Abstract
Early stage endometrial cancer is generally curable. However, progress in the treatment of advanced and recurrent endometrial cancer has been limited. This has led to a shift from the use of traditional chemotherapeutic agents and radiotherapy regimens to the promising area of targeted therapy, given the large number of druggable molecular alterations found in endometrial cancer. To maximize the effects of directed targeted therapy, careful molecular characterization of the endometrial tumor is necessary. This represents an important difference in the use of targeted therapy vs. traditional chemotherapy or radiation treatment. This review will discuss relevant pathways to target in endometrial cancer as well as the challenges that arise during development of a personalized oncology approach.
Introduction
In the United States, endometrial cancer remains the most common gynecologic malignancy. In 2011, the American Cancer Society estimated there would be 46,470 new cases and 8,120 deaths from endometrial cancer. In contrary to the progress observed in many other cancer types, the incidence of endometrial cancer has increased over the past 30 y.Citation1 This is in part related to the current epidemic of obesity, which is tightly linked to risk of endometrial cancer.Citation2 Fortunately, the majority of women with endometrial cancer are diagnosed at an early stage and may be cured by surgery with or without adjuvant radiotherapy.Citation3 However, the subset of patients with either advanced stage endometrial cancer at diagnosis or recurrent disease following surgery presents a significant therapeutic challenge. The use of chemotherapy has been well-studied in this group, with only modest outcomes among those patients that respond to front-line therapy. The optimal treatment approaches yield response rates from 40–78% in the primary advanced setting and 15–30% in the recurrent setting. Furthermore, among the optimal chemotherapy regimens for this group, median progression-free survival is only 6 mo and median overall survival reaches 12 mo.Citation4 Clearly, this is a population of cancer patients in great need for the development of novel approaches for therapy.
Opportunities for Targeted Therapy in Advanced and Recurrent Endometrial Cancer
As the era of targeted therapy and personalized cancer treatments dawns, endometrial cancer has great potential to benefit from novel agents currently under development. These therapies hold the potential for reduction in mortality from endometrial cancer, and perhaps reduction in morbidity associated with traditional cancer treatment by selectively targeting cancer cells. Targeted therapy may be achieved both through combination with traditional cytotoxic agents and through combinations of novel biologic agents which hit multiple relevant targets.
The molecular alterations of endometrial cancer are well documented in the literature and appear to directly relate to the histologic type. This knowledge has significantly helped advance the development of targeted therapy for endometrial cancer. In general, endometrioid carcinoma is the most common histology (80%) compared with non-endometrioid carcinomas (20%) which include serous carcinoma, clear cell carcinoma, and carcinosarcoma. summarizes the known molecular changes in both endometrioid and non-endometrioid endometrial carcinomas. Note that from this table there are a number of molecular alterations that represent potentially druggable targets. As our depth of understanding of the molecular pathways critical to the development and progression of endometrial cancer expands, so to will the list of potential targets. Undoubtedly, the NIH-sponsored The Cancer Genome Atlas (TCGA) will contribute to the identification of additional endometrial cancer molecular alterations that may be exploited therapeutically.
As the role of targeted therapy continues to grow in oncology, there are several unmet needs which must be addressed in endometrial cancer clinical trials. Arguably, the most pressing need is the selection of appropriate patients for treatment with a given specific targeted agent. The use of biologic agents in an unselected patient population has the potential to lead to incorrect classification of a drug as inactive, dooming it to be classified as a failure. The reasons for this are myriad, including lack of expression of the relevant target or presence of a mutation which confers resistance to the agent.Citation5 Furthermore, use of markers to predict unique toxicities and optimal drug dosing will also be important.Citation6 Such pre-selection of patients for treatment is a relatively new concept in oncology clinical trials, as most previously completed chemotherapy and radiation treatment trials used unselected patient populations. In the targeted therapy era, these issues and trial designs must be considered to maximize effective and timely drug development. The objective for this review is to provide an overview of current relevant pathways and the potential for biomarker driven trials in advanced and recurrent endometrial cancer.
Pathways of Interest in Endometrial Cancer
As our understanding of endometrial cancer grows, we begin to identify more potential pathways to target therapeutically. demonstrates a simplified schematic of select relevant pathways and sites of interaction and cross talk. The overlapping nature of these pathways in endometrial cancer makes the combination of multiple targeted agents or agents which target more than one pathway very attractive and feasible. At the time of this review, however, the majority of clinical investigations employing targeted agents in endometrial cancer consist primarily of single agent trials.
Phosphatidylinositol-3-kinase (PI3K)/AKT.
The PI3K/AKT pathway is well-known to play a central role in cell survival, growth, and avoidance of apoptosis in many different cancer types.Citation7,Citation8 Stimulation of the PI3K/AKT pathway occurs through the activity of myriad receptors including epidermal growth factor receptor (EGFR), insulin-like growth factor I receptor (IGFIR), and fibroblast growth factor receptor 2 (FGFR2). Furthermore, the tight linkage of the PI3K/AKT pathway to other key survival pathways such as the RAS/RAF/MEK pathway holds significant clinical implications for trial design.
In general, constitutive activation of the PI3K/AKT pathway in endometrial cancer occurs most commonly through loss of PTEN tumor suppressor activity or activating mutations in PIK3CA, which codes for the α catalytic subunit of PI3K. In addition, activating mutations in AKT and overexpression of tyrosine kinase receptors that stimulate the pathway are known to be important in subsequent PI3K/AKT dysregulation.Citation9–Citation21 The mammalian target of rapamycin (mTOR), a serine/threonine kinase, is a critical downstream target of the PI3K/AKT pathway. mTOR upregulation through AKT leads to subsequent activation of the protein S6 kinase (pS6K) which regulates protein translation and cell cycle progression.Citation7
Given the frequency of abnormalities in the PI3K/AKT pathway, this signaling pathway arguably presents one of the most promising targets for endometrial cancer. Furthermore, the known alterations in the pathway providing potential targets also present opportunities for patient selection and monitoring therapy response. At present, agents under exploration consist primarily of small molecule inhibitors of critical mediators of the pathway. As seen in the discussion that follows, it appears the inhibition of only one pathway node may not be sufficient to impact tumor growth. This is in part due to the significant pathway feedback loops as well as crosstalk between pathways. For example, the downstream proteins activated by mTOR also participate in a feedback loop that can lead to subsequent upregulation of AKT phosphorylation.Citation22 Thus, after confirmation of safety in the single agent setting, exploration of combination therapies is critical.
Potential biomarkers to predict response to therapy and guide eligibility for trials are extensive for this pathway. As shown in , many of the relevant molecular abnormalities in endometrial cancer are directly or indirectly related to the PI3K/AKT pathway. Primary candidates include mutations in PIK3CA, PTEN, AKT, as well as overexpression of phosphorylated mTOR and phosphorylated AKT (pAKT).Citation23–Citation25 Preliminary results from a group of patients with advanced solid tumors, including endometrial cancer, harboring PIK3CA mutations that were treated with PI3K/AKT/mTOR inhibitors alone or in combination with other agents, reveals higher than expected response rates (35%) in a highly pretreated group of patients.Citation26
Interestingly, the presence of KRAS mutations appeared to confer resistance in certain tumor types in the retrospective study by Janku and colleagues.Citation26 The importance of the possible interaction between PIK3CA and KRAS mutations has also been demonstrated in human cancer cells in response to treatment with everolimus in vitro. Cells harboring PIK3CA mutations were quite sensitive to everolimus, while those cells with concurrent PIK3CA and KRAS mutations were resistant.Citation27 This resistance may be explained by a negative feedback loop with the RAS/RAF/MEK pathway as suggested by Carracedo and colleagues. In a molecular analysis of patients with advanced cancer, they found upregulation of phospho-ERK in tumor samples obtained after treatment with everolimus on a Phase I trial. In subsequent cell line studies, MEK activation was discovered after everolimus treatment through a feedback loop involving pS6K. Further, inhibition of the RAS/RAF/MEK pathway enhanced the antitumor effect of everolimus in cell lines and in vitro.Citation28 Thus, it appears that consideration of both pathways will likely prove useful in future trial design.
mTOR inhibition. The protein kinase mTOR is composed of two potential complexes to target, mTORC1 and mTORC2, which have different downstream effectors.Citation29 Thus far, the majority of studies in advanced and recurrent endometrial cancer have focused on rapalogs, which primarily inhibit MTORC1. Newer agents which target mTORC2 or both complexes appear to have greater activity but the data are still emerging. Temsirolimus (CC1-779), a parenterally administered ester of rapamycin, was evaluated in a phase II trial of recurrent or metastatic endometrial cancer patients. Response rates were favorable in the chemotherapy naïve arm (n = 29), with 14% partial response (PR) and 69% stable disease (SD). Interestingly, response in the patients treated with one prior chemotherapy regimen (n = 25) had less dramatic results, with only 4% PR and 28% SD. Response occurred independent of PTEN mutation, PTEN loss, phosphorylated mTOR (p-mTOR), pAKT, or pS6K status in archival tumor specimens from original diagnosis. Of note, the study was not powered to determine the association between molecular status and response, so these results must be interpreted with caution.Citation30 These favorable response outcomes were also demonstrated in a single agent trial of temsirolimus in heavily pre-treated patients without any molecular correlates. Of 27 patients treated, 2 achieved PR and 12 SD.Citation31 Given this apparent activity, the Gynecologic Oncology Group (GOG) has included temsirolimus combined with carboplatin and paclitaxel as a treatment arm in an ongoing three arm trial exploring combination therapies in advanced endometrial cancer (GOG0086P; NCT:00977574).
