation68 Briefly, under isoflurane (3%) anesthesia, the right common carotid artery was isolated, double-ligated (Silkam, 5/0, B/BRAUN Aesculap) and cut. After 2 h of recovery with the dam, the rat pups were exposed to 2 h of systemic hypoxia at 8% of oxygen in a humidified chamber maintained at 35.5 °C. Sham-operated animals (same anesthetic and surgical procedures but without section of the common carotid artery) were used as controls.
Kainate-hypoxia (KaHx) stimulation
Hypoxia was performed for 30 min. in a sealed modular incubator chamber (Billups-Rothenberg, MIC-101) that had been flushed for 5 min with a gas mixture at 6% oxygen. At the start of the 30 min of hypoxia, all the (neurobasal) culture medium was replaced with bicarbonate-buffered saline (BBS) solution (116 mM NaCl, 5.4 mM KCl, 0.8 mM MgSO4, 1 mM NaH2PO4, 26.2 mM NaHCO3, 0.01 mM glycine, 1.8 mM CaCl2, 4.5 mg/mL glucose [reagents from Sigma-Aldrich]) previously deoxygenated for 1.5 h in the hypoxic chamber. Kainate (30 µM) (OPIKA-1, Ocean Produce International, 2020, stock solution at 20 mM in water) diluted in BBS was added immediately after the BBS, and the neuronal cultures were thus exposed to KaHx stimulation during the 30 min of hypoxia. Then, the BBS was removed and replaced with the previous neurobasal medium that had been reserved mixed with one-third volume of fresh medium. Thirty min in normoxic BBS was used as the control (ct) stimulation.
Pharmacological treatments
Primary cortical neuronal cultures were pre-treated for 1 h with 25 µM Q-VD-OPH (quinoline-Val-Asp(ome)-Ch2-O-phenoxy; MP Biomedicals, 030PH10903), 10 mM 3-methyladenine (3-MA; Sigma-Aldrich, M9281), 5 mM EGTA (Sigma-Aldrich, E0396) or 40 μM MK801 (Sigma-Aldrich, M107). For inhibition of lysosomal degradation, a cocktail of 10 μg/ml pepstatin A1 (PepA; Sigma-Aldrich, P5318) and 10 μg/ml E64 (Sigma-Aldrich, E3132) was applied for 6 h prior to KaHx stimulation.
Immunoblotting
Cortical neurons or striatal brain tissue were collected in lysis buffer (20 mM HEPES, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 2.5 mM EGTA, 0.1 mM dithiothreitol, 50 mM NaF [71519], 1 mM Na3VO4 [S6508], 1% Triton X-100 [93420]) (reagents from Sigma-Aldrich), and a protease inhibitor cocktail (Roche,11873580001). Proteins were separated by SDS-PAGE, transferred on nitrocellulose membrane and analyzed by immunoblotting. Antibodies were diluted in a blocking solution containing 0.1% casein (Sigma-Aldrich, C8654) and 0.1% Tween 20. The following primary antibodies were used for protein immunodetection: anti-TUBA/α-tubulin (sc-8035) mouse monoclonal, anti-ATG7 (sc-33211) rabbit polyclonal and anti-BECN1 (sc-11427) rabbit polyclonal from Santa Cruz Biotechnology; anti-LC3 (NB100-2220) rabbit polyclonal from Novus Biologicals; anti-cleaved CASP3/caspase-3 (9661) rabbit polyclonal and anti-BCL2 (2870) rabbit monoclonal from Cell Signalling Technology; anti-SQSTM1 (P0067) rabbit polyclonal from Sigma-Aldrich. After washes in PBS (150 mM NaCl, 8 mM K2HPO4, 1.99 mM KH2HPO4)-0.1% Tween, polyclonal goat anti-mouse or anti-rabbit IgG secondary antibodies were applied (IRDye 680 [LI-COR, B70920-02] or IRDye 800 [LI-COR, 926-32210]). The Odyssey Infrared Imaging System (LI-COR) was used to analyze protein bands using Odyssey v1.2 software (LI-COR). OD values were normalized against tubulin and expressed as a percentage of values obtained with neurons after control stimulation (100%).