Oral mTOR inhibition by everolimus (RAD001) in recurrent endometrial cancer achieved encouraging short and long-term clinical benefit with 43% of patients at 8 weeks and 21% of patients at 16 weeks achieving SD.Citation32 Meyer and colleagues recently reported attempts to correlate response on this trial to molecular status of the primary tumor, including PTEN protein expression, pS6K expression and KRAS mutation. Among the 28 patients with available tissue, none of these markers successfully predicted response, although there was a trend for KRAS mutation correlation with therapy resistance.Citation33 Ridaforolimus (AP23573), an mTOR inhibitor given parenterally or orally, yielded a 33% clinical benefit rate including two patients with a PR among patients with recurrent endometrial cancer.Citation34 An additional study of oral ridaforolimus in chemotherapy naïve endometrial cancer achieved a PR rate of 7.7% and SD rate of 58%. This study has correlative endpoints, including PTEN and PI3K, ongoing.Citation35 A randomized Phase II trial comparing oral ridaforolimus to traditional hormonal or chemotherapy revealed improvement in PFS of 2 mo among patients in the experimental arm.Citation36
The combination of mTOR inhibition with hormonal agents is actively being explored secondary to known activity of both agents in endometrial cancer. Furthermore, there are recent data that indicate the importance of the PI3K/AKT/mTOR pathway in mediating resistance to endocrine therapy in breast cancer.Citation37,Citation38 A study of everolimus and letrozole in recurrent endometrial cancer with less than two prior chemotherapeutic regimens achieved a response rate of 21% with one complete response (CR) and three PRs. Furthermore, four patients had SD and seven still remain on treatment.Citation39 A recently reported GOG study combining temsirolimus with alternating hormones, megestrol acetate and tamoxifen, was closed secondary to high levels of venous thrombosis and insufficient additional activity to warrant further study.Citation40
AKT inhibition. There are numerous small molecule inhibitors of AKT currently in development for endometrial cancer and solid tumors. MK-2206 is an allosteric AKT inhibitor which is currently under active investigation in multiple clinical trials, alone and in combination with chemotherapy or other targeted agents.Citation41 This agent is currently employed in a biomarker driven Phase II trial of recurrent endometrial cancer with less than two prior therapies. Patients are stratified based on presence or absence of a PIK3CA mutation prior to the initiation of therapy (NCT01312753).
PI3K inhibition. The inhibition of PI3K is an area of great interest in endometrial cancers and solid tumors. There are myriad agents currently in development in Phase I trials of advanced solid tumors. This includes agents which specifically target PI3K as well as dual inhibitors which target mTOR in combination with PI3K.Citation42 A Phase I trial of XL147 (SAR245408), a selective oral PI3K inhibitor, demonstrated durable clinical benefit among multiple solid tumors. XL147 is in Phase II trial for advanced and recurrent endometrial cancer (NCT01013324). Furthermore, a phase I trial combining this agent with carboplatin and paclitaxel is currently under dose expansion for endometrial cancer secondary to promising tumor response (NCT00756847). Other agents in this class have also demonstrated promising toxicity and early clinical response among endometrial cancer patients in Phase I trials,Citation43,Citation44 and many Phase II studies are in progress (clinicaltrials.gov).
Metformin. Metformin (N′-N′ dimethylguanide) is a well tolerated oral anti-diabetic agent that has recently demonstrated promising findings in reduction of cancer risk and antitumor activity.Citation45,Citation46 The theoretical mechanisms for the anticancer activity of metformin include activation of the AMPK pathway through LKB1 as well as reduction of circulating insulin levelsCitation47,Citation48 (). Given the role of obesity and a high prevalence of insulin resistance among women with endometrial cancer, metformin is of considerable interest.Citation49 Furthermore, this agent has been shown to induce cell cycle arrest and apoptosis,Citation50 as well as reverse progesterone resistance and induce progesterone receptor expression in endometrial cancer cell lines.Citation51,Citation52 Metformin has potential for use in early as well as advanced endometrial cancer in combination with hormonal therapy and as a single agent. A current Phase 0 study is ongoing to explore pharmacodynamic markers of metformin activity in endometrial cancer patients treated prior to primary surgical resection. Markers for proliferation, apoptosis, the insulin-like growth factor signaling pathway, and the PI3K/AKT pathway will be assessed in baseline (pre-treatment) and after a short course of metformin (NCT01205672).
RAS/RAF/MEK.
The RAS/RAF/MEK pathway shown in is involved in a variety of essential tumorigenic functions including angiogenesis, cell cycle regulation, proliferation, and survival.Citation53 As noted in , there is a relatively high prevalence of KRAS mutations in endometrial cancer, especially the endometrioid carcinomas, as well as other solid tumors.Citation54–Citation57 This has lead to the exploration of therapeutic agents to target key nodes of this pathway.Citation58,Citation59 MEK inhibitors are the most clinically developed, with emerging applications in melanoma and papillary thyroid carcinoma secondary to activating BRAF mutation rates of 20–80%Citation60 and 29–83%,Citation61 respectively. In addition, RAF kinase small molecule inhibitors are in clinical development alone and in combination for the treatment of advanced solid tumors, especially melanoma.Citation62
The GOG has opened a Phase II trial of the oral MEK inhibitor, AZD6244, in patients with endometrial cancer treated with 1–2 prior therapies (NCT 01011933). This trial has achieved an adequate response rate to proceed to the second stage of accrual and results are eagerly anticipated. The knowledge that RAS/RAF/MEK pathway activation in the presence of AKT blockade can promote cell survivalCitation63 is leading to development of trials that combine PI3K/AKT inhibition and MEK inhibition in endometrial cancer. This combination is further supported by the apparent negative feedback loop involving pS6K and MEK as previously discussed in the section on the PI3K/AKT pathway.Citation28
Based on data regarding successful MEK inhibition in melanoma and other solid tumors, there are several key biomarkers that should be assessed in trials incorporating these agents. Activating mutations in BRAF render tumors highly sensitive to MEK inhibition based on work in cell lines and in early clinical trials of melanoma.Citation64,Citation65 Interestingly, BRAF mutations are rare in endometrial cancers,Citation66,Citation67 so it will be relevant to identify alternative mutations that can be used to identify endometrial tumors as being potentially sensitive to MEK inhibitors. In addition, the presence of KRAS or NRAS mutation has the potential to affect response to MEK inhibition, although the data here are less clear.Citation58,Citation59 Demonstrating evidence of pathway activation (presence of phospho-ERK and phosphoEGFRCitation62) in the tumor may also be relevant.
IGF-IR.
As seen in , through the activation of IGF-IR, Insulin-like Growth Factor (IGF) is a mitogen for PI3K/AKT and RAS/RAF/MEK pathway activation.Citation68 IGF-IR is overexpressed in the precursor to endometrial cancer, endometrial hyperplasia, as well as in endometrial carcinoma.Citation69,Citation70 In vitro studies of the IGF-IR inhibitor, NVP-AEW541, demonstrated inhibition of proliferation and increase in apoptosis among treated endometrial cancer cell lines.Citation71 Due to the known activation of the IGF-IR pathway with inhibition of mTOR, there exists a rationale for combination of IGF-IR inhibitors and drugs targeting mTOR.Citation72
There are several IGF-IR inhibitors in clinical investigation, including IMC-A12, AMG479, H10H5, and OSI-906. These agents are very well tolerated, with the most common adverse events consisting of hyperglycemia, fatigue, nausea, and changes in body weight.Citation73,Citation74 As yet, there are no clinical trials that have explored this promising pathway in endometrial cancer. Potential biomarkers for this pathway may include IGF-I, IGF-BP1, IGF-BP2 and insulin receptor substrates-1 (IRS1) and -2 (IRS2).Citation75,Citation76
Angiogenesis.
The formation of new blood vessels through angiogenesis is a key factor in tumorigenesis. Angiogenesis allows for the supply of nutrients, oxygen and growth factors to the tumor and promotes tumor dissemination and metastasis.Citation77,Citation78 Regulation of angiogenesis occurs though a complex set of stimulatory and inhibitory factors that act on the endothelial cells lining the vessel (). When pro-angiogenic factors such as Vascular Endothelial Growth Factor (VEGF) are found in tumors, there is a resulting increase in unregulated division and growth of the endothelial cells.Citation79,Citation80 Furthermore, overexpression of VEGF is associated with poor prognostic factors in endometrial cancer such as deep myometrial invasion and lymph node metastasis.Citation81,Citation82
Targeting VEGF and related molecules. Despite the numerous potential targets for inhibition of angiogenesis shown (), the most significant developments of clinical agents in this field are those that target the VEGF ligands and receptors. Bevacizumab, a monoclonal antibody targeting VEGF-A, has been studied in a Phase II trial of recurrent endometrial cancer. This agent had favorable single agent activity demonstrating 13.5% response durable for a median of 6 mo and a median overall survival of 10.5 mo.Citation83 Multiple trials are ongoing of bevacizumab as a single agent or in combination in uterine cancer, including a trial combining bevacizumab with cisplatin for chemosensitization (NCT01005329).
VEGF-Trap is a decoy receptor for all VEGF isoforms that demonstrates high affinity VEGF binding and prevention of VEGF pathway activation. This agent is manufactured utilizing the ligand-binding domains from two VEGF receptors with a constant region of IgG1.Citation84 There is a Phase II trial of VEGF-Trap in recurrent endometrial cancer currently accruing through the GOG. This trial incorporates a number of translational endpoints to predict disease response which will be discussed at the end of this section (NCT004682826).
Small molecule inhibitors targeting one or more components of the VEGF pathway are of great interest in the treatment of recurrent or persistent endometrial cancer. Sunitinib targets VEGF receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), epidermal growth factor (EGF), and the stem cell factor (KIT) receptor.Citation85 This agent is under study as a single agent in endometrial cancer.
Targeting fibroblast growth factors (FGF). FGFs are involved with angiogenesis as well as other mechanisms important to tumorigenesis.Citation86 The discovery of activating mutations in the gene that encodes FGFR-2 in endometrial cancers makes this a rational target. One factor that may dampen enthusiasm for targeting FGFR-2 is that mutations only occur in approximately 16% of endometrial cancers.Citation11,Citation87,Citation88 It is not known if endometrial cancer metastases/recurrences have a greater percentage of these mutations. There are several agents which target FGFR-2 which are under development, although only two are being explored in endometrial cancer. Cedirinib, a small molecule inhibitor of VEGFR-2 and VEGFR-3, PDGFR, as well as FGFR-2,Citation89 has been active in multiple Phase II trials and is to undergo study in endometrial cancer by the GOG. Brivanib is a dual inhibitor of VEGFR2 and FGFRCitation90 that is also undergoing phase II evaluation for endometrial cancer in the GOG (NCT00888173). In addition, FP-1039 is a FGF1R:Fc soluble fusion protein that binds multiple FGF ligands.Citation91 This agent is under evaluation in a Phase II pilot study in patients with FGFR2 mutated endometrial cancer (NCT01244438).
Unique anti-angiogenic agents. Thalidomide is an anti-angiogenic agent acting through a variety of different mechanisms including inhibition of FGF-2. This agent demonstrated only a 25% clinical benefit rate with two PR and four SD among patients with recurrent endometrial cancer treated with less than two prior chemotherapeutic regimens. There was no correlation found between serum VEGF levels and response to thalidomide.Citation92
Thus far, measurements of biomarkers to predict response to anti-angiogenic agents have been unsuccessful. Tissue and/or serum expression of relevant ligands and receptors is a rational option to predict response, but the sheer number of potential markers to monitor is overwhelming (). Given the importance of VEGF, this ligand has been the most commonly studied in attempting to predict response. In most studies, VEGF expression in tissue or serum has not correlated with response to anti-angiogenic agents.Citation92–Citation95 The GOG performed an analysis of biomarkers to predict response to single agent bevacizumab in ovarian cancer and found a correlation between high CD31/microvessel density (MVD) count and lack of response.Citation96 This has not been confirmed in other tumor types.Citation94,Citation97 Assessing VEGF polymorphisms holds promise for appropriately targeting therapy, but published studies are limited by small numbers of patients.Citation98
Poly (ADP-ribose) polymerase (PARP) pathway.
The cell has a variety of mechanisms to repair DNA damage including direct repair, base excision, mismatch repair, and nucleotide excision repair. Resistance to cytotoxic chemotherapy is typically found in cancer cells with a high activity of DNA damage repair pathways. PARP participates in single-strand DNA damage repair through the base excision pathway.Citation99,Citation100 PARP inhibition results in cellular inability to repair DNA damage caused by single strand breaks. Further, when single strand breaks remain unrepaired, this leads to double strand breaks after the process of replication. Homologous recombination is the mechanism involved in the repair of these double strand breaks. In patients with defects in the homologous recombination pathway, PARP inhibition leaves the cells unable to repair DNA effectively, leading to genetic instability and cell death. Thus, PARP inhibition has been developed for use in anti-cancer therapy, especially in patients with impaired DNA repair mechanisms, such as breast and ovarian cancer patients with BRCA mutations.Citation100–Citation102 This concept, known as synthetic lethality, is currently being employed in a number of cancer types. Interestingly, PTEN loss leads to dysfunctional homologous recombination DNA repair, creating cellular susceptibility to PARP inhibition in vitro and in vivo in a variety of cell lines, including endometrial cancer.Citation100,Citation103–Citation105 Fifteen to twenty percent of endometrial cancers have high levels of microsatellite instability due to methylation and subsequent gene silencing of the promoter for MLH1, a gene that encodes a protein involved in DNA mismatch repair. The influence, if any, of such microsatellite instability on targeted therapy of endometrial cancer is currently unknown.
There are a variety of PARP inhibitors in clinical development, including olaparib, veliparib, and iniparib. These agents have been primarily explored in malignancies that harbor BRCA mutations such as ovarian and breast cancer.Citation101 Given the data presented, we anticipate that PARP inhibitors will be employed in a variety of cancers that harbor homologous recombination defects, including endometrial cancer. Currently, iniparib is being employed to treat recurrent carcinosarcoma of the uterus in combination with carboplatin and paclitaxel (NCT00687687). Sensitivity to PARP inhibitors can be predicted by the presence of mutations that confer impaired DNA damage repair.Citation106 A screen for homologous recombination defects would be another potential method to capture PARP inhibition sensitive patients.
EGFR family.
The EGFR family has garnered a great deal of interest as a cancer therapeutic target given that it activates important downstream pathways such as PI3K/AKT and ras/raf/MEK.Citation107–Citation109 There are 4 EGFR-specific cell-surface receptors that make up this group, EGFR (HER-1, ERBB1), HER-2 (ERBB2), HER-3 (ERBB3), and HER-4 (ERBB4). These receptors are present on endothelial cells within the tumor microenvironment, and activation increases angiogenesis through stimulation of endothelial cell proliferation.Citation110 Finally, activation of EGFR is known to stimulate cancer cell invasion while suppressing apoptosis.Citation107,Citation109 EGFR is overexpressed in a large proportion of endometrial cancer, regardless of histotype.Citation111 Unfortunately, studies of drugs targeting EGFR have demonstrated only modest success in the treatment of endometrial cancer. This may relate to fact that recent data in several solid tumors have indicated that EGFR overexpression is not sufficient to predict response to therapy. It appears that response is more closely correlated with the presence of EGFR mutation.Citation112 Current data indicate that the rate of EGFR mutation in the serous and carcinosarcoma subtypes is quite low, although further study is indicated to determine the prevalence of this mutation in the endometrioid subtype.Citation113,Citation114
Another modifier of response to EGFR directed therapy is KRAS mutation. The presence of KRAS mutations appears to negatively impact response in colorectal cancer patients.Citation115,Citation116 The data were so strong that an American Society of Clinical Oncology Provisional Clinical Opinion was published stating “all metastatic colorectal carcinoma patients who are candidates for anti-EGFR antibody therapy should have their tumor tested for KRAS mutations.”Citation117 In non-small cell lung cancer, EGFR mutations appear to correlate with response to gefitinib and erlotinib,Citation118,Citation119 while KRAS mutations are associated with lack of response.Citation120 Clearly, close examination of this pathway will be necessary to determine the appropriate cohort of patients to treat with these targeted agents.
Targeting EGFR. There are a variety of small molecule inhibitors of EGFR that have been explored in endometrial cancer. Erlotinib blocks the auto-phosphorylation of the tyrosine kinase portion of EGFR. In a group of recurrent endometrial cancers without prior chemotherapy, erlotinib had a 12.5% PR and 47% SD rate. EGFR expression was analyzed in all patients with 19/30 patients were positive. Of those patients with EGFR expression, three had PR and seven had SD. EGFR mutational status was also assessed among all responders with no mutations detected.Citation121
Gefitinib, a small molecule inhibitor that binds to the ATP-binding site of EGFR, was evaluated in recurrent or persistent endometrial cancer after 1–2 prior regimens. Unfortunately, response was quite low with only 1/29 patients demonstrating CR and 7/29 with SD. EGFR mutations, as well as expression of EGFR, phospho-EGFR, phospho-ERK were examined in tumors, but did not correlate with clinical response.Citation122
Monoclonal antibodies which bind the external domain of EGFR, including cetuximab and panitumumab, have also had success in the treatment of advanced solid tumors.Citation123 Cetuximab, a chimerized monoclonal antibody to EGFR, was employed to treat recurrent endometrial cancer patients previously treated with chemotherapy (NCT00392769). This trial recently completed accrual though the GOG and results are anticipated soon.
Targeting HER-2.* Poor prognosis in endometrial cancer has been associated with elevated immunohistochemical expression of HER-2, especially among the papillary serous subtype.Citation124–Citation126 Trastuzumab, a humanized monoclonal antibody to HER-2, was evaluated in a Phase II trial for advanced and recurrent endometrial cancer with overexpression or amplification of HER-2. Unfortunately, there were no clinical responses, and only 12 of 30 patients had SD. This trial closed early due to poor accrual.Citation127 A novel dual tyrosine kinase inhibitor of EGFR and HER-2, lapatinib,Citation128 was also studied in this disease cohort (NCT00096447). Although no data have been published from this GOG trial, second stage accrual was not opened, likely indicating low clinical activity. A more focused trial in advanced or recurrent uterine serous cancers is planned, comparing treatment with paclitaxel/carboplatin with or without herceptin only in patients with tumor expression of HER-2/neu by immunohistochemistry and HER-2/neu gene amplification documented by FISH (NCT01367002).
E-cadherin/β-catenin pathway.
The E-cadherin/catenin unit is responsible for the maintenance of normal cell architecture and cell differentiation.Citation129,Citation130 β-catenin and E-cadherin also are part of the Wnt signaling pathway, which has been implicated in tumorigenesis in numerous cancer types.Citation131–Citation134 Loss of E-cadherin and change in localization of β-catenin to the nucleus is associated with the epithelial-to-mesenchymal transition (EMT). EMT allows cells to obtain mesenchymal properties corresponding to increased motility, invasion, and metastasis.Citation135 Interestingly, EMT has also been associated with chemoresistance to oxaliplatin in colon cancerCitation135 and resistance to EGFR-targeted therapy in lung, pancreatic, and colorectal cancer cell lines.Citation136,Citation137 Accumulation of β-catenin in the nucleus secondary to mutations in CTNNB1, the gene which codes for β-catenin, and loss of E-cadherin expression is not uncommon in endometrial cancer.Citation129,Citation130,Citation133,Citation134,Citation138 No active agents target this pathway currently, but such agents might be potentially useful given this pathway's regulation of key processes in determining a cancer's clinical aggressiveness.
Combination agents.
The development of agents that target a variety of different pathways holds great promise in the treatment of endometrial cancer. Sorafenib is a multi-kinase inhibitor that blocks VEGFR, PDGFR, and KIT as well as raf, a key component of the ras/raf/MEK pathway. This agent demonstrated a 5% PR and 50% SD rate in a Phase II trial of recurrent endometrial cancer.Citation139 Dasatinib inhibits a large array of targets including the SRC family, BCR-ABL, C-KIT and PDGF. Furthermore, dasatinib is known to target the EphA2 receptor, a member of the Ephrin family that is involved in cell proliferation, survival, migration, and angiogenesis.Citation140 EphA2 was found to be over-expressed in a high proportion of endometrioid tumors and correlated with advanced disease and poor prognosis.Citation141 Due to this promising molecular data, development of dasatinib in clinical trials for endometrial cancer is ongoing.
Unmet Needs and Challenges for Targeted Therapy in Endometrial Cancer
There are several cautionary tales in the literature that demonstrate the importance of appropriate patient selection in trials evaluating biologic agents. Given the importance of EGFR mutations in lung cancer, gefitinib was evaluated in a Phase III study which demonstrated improvement in symptoms and a 10% response rate.Citation142 Interestingly, mutations were found in the tyrosine kinase domain of 89% of the responders.Citation143 Unfortunately, in a subsequent Phase III study in an unselected patient population, gefitinib showed no benefit compared with placebo.Citation144 This resulted in gefitinib's removal from the United States market. However, the recent IPASS study utilized a select group of patients based on likelihood of response to gefitinib and found a favorable profile in comparison to standard paclitaxel and carboplatin chemotherapy.Citation145
The use of trastuzamab in breast cancer provides example of the potential for success when the appropriate drug is utilized in the appropriate patient. In patients with HER-2/neu amplification in the breast carcinoma, the impact of this agent on response and overall survival has been clearly documented.Citation146,Citation147 This is a simple trial design that relies on the demonstration of a druggable target prior to the institution of the given targeted therapy.
As this field has continued to grow, the different designs for biomarker-driven clinical trials have expanded in turn. The multiple options for clinical trials integrating biomarkers are beyond the scope of this paper but may be found in the excellent review by Buyse and colleagues.Citation148 One particularly impressive example of novel trial design is seen in the recent Biomarker-Integrated Approaches of Targeted Therapy for Lung Cancer Elimination (BATTLE) trial in lung cancer. The Bayesian adaptive design utilized allowed biomarker-predicted responses in the trial to inform later treatment group assignment.Citation149
Low numbers of patients.