Lentiviral vectors
Downregulations of Atg genes were performed with pLKO lentiviral vectors (Open Biosystems/Thermo Scientific) expressing rat-specific shRNA sequences from TRC (the RNAi consortium) library as described previously:Citation28 a combination of TRCN0000092163 and TRCN0000092166 for Atg7 (GenBank NM_001012097) and TRCN0000033552 for Becn1 (GenBank NM_053739.2). Atg7 and Becn1 overexpressions were achieved by self-inactivating lentiviral transfer vectors under the control of the mouse Pgk1 (phosphoglycerate kinase 1) promoter (SIN-W-PGK) transducing cDNAs sequences encoding full-length rat Atg7 (GenBank NM_001012097) and Becn1 (GenBank NM_053739) created using PCR with specific primers (Atg7: nucleotides 16–35 [forward], 2092–2112 [reverse]; Becn1: nucleotides 225–243 [forward], 1549–1570 [reverse]) to which the myc-tag encoding sequence was added as explained previously.Citation28 Primary cortical neuron cultures were infected at DIV7 with 50 ng of the viral capsid protein p24/ml culture medium for each vector, and half the medium was replaced on DIV11. A pLKO vector containing scrambled shRNA (Open Biosystems/Thermo Scientific) in Atg knockdown experiments or a SIN-W-PGK empty vector in Atg upregulation experiments were used as control vectors.
mRFP-GFP-LC3 plasmid transfection and quantification
This plasmid permits the distinction between acidic and nonacidic LC3-positive structures, because GFP is acid-sensitive whereas RFP is not. Neurons on coverslips were transfected using Lipofectamine 2000 (Invitrogen, 11668-019) as described previously.Citation69 In brief, 2/3 of the medium was removed and replaced for 5 h by fresh Neurobasal medium (Life Technologies, 21103-049) plus transfection mix with the tandem mRFP-GFP-LC3-expressing plasmid ptfLC3 (Addgene, 21074) (1 μg of DNA for 6 μl of Lipofectamine). Confocal images were acquired using a Zeiss LSM 710 Meta confocal laser scanning microscope. Total LC3-positive dots (GFP+ RFP+ and GFP− RFP+ dots), early autophagosomes (GFP+ RFP+ dots) and autolysosomes (GFP− RFP+ dots) were analyzed using ImageJ software and expressed as a number of positive dots per neuron per µm2.
Quantification of cell death with propidium iodide
Cortical neurons on coverslips were washed in PBS containing 1 mM MgCl2 at 4 °C and incubated for 10 min in propidium iodide (PI, Sigma-Aldrich, P4170) diluted in PBS containing 1 mM MgCl2. After PBS washes neurons were fixed in 4% paraformaldehyde in 0.1 M PBS (pH 7.4) for 15 min. A Hoechst staining was performed to visualize the total number of nuclei. Coverslips were mounted with FluorSave (Calbiochem, 345-789-20). Cells were examined using a Zeiss Axioplan 2 imaging microscope. Results were expressed as a percentage of PI-positive nuclei relative to Hoechst-positive nuclei.
Lactate dehydrogenase release
Cell death was assayed by measurement of LDH released in the medium using the Cytotox 96 nonradioactive cytotoxicity assay kit (Promega, G1780) as previously described.Citation66 LDH measurements were normalized with respect to the values with control vector 6 h after the KaHx treatment condition.