Compared with cancers such as lung, colon, and breast, endometrial cancer has a relatively low number of patients to participate in clinical trials. This presents several issues. The most obvious is the ability to successfully test the expanding number of targeted agents that are currently available and relevant in this disease. The use of multi-institutional and cooperative group trials could potentially expedite trial accrual and timely therapy evaluation. However, as the use of biomarker-driven trials increases, the ability to ensure standard performance of the molecular testing across different institutions becomes a relevant concern. Currently in the United States, biomarkers intended to direct treatment must be validated in a clinical trial setting and performed in a laboratory which meets the recommendations of the Clinical Laboratory Improvement Amendments (CLIA) of 1988.Citation150 Such CLIA-certified clinical laboratories to perform these molecular diagnostics tests are becoming more prevalent in larger academic centers, but their availability in smaller centers is certainly not yet universal. This could potentially be addressed by encouraging patients with gynecologic cancer to pursue treatment at NCI-designated comprehensive cancer centers around the country.
The Phase 0 trial design has the promise to allow for the rapid evaluation of a large number of targeted agents among patients with early endometrial cancer that may then be applied to patients with advanced and/or recurrent disease. This design involves a small number of patients treated with a given agent for a short period prior to surgery, allowing for determination of pharmacodynamic effects in tissue, blood, and other tissues. An agent that appears active in this setting would warrant further study in larger clinical trials.Citation151 Currently, the MD Anderson Cancer Center is actively accruing to a Phase 0 trial of metformin given 7–14 d prior to surgical resection of primary endometrial cancer (NCT01205672).
Lack of tissue.
The lack of available tumor tissue, specifically from the metastasis or recurrence, is a clear issue for the use of biomarkers to direct targeted therapy. This issue is certainly not unique to endometrial cancer. Furthermore, this represents a clinical problem that was never manifest in traditional chemotherapy/radiotherapy trials. It is common practice to perform molecular characterization of a tumor using the available primary tumor from the hysterectomy surgical specimen. However, it is known that recurrent endometrial carcinomas are often quite different at the molecular level, especially when prior treatment with chemotherapy or radiation is considered.Citation152 The use of biopsies from sites of metastasis/recurrence at the time of study enrollment must be considered to maximize success in biomarker validation among patients with endometrial cancer.Citation5,Citation153 A relevant issue to biomarker-guided trial design arises with the procurement of tissue utilizing fine needle aspiration or core biopsy in the recurrent/progressive setting, which may yield only a small amount of tissue on which to perform molecular testing compared with the primary hysterectomy specimen ().
Role of tumor histology.
The role of histology in response to targeted agents has not been completely elucidated. In some cases, tumors with apparently similar histologies may have drastically different molecular features, leading to discordant response to therapy. In non-small cell lung cancer, there are clear clinical and pathologic features which are associated with response to single-agent tyrosine kinase inhibitor therapy. Patient characteristics such as female gender, Asian ethnicity, non-smoker, and adenocarcinoma histology were associated with response to gefitinib therapy. Not surprisingly, these clinical features were soon found to be associated with the presence of an EGFR mutation, thus explaining the higher response levels.Citation115,Citation143,Citation154
It is well-established that endometrioid-type endometrial carcinomas tend to have higher expression of hormone receptors estrogen receptor (ER) and progesterone receptor (PR). In endometrial cancer, hormonal response rates range from 11–56%. This response appears higher in those patients with expression of ER or PR.Citation152,Citation155 Interestingly, our experience in endometrial and ovarian cancer indicates that the expression of a hormone receptor by immunohistochemistry does not necessarily indicate response to hormonal agents, especially in non-endometrioid type tumors.Citation156 Thus, the importance of histologic type of endometrial carcinoma when considering targeted therapy should not be completely overlooked.
Tissue assays.
Most molecular assays of tumors have traditionally been developed using frozen cancer tissues, especially from primary tumors from which ample tumor is typically available. However, the use of frozen tissues is not feasible clinically. The adaptation of select molecular assays for use in formalin-fixed, paraffin-embedded tissues can be done, especially if these assays involve detection of point mutations in single exons or a small number of exons. Such assays include mutational analysis of BRAF, KRAS and NRAS. However, for many genes, such hotspot mutations do not exist, making sequencing the entire gene necessary. Sequencing of large genes, such as PTEN, may not be practical using formalin-fixed, paraffin-embedded tissues. In the case of PTEN, the commercially available immunohistochemical antibodies can be used as a surrogate method to determine PTEN functional loss in an endometrial cancer. However, for many genes, such clinically useful antibodies do not exist. Another challenge for assay development is the assessment of phosphorylated proteins, including pAKT and pERK, in formalin-fixed, paraffin-embedded endometrial cancers. Many such antibodies directed against phosphorylated proteins are useful in protein gel blots of frozen tissues, but do not work reliably in formalin-fixed, paraffin-embedded tissues. Finally, if recurrences or metastases will be examined, molecular tests need to be adapted to the use of very small amounts of tissue, in contrast to the abundance of tissue available from the primary surgical specimen ().
Conclusions
The use of tailored combinations of surgery, radiation, and chemotherapy initially led to marked improvements in survival in endometrial cancer. More recently, however, the improvements in disease-specific mortality have reached a plateau. Augmentation of current treatment regimens with additional cytotoxic chemotherapy seems unlikely to provide further benefit without significant toxicity. The use of targeted therapies appears to hold the promise of achieving greater levels of response and survival among women with advanced or recurrent endometrial cancer. However, rational trial design and biomarker-directed eligibility will be essential to ensure the success of these agents in the appropriate population.
Figures and Tables
Figure 1 Druggable signaling pathways in endometrial cancer. Abbreviations: EGF, epidermal growth factor; IGF, insulin-like growth factor; TGFα, transforming growth factor α; FGF, fibroblast growth factor; HER, human epidermal growth factor receptor; EGFR, epidermal growth factor receptor; IGFR1, insulin-like growth factor receptor 1; FGFR2, fibroblast growth factor receptor 2; ATP, adenosine triphosphate; AMP, adenosine monophosphate; VEGF, vascular endothelial growth factor, PDGF, platelet-derived growth factor; VEGFR, vascular endothelial growth factor receptor; PDGFR, platelet-derived growth factor receptor; GRB2, growth factor receptor-bound protein 2; RAS, rat sarcoma gene; Raf, V-raf-1 murine leukemia viral oncogene homolog 1; MEK, mitogen activated protein kinase kinase; ERK, mitogen activate protein kinase; SRC, V-src sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog; IRS, insulin receptor substrate; PI3K, phosphatidylinositol 3 Kinase; PTEN, phosphatase and tensin homolog deleted on chromosome 10; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol (3,4,5)-triphosphate; AKT, v-akt murine thymoma viral oncogene homolog 1; PKA, protein kinase A; LKB1, liver kinase B1; AMPK, adenosine monophosphate kinase; mTORc, mammalian target of rapamycin Complex.
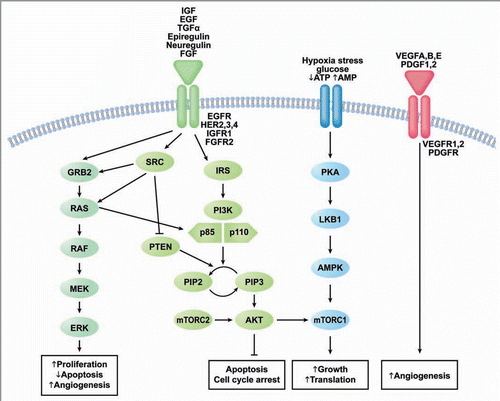
Figure 2 Comparison between tissue obtained from a primary hysterectomy specimen and tissue obtained from core needle biopsy at the time of recurrence. Recurrence tissues are much smaller, have less tumor cells, and commonly consist of tumor cells admixed with stroma. This latter feature may necessitate the use of laser capture microdissection prior to PCR-based molecular diagnostics testing.
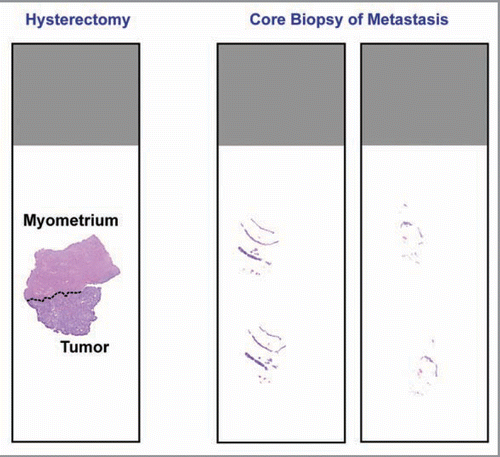
Table 1 Molecular characterization of endometrial cancers by histologic subtype
Table 2 Molecules of importance in angiogenesis
Acknowledgments
Funding from NIH 2P50 CA098258-06 SPORE in Uterine Cancer.
References
- Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin 2011; 61:212 - 236; PMID: 21685461; http://dx.doi.org/10.3322/caac.20121
- CDC. Overweight and Obesity 2011; Atlanta, GA Centers for Disease Control and Prevention
- Amant F, Moerman P, Neven P, Timmerman D, Van Limbergen E, Vergote I. Endometrial cancer. Lancet 2005; 366:491 - 505; PMID: 16084259; http://dx.doi.org/10.1016/S0140-6736(05)67063-8
- Fleming GF, Brunetto VL, Cella D, Look KY, Reid GC, Munkarah AR, et al. Phase III trial of doxorubicin plus cisplatin with or without paclitaxel plus filgrastim in advanced endometrial carcinoma: a Gynecologic Oncology Group Study. J Clin Oncol 2004; 22:2159 - 2166; PMID: 15169803; http://dx.doi.org/10.1200/JCO.2004.07.184
- Stewart DJ, Kurzrock R. Cancer: the road to Amiens. J Clin Oncol 2009; 27:328 - 333; PMID: 19064964; http://dx.doi.org/10.1200/JCO.2008.18.9621
- Huang RS, Ratain MJ. Pharmacogenetics and pharmacogenomics of anticancer agents. CA Cancer J Clin 2009; 59:42 - 55; PMID: 19147868; http://dx.doi.org/10.3322/caac.20002
- Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov 2005; 4:988 - 1004; PMID: 16341064; http://dx.doi.org/10.1038/nrd1902
- Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. J Clin Oncol 2009; 27:2278 - 2287; PMID: 19332717; http://dx.doi.org/10.1200/JCO.2008.20.0766
- Bigsby RM, Li AX, Bomalaski J, Stehman FB, Look KY, Sutton GP. Immunohistochemical study of HER-2/neu, epidermal growth factor receptor, and steroid receptor expression in normal and malignant endometrium. Obstet Gynecol 1992; 79:95 - 100; PMID: 1345772
- Dutt A, Salvesen HB, Greulich H, Sellers WR, Beroukhim R, Meyerson M. Somatic mutations are present in all members of the AKT family in endometrial carcinoma. Br J Cancer 2009; 101:1218 - 1219; author reply 20-1 PMID: 19738612; http://dx.doi.org/10.1038/sj.bjc.6605301
- Dutt A, Salvesen HB, Chen TH, Ramos AH, Onofrio RC, Hatton C, et al. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc Natl Acad Sci USA 2008; 105:8713 - 8717; PMID: 18552176; http://dx.doi.org/10.1073/pnas.0803379105
- Hecht JL, Mutter GL. Molecular and pathologic aspects of endometrial carcinogenesis. J Clin Oncol 2006; 24:4783 - 4791; PMID: 17028294; http://dx.doi.org/10.1200/JCO.2006.06.7173
- Shoji K, Oda K, Nakagawa S, Hosokawa S, Nagae G, Uehara Y, et al. The oncogenic mutation in the pleckstrin homology domain of AKT1 in endometrial carcinomas. Br J Cancer 2009; 101:145 - 148; PMID: 19491896; http://dx.doi.org/10.1038/sj.bjc.6605109
- Cohen Y, Shalmon B, Korach J, Barshack I, Fridman E, Rechavi G. AKT1 pleckstrin homology domain E17K activating mutation in endometrial carcinoma. Gynecol Oncol 2010; 116:88 - 91; PMID: 19853286; http://dx.doi.org/10.1016/j.ygyno.2009.09.038
- Bussaglia E, del Rio E, Matias-Guiu X, Prat J. PTEN mutations in endometrial carcinomas: a molecular and clinicopathologic analysis of 38 cases. Hum Pathol 2000; 31:312 - 317; PMID: 10746673; http://dx.doi.org/10.1016/S0046-8177(00)80244-0
- Kong D, Suzuki A, Zou TT, Sakurada A, Kemp LW, Wakatsuki S, et al. PTEN1 is frequently mutated in primary endometrial carcinomas. Nat Genet 1997; 17:143 - 144; PMID: 9326929; http://dx.doi.org/10.1038/ng1097-143
- Risinger JI, Hayes AK, Berchuck A, Barrett JC. PTEN/MMAC1 mutations in endometrial cancers. Cancer Res 1997; 57:4736 - 4738; PMID: 9354433
- Mutter GL, Lin MC, Fitzgerald JT, Kum JB, Baak JP, Lees JA, et al. Altered PTEN expression as a diagnostic marker for the earliest endometrial precancers. J Natl Cancer Inst 2000; 92:924 - 930; PMID: 10841828; http://dx.doi.org/10.1093/jnci/92.11.924
- Lax SF. Molecular genetic pathways in various types of endometrial carcinoma: from a phenotypical to a molecular-based classification. Virchows Arch 2004; 444:213 - 223; PMID: 14747944; http://dx.doi.org/10.1007/s00428-003-0947-3
- Velasco A, Bussaglia E, Pallares J, Dolcet X, Llobet D, Encinas M, et al. PIK3CA gene mutations in endometrial carcinoma: correlation with PTEN and K-RAS alterations. Hum Pathol 2006; 37:1465 - 1472; PMID: 16949921; http://dx.doi.org/10.1016/j.humpath.2006.05.007
- Hayes MP, Wang H, Espinal-Witter R, Douglas W, Solomon GJ, Baker SJ, et al. PIK3CA and PTEN mutations in uterine endometrioid carcinoma and complex atypical hyperplasia. Clin Cancer Res 2006; 12:5932 - 5935; PMID: 17062663; http://dx.doi.org/10.1158/1078-0432.CCR-06-1375
- O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 2006; 66:1500 - 1508; PMID: 16452206; http://dx.doi.org/10.1158/0008-5472.CAN-05-2925
- Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004; 304:554; PMID: 15016963; http://dx.doi.org/10.1126/science.1096502
- Bartlett JM. Biomarkers and patient selection for PI3K/Akt/mTOR targeted therapies: current status and future directions. Clin Breast Cancer 2010; 10:Suppl 3 S86 - S95; PMID: 21115427; http://dx.doi.org/10.3816/CBC.2010.s.017
- Oda K, Stokoe D, Taketani Y, McCormick F. High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res 2005; 65:10669 - 10673; PMID: 16322209; http://dx.doi.org/10.1158/0008-5472.CAN-05-2620
- Janku F, Tsimberidou AM, Garrido-Laguna I, Wang X, Luthra R, Hong DS, et al. PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Mol Cancer Ther 2011; 10:558 - 565; PMID: 21216929; http://dx.doi.org/10.1158/1535-7163.MCT-10-0994
- Di Nicolantonio F, Arena S, Tabernero J, Grosso S, Molinari F, Macarulla T, et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J Clin Invest 2010; 120:2858 - 2866; PMID: 20664172; http://dx.doi.org/10.1172/JCI37539
- Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest 2008; 118:3065 - 3074; PMID: 18725988
- Watanabe R, Wei L, Huang J. mTOR signaling, function, novel inhibitors, and therapeutic targets. J Nucl Med 2011; 52:497 - 500; PMID: 21421716
- Oza AM, Elit L, Tsao MS, Kamel-Reid S, Biagi J, Provencher DM, et al. Study of temsirolimus in women with recurrent or metastatic endometrial cancer: a trial of the ncic clinical trials group. J Clin Oncol 2011; 29:3278 - 3285; PMID: 21788564; http://dx.doi.org/10.1200/JCO.2010.34.1578
- Oza AM, Elit L, Provencher D, Biagi JJ, Panasci L, Sederias J, et al. A phase II study of temsirolimus (CCI-779) in patients with metastatic and/or locally advanced recurrent endometrial cancer previously treated with chemotherapy: NCIC CTG IND 160b. J Clin Oncol 2008; 26; suppl; abstr 5516 PMID: 18591547
- Slomovitz BM, Lu KH, Johnston T, Coleman RL, Munsell M, Broaddus RR, et al. A phase 2 study of the oral mammalian target of rapamycin inhibitor, everolimus, in patients with recurrent endometrial carcinoma. Cancer 2010; 116:5415 - 5419; PMID: 20681032; http://dx.doi.org/10.1002/cncr.25515
- Meyer PN, Slomovitz BM, Djordjevic B, Galbincea JM, Johnston TA, Munsell M, et al. The search continues: looking for predictive biomarkers for response mTOR inhibition in endometrial cancer. J Clin Oncol 2011; 29; suppl; abstr 5016 PMID: 21135273
- Colombo N, McMeekin DS, Schwartz P, Kostka J, Sessa C, Gehrig P, et al. A phase II trial of the mTOR inhibitor AP23573 as a single agent in advanced endometrial cancer. J Clin Oncol 2007; 25:suppl; abstr 5516 18S; PMID: 17617526
- Mackay H, Welch S, Tsao MS, Biagi JJ, Elit L, Ghatage P, et al. Phase II study of oral ridaforolimus in patients with metastatic and/or locally advanced recurrent endometrial cancer: NCIC CTG IND 192. J Clin Oncol 2011; 29 Suppl: abstr 5013
- Oza AM, Poveda A, Clamp AR, Pignata S, Scambia G, Del Campo J, et al. A randomized phase II (RP2) trial of ridaforolimus (R) compared with progestin (P) or chemotherapy (C) in female adult patients with advanced endometrial carcinoma. J Clin Oncol 2011; 29; suppl: abstr 5009 PMID: 21788564
- Sheri A, Martin LA, Johnston S. Targeting endocrine resistance: is there a role for mTOR inhibition?. Clin Breast Cancer 2010; 10:Suppl 3 S79 - S85; PMID: 21115426; http://dx.doi.org/10.3816/CBC.2010.s.016
- Butt AJ. Overcoming resistance: Targeting the PI3K/mTOR pathway in endocrine refractory breast cancer. Cancer Biol Ther 2011; 11:947 - 949; PMID: 21577052; http://dx.doi.org/10.4161/cbt.11.11.15953
- Slomovitz BM, Brown J, Johnston TA, Mura D, Levenback C, Wolf J, et al. A phase II study of everolimus and letrozole in patients with recurrent endometrial cancer. J Clin Oncol 2011; 29 Suppl: abstr 5012
- Fleming GF, Filiaci VL, Hanjani P, Burke JJ, Davidson SA, Leslie KK, et al. Hormone therapy plus temsirolimus for endometrial carcinoma (EC): gynecologic oncologic group trial #248. J Clin Oncol 2011; 29 Suppl: abstr 5014
- Pal SK, Reckamp K, Yu H, Figlin RA. Akt inhibitors in clinical development for the treatment of cancer. Expert Opin Investig Drugs 2010; 19:1355 - 1366; PMID: 20846000; http://dx.doi.org/10.1517/13543784.2010.520701
- Ciraolo E, Morello F, Hirsch E. Present and future of pi3k pathway inhibition in cancer: perspectives and limitations. Curr Med Chem 2011; 18:2674 - 2685; PMID: 21649577; http://dx.doi.org/10.2174/092986711796011193
- Grana B, Burris HA, Rodon Ahnert J, Abdul Razak R, De Jonge MJ, Eskens F, et al. Oral PI3 kinase inhbitor BKM120 monotherapy in patients (pts) with advanced solid tumors: an update on safety and efficacy. J Clin Oncol 2011; 29 Suppl: abstr 3043