Intrastriatal injection of lentivirus
Lentiviral vectors were stereotaxically injected in the right striatum of 1- or 2-d old rat pups (Fig. S5). Rats were anesthetized with isoflurane (3%) and positioned in a stereotaxic frame. A midline scalp incision and a small hole were made in the skull. A 5.0-µl Hamilton syringe was used for injection at a site specified by the following stereotaxic coordinates from Bregma: 0.5-mm anterior, 2.5-mm right and 3-mm depth from the skull surface. One microliter of lentiviral suspension was infused over 5 min with an automatic injector. The needle stayed in place for 2 min before a slow removal. Lentiviral suspensions were prepared by mixing SIN-W-PGK vector encoding YFP (to visualize the infected area) and pLKO vectors encoding control or Becn1 shRNA at 1:1 ratio and with a particle content normalized to 125,000 µg p24/ml for each lentivirus. For immunoblotting studies infected striatal tissue was dissected under a stereomicroscope with fluorescence (Leica MZ16FA).
Measurement of the surviving striatum after neonatal hypoxia-ischemia
The lesion volumes were measured 24 h after the insult. Perfused and frozen brains were entirely cut into 50-μm coronal sections disposed in series. In a series of cresyl violet-stained sections spaced at 500 μm, the contralateral striatum and the intact ipsilateral striatum areas were measured using the Neurolucida program (MicroBrightField) and the corresponding volumes were then calculated with Neuroexplorer (MicroBrightField). The surviving striatum was then expressed as a percentage of contralateral striatum volume.
Immunostaining
Immunostainings were performed on primary cortical neurons that had been cultured on coverslips and fixed with 4% paraformaldehyde for 15 min, and on 18-μm cryostat brain sections from rat pups that had been perfused intracardially with the same fixative. Blocking and permeabilization were done in PBS with 15% donkey serum and 0.3% Triton X-100 for 30–45 min. Primary antibodies were diluted in PBS with 1.5% donkey serum and 0.1% Triton X-100 overnight at 4 °C. Secondary antibodies were diluted in PBS for 2 h at room temperature.
The following primary antibodies were used: anti-CTSB/cathepsin B (06-480) rabbit polyclonal antibody from Upstate Biotechnology; anti-RBFOX3/NeuN (MAB377) mouse monoclonal antibody and anti-MAP2 rabbit (AB5622) from Chemicon; anti-LAMP1 (428017, for rat) from Calbiochem. Anti-LC3 was a generous gift from Prof Yasuo Uchiyama (Tokyo, Japan).
For immunofluorescence labeling, Alexa Fluor 488 or 594 donkey-anti-rabbit or mouse secondary antibodies (Invitrogen, A21202, A21203, A21206, A21207) were used and sections or coverslips were mounted with FluorSave after a Hoechst staining. A LSM 510 Meta confocal microscope (Carl Zeiss) was used for confocal laser microscopy. Images were processed with LSM 510 software and mounted using Adobe Photoshop.
For immunoperoxidase labeling on rat brain sections, endogenous peroxidases were quenched in 0.3% H2O2 in methanol for 20 min before incubation in blocking medium. After the incubation in biotinylated secondary antibody (Jackson Immunoresearch, 715-065-151, 711-065-152, 705-065-003), avidin-biotinylated peroxidase complex (VECTASTAIN Elite ABC Kit Vector, PK-6200) was added for 2 h at room temperature and then developed by incubation with diaminobenzidene (DAB, Roche, 11718096001) substrate solution. The sections were dehydrated in graded alcohols and mounted in Eukitt (O. Kindler GmbH, 1330-20-7). Sections were analyzed using a Zeiss Axioplan 2 imaging microscope.
N-Acetylhexosaminidase histochemistry
Experiments were performed as described previously.Citation12 Briefly, sections were incubated for 1 h at 37 °C in Fast Red Violet salt solution (0.63 M ethylene glycol monomethyl ether, 1% polyvinyl pyrrolidone, 0.1 M citric acid-citrate buffer, pH 4.4, 1.6 M sodium chloride, 2.7 mM 5-chloro-4-benzamido-2-methylbenzene-diazonium chloride (Fast Red Violet LB salt; Sigma-Aldrich, F3381) using 0.5 mmol/L naphthol-AS-BI-N-acetyl-D-glucosaminide (Sigma-Aldrich, N4006) as the substrate. The sections were then fixed in 10% formaldehyde, dehydrated in graded alcohols, and mounted in Eukitt medium. To verify the staining specificity, controls with omission of the substrate or at basic pH (pH 10) were done in parallel. Image acquisition was performed with an Axiovision Zeiss microscope.