- Burris H, Rodon J, Sharma S, Herbst RS, Tabernero J, Infante JR, et al. First-in-human phase I study of the oral PI3K inhibitor BEZ235 in patients (pts) with advaned solid tumors. J Clin Oncol 2010; 28:15s
- Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005; 330:1304 - 1305; PMID: 15849206; http://dx.doi.org/10.1136/bmj.38415.708634.F7
- Li D. Metformin as an antitumor agent in cancer prevention and treatment. J Diabetes 2011; In press PMID: 21631893; http://dx.doi.org/10.1111/j.17530407.2011.00119.x
- Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res 2006; 66:10269 - 10273; PMID: 17062558; http://dx.doi.org/10.1158/0008-5472.CAN-06-1500
- Ben Sahra I, Le Marchand-Brustel Y, Tanti JF, Bost F. Metformin in cancer therapy: a new perspective for an old antidiabetic drug?. Mol Cancer Ther 2010; 9:1092 - 1099; PMID: 20442309; http://dx.doi.org/10.1158/1535-7163.MCT-09-1186
- Burzawa JK, Schmeler KM, Soliman PT, Meyer LA, Bevers MW, Pustilnik TL, et al. Prospective evaluation of insulin resistance among endometrial cancer patients. Am J Obstet Gynecol 2011; 204:355 - 357; PMID: 21324431; http://dx.doi.org/10.1016/j.ajog.2010.11.033
- Cantrell LA, Zhou C, Mendivil A, Malloy KM, Gehrig PA, Bae-Jump VL. Metformin is a potent inhibitor of endometrial cancer cell proliferation-implications for a novel treatment strategy. Gynecol Oncol 2010; 116:92 - 98; PMID: 19822355; http://dx.doi.org/10.1016/j.ygyno.2009.09.024
- Zhang Z, Dong L, Sui L, Yang Y, Liu X, Yu Y, et al. Metformin reverses progestin resistance in endometrial cancer cells by downregulating GloI expression. Int J Gynecol Cancer 2011; 21:213 - 221; PMID: 21270604; http://dx.doi.org/10.1097/IGC.0b013e318207dac7
- Xie Y, Wang YL, Yu L, Hu Q, Ji L, Zhang Y, Liao QP. Metformin promotes progesterone receptor expression via inhibition of mammalian target of rapamycin (mTOR) in endometrial cancer cells. J Steroid Biochem Mol Biol 2011; 126:113 - 120; PMID: 21168492; http://dx.doi.org/10.1016/j.jsbmb.2010.12.006
- Montagut C, Settleman J. Targeting the RAF-MEK-ERK pathway in cancer therapy. Cancer Lett 2009; 283:125 - 134; PMID: 19217204; http://dx.doi.org/10.1016/j.canlet.2009.01.022
- Koul A, Willen R, Bendahl PO, Nilbert M, Borg A. Distinct sets of gene alterations in endometrial carcinoma implicate alternate modes of tumorigenesis. Cancer 2002; 94:2369 - 2379; PMID: 12015762; http://dx.doi.org/10.1002/cncr.10498
- Semczuk A, Berbec H, Kostuch M, Cybulski M, Wojcierowski J, Baranowski W. K-ras gene point mutations in human endometrial carcinomas: correlation with clinicopathological features and patients' outcome. J Cancer Res Clin Oncol 1998; 124:695 - 700; PMID: 9879831; http://dx.doi.org/10.1007/s004320050234
- Sasaki H, Nishii H, Takahashi H, Tada A, Furusato M, Terashima Y, et al. Mutation of the Ki-ras protooncogene in human endometrial hyperplasia and carcinoma. Cancer Res 1993; 53:1906 - 1910; PMID: 8467512
- Duggan BD, Felix JC, Muderspach LI, Tsao JL, Shibata DK. Early mutational activation of the c-Ki-ras oncogene in endometrial carcinoma. Cancer Res 1994; 54:1604 - 1607; PMID: 8137266
- Friday BB, Adjei AA. Advances in targeting the Ras/Raf/MEK/Erk mitogen-activated protein kinase cascade with MEK inhibitors for cancer therapy. Clin Cancer Res 2008; 14:342 - 346; PMID: 18223206; http://dx.doi.org/10.1158/1078-0432.CCR-07-4790
- Chapman MS, Miner JN. Novel mitogen-activated protein kinase kinase inhibitors. Expert Opin Investig Drugs 2011; 20:209 - 220; PMID: 21235429; http://dx.doi.org/10.1517/13543784.2011.548803
- Thomas NE. BRAF somatic mutations in malignant melanoma and melanocytic naevi. Melanoma Res 2006; 16:97 - 103; PMID: 16567964; http://dx.doi.org/10.1097/01.cmr.0000215035.38436.87
- Li Y, Nakamura M, Kakudo K. Targeting of the BRAF gene in papillary thyroid carcinoma [review]. Oncol Rep 2009; 22:671 - 681; PMID: 19724843
- Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007; 26:3291 - 3310; PMID: 17496923; http://dx.doi.org/10.1038/sj.onc.1210422
- Ramjaun AR, Downward J. Ras and phosphoinositide 3-kinase: partners in development and tumorigenesis. Cell Cycle 2007; 6:2902 - 2905; PMID: 17993782; http://dx.doi.org/10.4161/cc.6.23.4996
- Dummer R, Robert C, Chapman PB, Sosman JA, Middleton M, Bastholt L, et al. AZD6244 (ARRY-142886) vs temozolomide (TMZ) in patients (pts) with advanced melanoma: An open-label, randomized, multicenter, phase II study. J Clin Oncol 2008; 26(Suppl; abstr 9033)
- Patel SP, Lazar AJ, Mahoney S, Vaughn C, Gonzalez N, Papadopoulos NE, et al. Clinical responses to AZD6244 (ARRY-142886)-based combination therapy stratified by gene mutations in patients with metastatic melanoma. ASCO Molecular Markers 2010; In press
- Salvesen HB, Kumar R, Stefansson I, Angelini S, MacDonald N, Smeds J, et al. Low frequency of BRAF and CDKN2A mutations in endometrial cancer. Int J Cancer 2005; 115:930 - 934; PMID: 15723290; http://dx.doi.org/10.1002/ijc.20702
- Kang S, Lee JM, Jeon ES, Lee S, Kim H, Kim HS, et al. RASSF1A hypermethylation and its inverse correlation with BRAF and/or KRAS mutations in MSI-associated endometrial carcinoma. Int J Cancer 2006; 119:1316 - 1321; PMID: 16619251; http://dx.doi.org/10.1002/ijc.21991
- Fürstenberger G, Senn HJ. Insulin-like growth factors and cancer. Lancet Oncol 2002; 3:298 - 302; PMID: 12067807; http://dx.doi.org/10.1016/S1470-2045(02)00731-3
- McCampbell AS, Broaddus RR, Loose DS, Davies PJ. Overexpression of the insulin-like growth factor I receptor and activation of the AKT pathway in hyper-plastic endometrium. Clin Cancer Res 2006; 12:6373 - 6378; PMID: 17085648; http://dx.doi.org/10.1158/1078-0432.CCR-06-0912
- Westin SN, Broaddus RR, Deng L, McCampbell A, Lu KH, Lacour RA, et al. Molecular clustering of endometrial carcinoma based on estrogen-induced gene expression. Cancer Biol Ther 2009; 8:2126 - 2135; PMID: 19755863; http://dx.doi.org/10.4161/cbt.8.22.9740
- Attias-Geva Z, Bentov I, Fishman A, Werner H, Bruchim I. Insulin-like growth factor-I receptor inhibition by specific tyrosine kinase inhibitor NVP-AEW541 in endometrioid and serous papillary endometrial cancer cell lines. Gynecol Oncol 2011; 121:383 - 389; PMID: 21295335; http://dx.doi.org/10.1016/j.ygyno.2011.01.008
- Rozengurt E, Sinnett-Smith J, Kisfalvi K. Crosstalk between insulin/insulin-like growth factor-1 receptors and G protein-coupled receptor signaling systems: a novel target for the antidiabetic drug metformin in pancreatic cancer. Clin Cancer Res 2010; 16:2505 - 2511; PMID: 20388847; http://dx.doi.org/10.1158/1078-0432.CCR-09-2229
- Tolcher AW, Sarantopoulos J, Patnaik A, Papadopoulos K, Lin CC, Rodon J, et al. Phase I, pharmacokinetic, and pharmacodynamic study of AMG 479, a fully human monoclonal antibody to insulin-like growth factor receptor 1. J Clin Oncol 2009; 27:5800 - 5807; PMID: 19786654; http://dx.doi.org/10.1200/JCO.2009.23.6745
- McKian KP, Haluska P. Cixutumumab. Expert Opin Investig Drugs 2009; 18:1025 - 1033; PMID: 19548856; http://dx.doi.org/10.1517/13543780903055049
- Zha J, O'Brien C, Savage H, Huw LY, Zhong F, Berry L, et al. Molecular predictors of response to a humanized anti-insulin-like growth factor-I receptor monoclonal antibody in breast and colorectal cancer. Mol Cancer Ther 2009; 8:2110 - 2121; PMID: 19671761; http://dx.doi.org/10.1158/1535-7163.MCT-09-0381
- McCampbell AS, Harris HA, Crabtree JS, Winneker RC, Walker CL, Broaddus RR. Loss of inhibitory insulin receptor substrate-1 phosphorylation is an early event in mammalian target of rapamycin-dependent endometrial hyperplasia and carcinoma. Cancer Prev Res (Phila) 2010; 3:290 - 300; PMID: 20179297; http://dx.doi.org/10.1158/1940-6207.CAPR-09-0199
- Folkman J. What is the evidence that tumors are angiogenesis dependent?. J Natl Cancer Inst 1990; 82:4 - 6; PMID: 1688381; http://dx.doi.org/10.1093/jnci/82.1.4
- Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol 2005; 23:1011 - 1027; PMID: 15585754; http://dx.doi.org/10.1200/JCO.2005.06.081
- Dvorak HF, Brown LF, Detmar M, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol 1995; 146:1029 - 1039; PMID: 7538264
- Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996; 86:353 - 364; PMID: 8756718; http://dx.doi.org/10.1016/S0092-8674(00)80108-7
- Hirai M, Nakagawara A, Oosaki T, Hayashi Y, Hirono M, Yoshihara T. Expression of vascular endothelial growth factors (VEGF-A/VEGF-1 and VEGF-C/VEGF-2) in postmenopausal uterine endometrial carcinoma. Gynecol Oncol 2001; 80:181 - 188; PMID: 11161857; http://dx.doi.org/10.1006/gyno.2000.6056
- Kamat AA, Merritt WM, Coffey D, Lin YG, Patel PR, Broaddus R, et al. Clinical and biological significance of vascular endothelial growth factor in endometrial cancer. Clin Cancer Res 2007; 13:7487 - 7495; PMID: 18094433; http://dx.doi.org/10.1158/1078-0432.CCR-07-1017
- Aghajanian C, Sill MW, Darcy KM, Greer B, McMeekin DS, Rose PG, et al. Trial of bevacizumab in recurrent or persistent endometrial cancer: a gynecologic oncology group study. J Clin Oncol 2011; 29:2259 - 2265; PMID: 21537039; http://dx.doi.org/10.1200/JCO.2010.32.6397
- Chu QS. Aflibercept (AVE0005): an alternative strategy for inhibiting tumour angiogenesis by vascular endothelial growth factors. Expert Opin Biol Ther 2009; 9:263 - 271; PMID: 19236257; http://dx.doi.org/10.1517/14712590802666397
- Escudier B. Sunitinib for the management of advanced renal cell carcinoma. Expert Rev Anticancer Ther 2010; 10:305 - 317; PMID: 20214511; http://dx.doi.org/10.1586/era.10.26
- Korc M, Friesel RE. The role of fibroblast growth factors in tumor growth. Curr Cancer Drug Targets 2009; 9:639 - 651; PMID: 19508171; http://dx.doi.org/10.2174/156800909789057006
- Byron SA, Pollock PM. FGFR2 as a molecular target in endometrial cancer. Future Oncol 2009; 5:27 - 32; PMID: 19243295; http://dx.doi.org/10.2217/14796694.5.1.27
- Pollock PM, Gartside MG, Dejeza LC, Powell MA, Mallon MA, Davies H, et al. Frequent activating FGFR2 mutations in endometrial carcinomas parallel germline mutations associated with craniosynostosis and skeletal dysplasia syndromes. Oncogene 2007; 26:7158 - 7162; PMID: 17525745; http://dx.doi.org/10.1038/sj.onc.1210529
- Brave SR, Ratcliffe K, Wilson Z, James NH, Ashton S, Wainwright A, et al. Assessing the activity of cediranib, a vegfr-2/3 tyrosine kinase inhibitor, against vegfr-1 and members of the structurally related pdgfr family. Mol Cancer Ther 2011; 10:861 - 873; PMID: 21441409; http://dx.doi.org/10.1158/1535-7163.MCT-10-0976
- Marathe PH, Kamath AV, Zhang Y, D'Arienzo C, Bhide R, Fargnoli J. Preclinical pharmacokinetics and in vitro metabolism of brivanib (BMS-540215), a potent VEGFR2 inhibitor and its alanine ester prodrug brivanib alaninate. Cancer Chemother Pharmacol 2009; 65:55 - 66; PMID: 19396600; http://dx.doi.org/10.1007/s00280-009-1002-0
- Marshall ME, Hinz TK, Kono SA, Singleton KR, Bichon B, Ware KE, et al. Fibroblast growth factor receptors are components of autocrine signaling networks in head and neck squamous cell carcinoma cells. Clin Cancer Res 2011; 17:5016 - 5025; PMID: 21673064; http://dx.doi.org/10.1158/1078-0432.CCR-11-0050
- McMeekin DS, Sill MW, Benbrook D, Darcy KM, Stearns-Kurosawa DJ, Eaton L, et al. A phase II trial of thalidomide in patients with refractory endometrial cancer and correlation with angiogenesis biomarkers: a Gynecologic Oncology Group study. Gynecol Oncol 2007; 105:508 - 516; PMID: 17306350; http://dx.doi.org/10.1016/j.ygyno.2007.01.019
- Jubb AM, Oates AJ, Holden S, Koeppen H. Predicting benefit from anti-angiogenic agents in malignancy. Nat Rev Cancer 2006; 6:626 - 635; PMID: 16837971; http://dx.doi.org/10.1038/nrc1946
- Jubb AM, Hurwitz HI, Bai W, Holmgren EB, Tobin P, Guerrero AS, et al. Impact of vascular endothelial growth factor-A expression, thrombospondin-2 expression, and microvessel density on the treatment effect of bevacizumab in metastatic colorectal cancer. J Clin Oncol 2006; 24:217 - 227; PMID: 16365183; http://dx.doi.org/10.1200/JCO.2005.01.5388
- Hasselbalch B, Eriksen JG, Broholm H, Christensen IJ, Grunnet K, Horsman MR, et al. Prospective evaluation of angiogenic, hypoxic and EGFR-related biomarkers in recurrent glioblastoma multiforme treated with cetuximab, bevacizumab and irinotecan. APMIS 2010; 118:585 - 594; PMID: 20666740
- Han ES, Burger RA, Darcy KM, Sill MW, Randall LM, Chase D, et al. Predictive and prognostic angiogenic markers in a gynecologic oncology group phase II trial of bevacizumab in recurrent and persistent ovarian or peritoneal cancer. Gynecol Oncol 2010; 119:484 - 490; PMID: 20870280; http://dx.doi.org/10.1016/j.ygyno.2010.08.016
- Wedam SB, Low JA, Yang SX, Chow CK, Choyke P, Danforth D, et al. Antiangiogenic and antitumor effects of bevacizumab in patients with inflammatory and locally advanced breast cancer. J Clin Oncol 2006; 24:769 - 777; PMID: 16391297; http://dx.doi.org/10.1200/JCO.2005.03.4645
- Schultheis AM, Lurje G, Rhodes KE, Zhang W, Yang D, Garcia AA, et al. Polymorphisms and clinical outcome in recurrent ovarian cancer treated with cyclophosphamide and bevacizumab. Clin Cancer Res 2008; 14:7554 - 7563; PMID: 19010874; http://dx.doi.org/10.1158/1078-0432.CCR-08-0351
- Gien LT, Mackay HJ. The emerging role of parp inhibitors in the treatment of epithelial ovarian cancer. J Oncol 2010; In press PMID: 20049345; http://dx.doi.org/10.1155/2010/151750
- Dedes KJ, Wilkerson PM, Wetterskog D, Weigelt B, Ashworth A, Reis-Filho JS. Synthetic lethality of PARP inhibition in cancers lacking BRCA1 and BRCA2 mutations. Cell Cycle 2011; 10:1192 - 1199; PMID: 21487248; http://dx.doi.org/10.4161/cc.10.8.15273
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 2009; 361:123 - 134; PMID: 19553641; http://dx.doi.org/10.1056/NEJMoa0900212
- Madhusudan S, Middleton MR. The emerging role of DNA repair proteins as predictive, prognostic and therapeutic targets in cancer. Cancer Treat Rev 2005; 31:603 - 617; PMID: 16298073; http://dx.doi.org/10.1016/j.ctrv.2005.09.006
- Dedes KJ, Wetterskog D, Mendes-Pereira AM, Natrajan R, Lambros MB, Geyer FC, et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci Transl Med 2010; 2:53ra75; PMID: 20944090; http://dx.doi.org/10.1126/scitranslmed.3001538
- McEllin B, Camacho CV, Mukherjee B, Hahm B, Tomimatsu N, Bachoo RM, et al. PTEN loss compromises homologous recombination repair in astrocytes: implications for glioblastoma therapy with temozolomide or poly(ADP-ribose) polymerase inhibitors. Cancer Res 201070:5457 - 5464; PMID: 20530668; http://dx.doi.org/10.1158/0008-5472.CAN-09-4295
- Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med 2009; 1:315 - 322; PMID: 20049735; http://dx.doi.org/10.1002/emmm.200900041
- Gelmon KA, Hirte HW, Robidoux A, Tonkin KS, Tischkowitz M, Swenerton K, et al. Can we define tumors that will respond to PARP inhibitors? A phase II correlative study of olaparib in advanced serous ovarian cancer and triple-negative breast cancer. J Clin Oncol 2010; 28:Suppl: abstr 3002 15s
- Mendelsohn J, Baselga J. The EGF receptor family as targets for cancer therapy. Oncogene 2000; 19:6550 - 6565; PMID: 11426640; http://dx.doi.org/10.1038/sj.onc.1204082
- Mendelsohn J. Targeting the epidermal growth factor receptor for cancer therapy. J Clin Oncol 2002; 20:1S - 13S; PMID: 12235219
- Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006; 366:2 - 16; PMID: 16377102; http://dx.doi.org/10.1016/j.gene.2005.10.018
- De Luca A, Carotenuto A, Rachiglio A, Gallo M, Maiello MR, Aldinucci D, et al. The role of the EGFR signaling in tumor microenvironment. J Cell Physiol 2008; 214:559 - 567; PMID: 17894407; http://dx.doi.org/10.1002/jcp.21260
- Khalifa MA, Mannel RS, Haraway SD, Walker J, Min KW. Expression of EGFR, HER-2/neu, P53, and PCNA in endometrioid, serous papillary, and clear cell endometrial adenocarcinomas. Gynecol Oncol 1994; 53:84 - 92; PMID: 7909788; http://dx.doi.org/10.1006/gyno.1994.1092
- Cheng L, Zhang S, Alexander R, Yao Y, MacLennan GT, Pan CX, et al. The landscape of EGFR pathways and personalized management of non-small-cell lung cancer. Future Oncol 2011; 7:519 - 541; PMID: 21463141; http://dx.doi.org/10.2217/fon.11.25
- Hayes MP, Douglas W, Ellenson LH. Molecular alterations of EGFR and PIK3CA in uterine serous carcinoma. Gynecol Oncol 2009; 113:370 - 373; PMID: 19272638; http://dx.doi.org/10.1016/j.ygyno.2008.12.021
- Murray S, Bobos M, Angouridakis N, Nikolaou A, Linardou H, Razis E, Fountzilas G. Screening for egfr mutations in patients with head and neck cancer treated with gefitinib on a compassionate-use program: a hellenic cooperative oncology group study. J Oncol 2010; 709678; PMID: 21274259; http://dx.doi.org/10.1155/2010/709678
- Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med 2008; 358:1160 - 1174; PMID: 18337605; http://dx.doi.org/10.1056/NEJMra0707704
- Normanno N, Tejpar S, Morgillo F, De Luca A, Van Cutsem E, Ciardiello F. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat Rev Clin Oncol 2009; 6:519 - 527; PMID: 19636327; http://dx.doi.org/10.1038/nrclinonc.2009.111
- Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, et al. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol 2009; 27:2091 - 2096; PMID: 19188670; http://dx.doi.org/10.1200/JCO.2009.21.9170
- Jackman DM, Miller VA, Cioffredi LA, Yeap BY, Janne PA, Riely GJ, et al. Impact of epidermal growth factor receptor and KRAS mutations on clinical outcomes in previously untreated non-small cell lung cancer patients: results of an online tumor registry of clinical trials. Clin Cancer Res 2009; 15:5267 - 5273; PMID: 19671843; http://dx.doi.org/10.1158/1078-0432.CCR-09-0888
- Rizvi NA, Rusch V, Pao W, Chaft JE, Ladanyi M, Miller VA, et al. Molecular characteristics predict clinical outcomes: prospective trial correlating response to the egfr tyrosine kinase inhibitor gefitinib with the presence of sensitizing mutations in the tyrosine binding domain of the egfr gene. Clin Cancer Res 2011; 17:3500 - 3506; PMID: 21558399; http://dx.doi.org/10.1158/1078-0432.CCR-10-2102
- Engelman JA, Janne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res 2008; 14:2895 - 2899; PMID: 18483355; http://dx.doi.org/10.1158/1078-0432.CCR-07-2248