Statistical analysis
Data are expressed as mean values ± SEM. Data were analyzed statistically using JB STAT software. The n values were sufficiently high for the statistical power to be greater than 0.8 in all experiments. We first tested each group of data for distribution normality using Shapiro-Wilk tests. In cases of normal distribution, we used a Welch’s ANOVA test (one-way ANOVA with unequal variances) followed by a post-hoc Tukey-Kramer test to compare the different treatments. When the distribution was not normal, we used a Kruskal-Wallis test (non-parametric analog of the one-way ANOVA) followed by a post-hoc Steel-Dwass test to compare the different treatments. P < 0.05 was chosen as the threshold for statistical significance.
| Abbreviations: | ||
| BBS | = | bicarbonate-buffered saline |
| Hx | = | hypoxia |
| HI | = | hypoxia-ischemia |
| Ka | = | kainate |
| LAMP1 | = | lysosomal-associated membrane protein 1 |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| 3-MA | = | 3-methyladenine |
| PepA | = | pepstatin A |
| PI | = | propidium iodide |
| Q-VD-OPH | = | quinoline-Val-Asp(ome)-Ch2-O-phenoxy |
| ROS | = | reactive oxygen species |
| shRNA | = | short hairpin RNA |
| STS | = | staurosporine |
Additional material
Download Zip (191.9 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the Cellular Imaging Facility (University of Lausanne, Switzerland) for experimental support. This research was supported by grants from the Swiss National Science Foundation (310030-130769), from the Fondation Emma Muschamp and from the Faculté de Biologie et de Médecine (Lausanne).
References
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445 - 544; http://dx.doi.org/10.4161/auto.19496; PMID: 22966490
- Clarke PGH. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 1990; 181:195 - 213; http://dx.doi.org/10.1007/BF00174615; PMID: 2186664
- Puyal J, Ginet V, Clarke PGH. Multiple interacting cell death mechanisms in the mediation of excitotoxicity and ischemic brain damage: a challenge for neuroprotection. Prog Neurobiol 2013; 105:24 - 48; http://dx.doi.org/10.1016/j.pneurobio.2013.03.002; PMID: 23567504
- Clarke PGH, Puyal J. Autophagic cell death exists. Autophagy 2012; 8:867 - 9; http://dx.doi.org/10.4161/auto.20380; PMID: 22652592
- Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 2008; 9:1004 - 10; http://dx.doi.org/10.1038/nrm2529; PMID: 18971948
- Puyal J, Ginet V, Grishchuk Y, Truttmann AC, Clarke PG. Neuronal autophagy as a mediator of life and death: contrasting roles in chronic neurodegenerative and acute neural disorders. Neuroscientist 2012; 18:224 - 36; http://dx.doi.org/10.1177/1073858411404948; PMID: 21525331
- Wei K, Wang P, Miao CY. A double-edged sword with therapeutic potential: an updated role of autophagy in ischemic cerebral injury. CNS Neurosci Ther 2012; 18:879 - 86; http://dx.doi.org/10.1111/cns.12005; PMID: 22998350
- Yuan J, Kroemer G. Alternative cell death mechanisms in development and beyond. Genes Dev 2010; 24:2592 - 602; http://dx.doi.org/10.1101/gad.1984410; PMID: 21123646
- Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis 2008; 32:329 - 39; http://dx.doi.org/10.1016/j.nbd.2008.07.022; PMID: 18760364