- Oza AM, Eisenhauer EA, Elit L, Cutz JC, Sakurada A, Tsao MS, et al. Phase II study of erlotinib in recurrent or metastatic endometrial cancer: NCIC IND-148. J Clin Oncol 2008; 26:4319 - 4325; PMID: 18591547; http://dx.doi.org/10.1200/JCO.2007.15.8808
- Leslie KK, Sill MW, Darcy KM, Baron AT, Wilken JA, Godwin AK, et al. Efficacy and safety of gefitinib and potential prognostic value of soluble EGFR, EGFR mutations, and tumor markers in a Gynecologic Oncology Group phase II trial of persistent or recurrent endometrial cancer. J Clin Oncol 2009; 27 Suppl: abstr e16542
- You B, Chen EX. Anti-EGFR monoclonal antibodies for treatment of colorectal cancers: development of cetuximab and panitumumab. J Clin Pharmacol 2011 In press PMID: 21427284
- Grushko TA, Filiaci VL, Mundt AJ, Ridderstrale K, Olopade OI, Fleming GF. An exploratory analysis of HER-2 amplification and overexpression in advanced endometrial carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol 2008; 108:3 - 9; PMID: 17945336; http://dx.doi.org/10.1016/j.ygyno.2007.09.007
- Santin AD, Bellone S, Gokden M, Palmieri M, Dunn D, Agha J, et al. Overexpression of HER-2/neu in uterine serous papillary cancer. Clin Cancer Res 2002; 8:1271 - 1279; PMID: 12006548
- Slomovitz BM, Broaddus RR, Burke TW, Sneige N, Soliman PT, Wu W, et al. Her-2/neu overexpression and amplification in uterine papillary serous carcinoma. J Clin Oncol 2004; 22:3126 - 3132; PMID: 15284264; http://dx.doi.org/10.1200/JCO.2004.11.154
- Fleming GF, Sill MW, Darcy KM, McMeekin DS, Thigpen JT, Adler LM, et al. Phase II trial of trastuzumab in women with advanced or recurrent, HER2-positive endometrial carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol 2010; 116:15 - 20; PMID: 19840887; http://dx.doi.org/10.1016/j.ygyno.2009.09.025
- Medina PJ, Goodin S. Lapatinib: a dual inhibitor of human epidermal growth factor receptor tyrosine kinases. Clin Ther 2008; 30:1426 - 1447; PMID: 18803986; http://dx.doi.org/10.1016/j.clinthera.2008.08.008
- Holcomb K, Delatorre R, Pedemonte B, McLeod C, Anderson L, Chambers J. E-cadherin expression in endometrioid, papillary serous, and clear cell carcinoma of the endometrium. Obstet Gynecol 2002; 100:1290 - 1295; PMID: 12468176; http://dx.doi.org/10.1016/S0029-7844(02)02391-8
- Moreno-Bueno G, Hardisson D, Sarrio D, Sanchez C, Cassia R, Prat J, et al. Abnormalities of E-and P-cadherin and catenin (beta-, gamma-catenin, and p120ctn) expression in endometrial cancer and endometrial atypical hyperplasia. J Pathol 2003; 199:471 - 478; PMID: 12635138; http://dx.doi.org/10.1002/path.1310
- Polakis P. Wnt signaling and cancer. Genes Dev 2000; 14:1837 - 1851; PMID: 10921899
- Taipale J, Beachy PA. The Hedgehog and Wnt signalling pathways in cancer. Nature 2001; 411:349 - 354; PMID: 11357142; http://dx.doi.org/10.1038/35077219
- Schlosshauer PW, Ellenson LH, Soslow RA. Beta-catenin and E-cadherin expression patterns in high-grade endometrial carcinoma are associated with histological subtype. Mod Pathol 2002; 15:1032 - 1037; PMID: 12379748; http://dx.doi.org/10.1097/01.MP.0000028573.34289.04
- Schlosshauer PW, Pirog EC, Levine RL, Ellenson LH. Mutational analysis of the CTNNB1 and APC genes in uterine endometrioid carcinoma. Mod Pathol 2000; 13:1066 - 1071; PMID: 11048799; http://dx.doi.org/10.1038/modpathol.3880196
- Yang AD, Fan F, Camp ER, van Buren G, Liu W, Somcio R, et al. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res 2006; 12:4147 - 4153; PMID: 16857785; http://dx.doi.org/10.1158/1078-0432.CCR-06-0038
- Witta SE, Gemmill RM, Hirsch FR, Coldren CD, Hedman K, Ravdel L, et al. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res 2006; 66:944 - 950; PMID: 16424029; http://dx.doi.org/10.1158/0008-5472.CAN-05-1988
- Buck E, Eyzaguirre A, Barr S, Thompson S, Sennello R, Young D, et al. Loss of homotypic cell adhesion by epithelial-mesenchymal transition or mutation limits sensitivity to epidermal growth factor receptor inhibition. Mol Cancer Ther 2007; 6:532 - 541; PMID: 17308052; http://dx.doi.org/10.1158/1535-7163.MCT-06-0462
- Moreno-Bueno G, Hardisson D, Sanchez C, Sarrio D, Cassia R, Garcia-Rostan G, et al. Abnormalities of the APC/beta-catenin pathway in endometrial cancer. Oncogene 2002; 21:7981 - 7990; PMID: 12439748; http://dx.doi.org/10.1038/sj.onc.1205924
- Nimeiri HS, Oza AM, Morgan RJ, Huo D, Elit L, Knost J, et al. Sorafenib (SOR) in patients (pts) with advanced/recurrent uterine carcinoma (UCA) or carcinosarcoma (CS): a phase II trial of the University of Chicago, PMH, and California Phase II Consortia. J Clin Oncol 2008; suppl: abstr 5585 26
- Kim LC, Rix U, Haura EB. Dasatinib in solid tumors. Expert Opin Investig Drugs 2010; 19:415 - 425; PMID: 20113198; http://dx.doi.org/10.1517/13543781003592097
- Kamat AA, Coffey D, Merritt WM, Nugent E, Urbauer D, Lin YG, et al. EphA2 overexpression is associated with lack of hormone receptor expression and poor outcome in endometrial cancer. Cancer 2009; 115:2684 - 2692; PMID: 19396818; http://dx.doi.org/10.1002/cncr.24335
- Kris MG, Natale RB, Herbst RS, Lynch TJ Jr, Prager D, Belani CP, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA 2003; 290:2149 - 2158; PMID: 14570950; http://dx.doi.org/10.1001/jama.290.16.2149
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004; 350:2129 - 2139; PMID: 15118073; http://dx.doi.org/10.1056/NEJMoa040938
- Thatcher N, Chang A, Parikh P, Rodrigues Pereira J, Ciuleanu T, von Pawel J, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet 2005; 366:1527 - 1537; PMID: 16257339; http://dx.doi.org/10.1016/S0140-6736(05)67625-8
- Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009; 361:947 - 957; PMID: 19692680; http://dx.doi.org/10.1056/NEJMoa0810699
- Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001; 344:783 - 792; PMID: 11248153; http://dx.doi.org/10.1056/NEJM200103153441101
- Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE Jr., Davidson NE, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med 2005; 353:1673 - 1684; PMID: 16236738; http://dx.doi.org/10.1056/NEJMoa052122
- Buyse M, Michiels S, Sargent DJ, Grothey A, Matheson A, de Gramont A. Integrating biomarkers in clinical trials. Expert Rev Mol Diagn 2011; 11:171 - 182; PMID: 21405968; http://dx.doi.org/10.1586/erm.10.120
- Kim ES, Herbst RS, Lee JJ, Blumenschein GR Jr., Tsao A, Alden CM, et al. The BATTLE trial (Biomarker-integrated Approaches of Targeted Therapy for Lung Cancer Elimination): personalizing therapy for lung cancer 101st Annual Meeting of the American Association for Cancer Research Washington, DC 2010;
- Services UDoHaH. Clinical Laboratory Improvement Amendments (CLIA) 2010;
- Pocard M, Soria JC, Aldaz-Carroll L, Bellet D. Phase 0 clinical trials in oncology: an exploratory methodology for constructing a study with patients undergoing surgery for metastatic disease. J Clin Oncol 2010; 28:4551 - 4553; PMID: 20837954; http://dx.doi.org/10.1200/JCO.2010.29.2870
- Singh M, Zaino RJ, Filiaci VJ, Leslie KK. Relationship of estrogen and progesterone receptors to clinical outcome in metastatic endometrial carcinoma: a Gynecologic Oncology Group Study. Gynecol Oncol 2007; 106:325 - 333; PMID: 17532033; http://dx.doi.org/10.1016/j.ygyno.2007.03.042
- Wistuba II, Gelovani JG, Jacoby JJ, Davis SE, Herbst RS. Methodological and practical challenges for personalized cancer therapies. Nat Rev Clin Oncol 2011; 8:135 - 141; PMID: 21364686; http://dx.doi.org/10.1038/nrclinonc.2011.2
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304:1497 - 1500; PMID: 15118125; http://dx.doi.org/10.1126/science.1099314
- Decruze SB, Green JA. Hormone therapy in advanced and recurrent endometrial cancer: a systematic review. Int J Gynecol Cancer 2007; 17:964 - 978; PMID: 17442022; http://dx.doi.org/10.1111/j.1525-1438.2007.00897.x
- Schlumbrecht MP, Xie SS, Shipley GL, Urbauer DL, Broaddus RR. Molecular clustering based on ERalpha and EIG121 predicts survival in high-grade serous carcinoma of the ovary/peritoneum. Mod Pathol 2011; 24:453 - 462; PMID: 21102415; http://dx.doi.org/10.1038/modpathol.2010.211
- Urick ME, Rudd ML, Godwin AK, Sgroi D, Merino M, Bell DW. PIK3R1 (p85alpha) is somatically mutated at high frequency in primary endometrial cancer. Cancer Res 2011; 71:4061 - 4067; PMID: 21478295; http://dx.doi.org/10.1158/0008-5472.CAN-11-0549
- Lax SF, Kendall B, Tashiro H, Slebos RJ, Hedrick L. The frequency of p53, K-ras mutations, and microsatellite instability differs in uterine endometrioid and serous carcinoma: evidence of distinct molecular genetic pathways. Cancer 2000; 88:814 - 824; PMID: 10679651; http://dx.doi.org/10.1002/(SICI)1097-0142(20000215)88:4<814::AID-CNCR12>3.0.CO;2-U
- MacDonald ND, Salvesen HB, Ryan A, Iversen OE, Akslen LA, Jacobs IJ. Frequency and prognostic impact of microsatellite instability in a large population-based study of endometrial carcinomas. Cancer Res 2000; 60:1750 - 1752; PMID: 10749149
- Basil JB, Goodfellow PJ, Rader JS, Mutch DG, Herzog TJ. Clinical significance of microsatellite instability in endometrial carcinoma. Cancer 2000; 89:1758 - 1764; PMID: 11042571; http://dx.doi.org/10.1002/1097-0142(20001015)89:8<1758::AID-CNCR16>3.0.CO;2-A