- Wang P, Guan YF, Du H, Zhai QW, Su DF, Miao CY. Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy 2012; 8:77 - 87; http://dx.doi.org/10.4161/auto.8.1.18274; PMID: 22113203
- Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, et al. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol 2008; 172:454 - 69; http://dx.doi.org/10.2353/ajpath.2008.070876; PMID: 18187572
- Puyal J, Vaslin A, Mottier V, Clarke PG. Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann Neurol 2009; 66:378 - 89; http://dx.doi.org/10.1002/ana.21714; PMID: 19551849
- Wen YD, Sheng R, Zhang LS, Han R, Zhang X, Zhang XD, Han F, Fukunaga K, Qin ZH. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy 2008; 4:762 - 9; PMID: 18567942
- Xing S, Zhang Y, Li J, Zhang J, Li Y, Dang C, Li C, Fan Y, Yu J, Pei Z, et al. Beclin 1 knockdown inhibits autophagic activation and prevents the secondary neurodegenerative damage in the ipsilateral thalamus following focal cerebral infarction. Autophagy 2012; 8:63 - 76; http://dx.doi.org/10.4161/auto.8.1.18217; PMID: 22108007
- Rice JE 3rd, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 1981; 9:131 - 41; http://dx.doi.org/10.1002/ana.410090206; PMID: 7235629
- Ginet V, Puyal J, Clarke PGH, Truttmann AC. Enhancement of autophagic flux after neonatal cerebral hypoxia-ischemia and its region-specific relationship to apoptotic mechanisms. Am J Pathol 2009; 175:1962 - 74; http://dx.doi.org/10.2353/ajpath.2009.090463; PMID: 19815706
- Vincent P, Mulle C. Kainate receptors in epilepsy and excitotoxicity. Neuroscience 2009; 158:309 - 23; http://dx.doi.org/10.1016/j.neuroscience.2008.02.066; PMID: 18400404
- Wang Q, Yu S, Simonyi A, Sun GY, Sun AY. Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol Neurobiol 2005; 31:3 - 16; http://dx.doi.org/10.1385/MN:31:1-3:003; PMID: 15953808
- Dong XX, Wang YR, Qin S, Liang ZQ, Liu BH, Qin ZH, Wang Y. p53 mediates autophagy activation and mitochondria dysfunction in kainic acid-induced excitotoxicity in primary striatal neurons. Neuroscience 2012; 207:52 - 64; http://dx.doi.org/10.1016/j.neuroscience.2012.01.018; PMID: 22330834
- Shacka JJ, Lu J, Xie ZL, Uchiyama Y, Roth KA, Zhang J. Kainic acid induces early and transient autophagic stress in mouse hippocampus. Neurosci Lett 2007; 414:57 - 60; http://dx.doi.org/10.1016/j.neulet.2006.12.025; PMID: 17223264
- Azad MB, Chen Y, Gibson SB. Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid Redox Signal 2009; 11:777 - 90; http://dx.doi.org/10.1089/ars.2008.2270; PMID: 18828708
- Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ 2009; 16:1040 - 52; http://dx.doi.org/10.1038/cdd.2009.49; PMID: 19407826
- Szumiel I. Autophagy, reactive oxygen species and the fate of mammalian cells. Free Radic Res 2011; 45:253 - 65; http://dx.doi.org/10.3109/10715762.2010.525233; PMID: 20964552
- Scherz-Shouval R, Weidberg H, Gonen C, Wilder S, Elazar Z, Oren M. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc Natl Acad Sci U S A 2010; 107:18511 - 6; http://dx.doi.org/10.1073/pnas.1006124107; PMID: 20937856
- Wen Y, Zhai RG, Kim MD. The role of autophagy in Nmnat-mediated protection against hypoxia-induced dendrite degeneration. Mol Cell Neurosci 2013; 52:140 - 51; http://dx.doi.org/10.1016/j.mcn.2012.11.008; PMID: 23159780
- Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, Baehrecke EH, Lenardo M. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci U S A 2006; 103:4952 - 7; http://dx.doi.org/10.1073/pnas.0511288103; PMID: 16547133
- Kubota C, Torii S, Hou N, Saito N, Yoshimoto Y, Imai H, Takeuchi T. Constitutive reactive oxygen species generation from autophagosome/lysosome in neuronal oxidative toxicity. J Biol Chem 2010; 285:667 - 74; http://dx.doi.org/10.1074/jbc.M109.053058; PMID: 19850931
- Grishchuk Y, Ginet V, Truttmann AC, Clarke PG, Puyal J. Beclin 1-independent autophagy contributes to apoptosis in cortical neurons. Autophagy 2011; 7:1115 - 31; http://dx.doi.org/10.4161/auto.7.10.16608; PMID: 21646862
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3:452 - 60; PMID: 17534139
- Borsello T, Croquelois K, Hornung JP, Clarke PG. N-methyl-d-aspartate-triggered neuronal death in organotypic hippocampal cultures is endocytic, autophagic and mediated by the c-Jun N-terminal kinase pathway. Eur J Neurosci 2003; 18:473 - 85; http://dx.doi.org/10.1046/j.1460-9568.2003.02757.x; PMID: 12911744
- Sadasivan S, Zhang Z, Larner SF, Liu MC, Zheng W, Kobeissy FH, Hayes RL, Wang KK. Acute NMDA toxicity in cultured rat cerebellar granule neurons is accompanied by autophagy induction and late onset autophagic cell death phenotype. BMC Neurosci 2010; 11:21; http://dx.doi.org/10.1186/1471-2202-11-21; PMID: 20167092
- Matyja E, Taraszewska A, Nagańska E, Rafałowska J. Autophagic degeneration of motor neurons in a model of slow glutamate excitotoxicity in vitro. Ultrastruct Pathol 2005; 29:331 - 9; http://dx.doi.org/10.1080/01913120500214333; PMID: 16257859
- Chang CF, Huang HJ, Lee HC, Hung KC, Wu RT, Lin AM. Melatonin attenuates kainic acid-induced neurotoxicity in mouse hippocampus via inhibition of autophagy and α-synuclein aggregation. J Pineal Res 2012; 52:312 - 21; http://dx.doi.org/10.1111/j.1600-079X.2011.00945.x; PMID: 22212051
- Wang Y, Han R, Liang ZQ, Wu JC, Zhang XD, Gu ZL, Qin ZH. An autophagic mechanism is involved in apoptotic death of rat striatal neurons induced by the non-N-methyl-D-aspartate receptor agonist kainic acid. Autophagy 2008; 4:214 - 26; PMID: 18094625
- Wang Y, Gu ZL, Cao Y, Liang ZQ, Han R, Bennett MC, Qin ZH. Lysosomal enzyme cathepsin B is involved in kainic acid-induced excitotoxicity in rat striatum. Brain Res 2006; 1071:245 - 9; http://dx.doi.org/10.1016/j.brainres.2005.10.074; PMID: 16409994
- Adhami F, Liao G, Morozov YM, Schloemer A, Schmithorst VJ, Lorenz JN, Dunn RS, Vorhees CV, Wills-Karp M, Degen JL, et al. Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am J Pathol 2006; 169:566 - 83; http://dx.doi.org/10.2353/ajpath.2006.051066; PMID: 16877357
- Cui D, Wang L, Qi A, Zhou Q, Zhang X, Jiang W. Propofol prevents autophagic cell death following oxygen and glucose deprivation in PC12 cells and cerebral ischemia-reperfusion injury in rats. PLoS One 2012; 7:e35324; http://dx.doi.org/10.1371/journal.pone.0035324; PMID: 22509406
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 2005; 1:112 - 9; http://dx.doi.org/10.1038/nchembio711; PMID: 16408008
- Nitatori T, Sato N, Waguri S, Karasawa Y, Araki H, Shibanai K, Kominami E, Uchiyama Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J Neurosci 1995; 15:1001 - 11; PMID: 7869078
- Tian F, Deguchi K, Yamashita T, Ohta Y, Morimoto N, Shang J, Zhang X, Liu N, Ikeda Y, Matsuura T, et al. In vivo imaging of autophagy in a mouse stroke model. Autophagy 2010; 6:1107 - 14; http://dx.doi.org/10.4161/auto.6.8.13427; PMID: 20930570
- Zhu C, Wang X, Xu F, Bahr BA, Shibata M, Uchiyama Y, Hagberg H, Blomgren K. The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia-ischemia. Cell Death Differ 2005; 12:162 - 76; http://dx.doi.org/10.1038/sj.cdd.4401545; PMID: 15592434
- Xu F, Li J, Ni W, Shen YW, Zhang XP. Peroxisome proliferator-activated receptor-γ agonist 15d-prostaglandin J2 mediates neuronal autophagy after cerebral ischemia-reperfusion injury. PLoS ONE 2013; 8:e55080; http://dx.doi.org/10.1371/journal.pone.0055080
- Puyal J, Clarke PGH. Targeting autophagy to prevent neonatal stroke damage. Autophagy 2009; 5:1060 - 1; http://dx.doi.org/10.4161/auto.5.7.9728; PMID: 19713756
- Liu Y, Shoji-Kawata S, Sumpter RM Jr., Wei Y, Ginet V, Zhang L, Posner B, Tran KA, Green DR, Xavier RJ, et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci U S A 2013; 110:20364 - 71; http://dx.doi.org/10.1073/pnas.1319661110; PMID: 24277826
- Pérez-Carrión MD, Pérez-Martínez FC, Merino S, Sánchez-Verdú P, Martínez-Hernández J, Luján R, Ceña V. Dendrimer-mediated siRNA delivery knocks down Beclin 1 and potentiates NMDA-mediated toxicity in rat cortical neurons. J Neurochem 2012; 120:259 - 68; http://dx.doi.org/10.1111/j.1471-4159.2011.07556.x; PMID: 22035151
- Carloni S, Buonocore G, Longini M, Proietti F, Balduini W. Inhibition of rapamycin-induced autophagy causes necrotic cell death associated with Bax/Bad mitochondrial translocation. Neuroscience 2012; 203:160 - 9; http://dx.doi.org/10.1016/j.neuroscience.2011.12.021; PMID: 22209856
- Carloni S, Girelli S, Scopa C, Buonocore G, Longini M, Balduini W. Activation of autophagy and Akt/CREB signaling play an equivalent role in the neuroprotective effect of rapamycin in neonatal hypoxia-ischemia. Autophagy 2010; 6:366 - 77; http://dx.doi.org/10.4161/auto.6.3.11261; PMID: 20168088
- Papadakis M, Hadley G, Xilouri M, Hoyte LC, Nagel S, McMenamin MM, Tsaknakis G, Watt SM, Drakesmith CW, Chen R, et al. Tsc1 (hamartin) confers neuroprotection against ischemia by inducing autophagy. Nat Med 2013; 19:351 - 7; http://dx.doi.org/10.1038/nm.3097; PMID: 23435171
- Sheng R, Zhang LS, Han R, Liu XQ, Gao B, Qin ZH. Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy 2010; 6:482 - 94; http://dx.doi.org/10.4161/auto.6.4.11737; PMID: 20400854
- Hughes KJ, Kennedy BK. Cell biology. Rapamycin paradox resolved. Science 2012; 335:1578 - 9; http://dx.doi.org/10.1126/science.1221365; PMID: 22461595
- Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, Ong CN, Codogno P, Shen HM. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem 2010; 285:10850 - 61; http://dx.doi.org/10.1074/jbc.M109.080796; PMID: 20123989
- Tiede LM, Cook EA, Morsey B, Fox HS. Oxygen matters: tissue culture oxygen levels affect mitochondrial function and structure as well as responses to HIV viroproteins. Cell Death Dis 2011; 2:e246; http://dx.doi.org/10.1038/cddis.2011.128; PMID: 22190005
- Oh S, Shin CS, Kim HS. The time course of NMDA- and Kainate-induced cGMP elevation and glutamate release in cultured neuron. Arch Pharm Res 1995; 18:153 - 8; http://dx.doi.org/10.1007/BF02979187
- Chen J, Li C, Pei DS, Han D, Liu XM, Jiang HX, Wang XT, Guan QH, Wen XR, Hou XY, et al. GluR6-containing KA receptor mediates the activation of p38 MAP kinase in rat hippocampal CA1 region during brain ischemia injury. Hippocampus 2009; 19:79 - 89; http://dx.doi.org/10.1002/hipo.20479; PMID: 18680160
- Lowry ER, Kruyer A, Norris EH, Cederroth CR, Strickland S. The GluK4 kainate receptor subunit regulates memory, mood, and excitotoxic neurodegeneration. Neuroscience 2013; 235:215 - 25; http://dx.doi.org/10.1016/j.neuroscience.2013.01.029; PMID: 23357115
- Schurr A, Rigor BM. Quinolinate potentiates the neurotoxicity of excitatory amino acids in hypoxic neuronal tissue in vitro. Brain Res 1993; 617:76 - 80; http://dx.doi.org/10.1016/0006-8993(93)90615-T; PMID: 8397046
- Schurr A, Payne RS, Rigor BM. Protection by MK-801 against hypoxia-, excitotoxin-, and depolarization-induced neuronal damage in vitro. Neurochem Int 1995; 26:519 - 25; http://dx.doi.org/10.1016/0197-0186(94)00148-N; PMID: 7492949
- Mishra OP, Kubin JA, McGowan JE, Delivoria-Papadopoulos M. Kainate receptor modification in the fetal guinea pig brain during hypoxia. Neurochem Res 1995; 20:1171 - 7; http://dx.doi.org/10.1007/BF00995380; PMID: 8746802
- Panigrahy A, White WF, Rava LA, Kinney HC. Developmental changes in [3H]kainate binding in human brainstem sites vulnerable to perinatal hypoxia-ischemia. Neuroscience 1995; 67:441 - 54; http://dx.doi.org/10.1016/0306-4522(95)00016-C; PMID: 7675177
- Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch 2010; 460:525 - 42; http://dx.doi.org/10.1007/s00424-010-0809-1; PMID: 20229265
- Wang Y, Qin ZH. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis 2010; 15:1382 - 402; http://dx.doi.org/10.1007/s10495-010-0481-0; PMID: 20213199
- Sakaida I, Kyle ME, Farber JL. Autophagic degradation of protein generates a pool of ferric iron required for the killing of cultured hepatocytes by an oxidative stress. Mol Pharmacol 1990; 37:435 - 42; PMID: 2314391
- Seyfried D, Han Y, Zheng Z, Day N, Moin K, Rempel S, Sloane B, Chopp M. Cathepsin B and middle cerebral artery occlusion in the rat. J Neurosurg 1997; 87:716 - 23; http://dx.doi.org/10.3171/jns.1997.87.5.0716; PMID: 9347980
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885 - 9; http://dx.doi.org/10.1038/nature04724; PMID: 16625204
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880 - 4; http://dx.doi.org/10.1038/nature04723; PMID: 16625205
- Vaslin A, Puyal J, Borsello T, Clarke PG. Excitotoxicity-related endocytosis in cortical neurons. J Neurochem 2007; 102:789 - 800; http://dx.doi.org/10.1111/j.1471-4159.2007.04564.x; PMID: 17437546
- Levine S. Anoxic-ischemic encephalopathy in rats. Am J Pathol 1960; 36:1 - 17; PMID: 14416289
- Ginet V, Puyal J, Magnin G, Clarke PG, Truttmann AC. Limited role of the c-Jun N-terminal kinase pathway in a neonatal rat model of cerebral hypoxia-ischemia. J Neurochem 2009; 108:552 - 62; http://dx.doi.org/10.1111/j.1471-4159.2008.05797.x; PMID: 19046406
- Vaslin A, Puyal J, Clarke PGH. Excitotoxicity-induced endocytosis confers drug targeting in cerebral ischemia. Ann Neurol 2009; 65:337 - 47; http://dx.doi.org/10.1002/ana.21584; PMID: 19334077