
Abstract
Epidermal growth factor receptor (EGFR) mutations in patients with non-small cell lung cancer (NSCLC) usually develop disease progression after a median of 10 to 14 mo on tyrosine kinase inhibitor (TKI). Several mechanisms of resistance to TKI have been described, threonine-methionine substitution at position 790 (T790M), mesenchymal–epithelial transition factor (MET) amplification, overexpression of hepatocyte growth factor (HGF), upregulation of insulin-like growth factor (IGF) receptor signaling, transformation to small cell lung cancer, and so on. A variety of different therapeutic approaches aimed at overcoming resistance are motivated, irreversible EGFR inhibitors, combination with EGFR targeted antibodies, mesenchymal–epithelial transition factor (MET) inhibitors, HGF inhibitors, and so forth. Nevertheless, the results were not optimistic. Here we report a case of reversion of erlotinib-acquired resistance twice, and had a good improvement of outcomes every time. There are some possible reasons for this phenomenon. Considering this report, the patients who acquired resistance after retreatment of EGFR-TKI, using EGFR-TKI repeatedly may be a choice selectively.
Keywords: :
Introduction
The discovery of epidermal growth factor receptor (EGFR) mutations in 2004 changed the standard approach to evaluating non-small cell lung cancer (NSCLC) and established a new paradigm of tumor genotyping in clinical practice.Citation1 NSCLC patients who harbor activating mutations (most frequently exon 19 deletions and exon 21 point mutation) in the EGFR gene are a clinically distinct entity with a much better prognosis compared with patients with non-mutant (Mut−) NSCLC in the treatment of EGFR tyrosine kinase inhibitors (EGFR-TKIs), while patients with EGFR-mutant (EGFR-Mut+) NSCLC develop disease progression after a median of 10 to 14 mo on TKI.Citation2
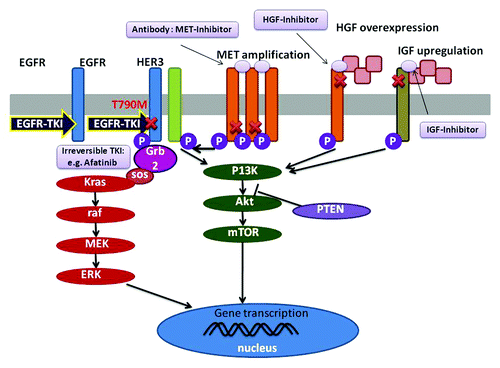
Since acquired resistance to EGFR-targeted therapies was first described in 2005,Citation3 several mechanisms of resistance to TKI have been described, and a variety of different therapeutic approaches aimed at overcoming resistance are motivated (). A threonine-methionine substitution at position 790 (T790M) is the most common resistance mutation, which is located in the adenosine 5′-triphosphate (ATP)-binding pocket of the catalytic region to which EGFR-TKIs bind and causes higher affinity to ATP and lower affinity to EGFR-TKIs. The irreversible EGFR-TKI, such as afatinib, neratinib, could bind to EGFR-T790M mutants, while the overall survival (OS) shows no benefits in the study of afatinib vs. placebo.Citation4 Combination of EGFR-targeted antibodies and secondary EGFR-TKI might be a new strategy to overcome the T790M mediated resistance.Citation5 Besides, mesenchymal–epithelial transition factor (MET) amplification, overexpression of hepatocyte growth factor (HGF), upregulation of insulin-like growth factor (IGF) receptor signaling, K-ras mutation, which activate downstream signals of EGFR, are all possible second mutations causing EGFR-TKI resistance. To conquer this kind of resistance, addition of MET-inhibitor or HGF-inhibitor, inhibition of parallel pathway is a feasible strategy. In addition, transformation to small cell cancer is another possible reason contributing to the acquired resistance; in this case, we might need to change the antineoplastic approach, such as chemotherapy.Citation6 Nevertheless, the results were not optimistic, which may be related to the elusive understanding of these sensitive or resistant mechanisms, the optimum doses, or the insufferable severe adverse effects. A new strategy to overcome EGFR-TKI resistance has been an urgent problem to solve. Here we report a case of reversion of erlotinib-acquired resistance twice.
Figure 1. The mechanisms of acquired resistance of EGFR-TKIs. The secondary T790M mutation of EGFR leads to decrease the affinity to EGFR-TKIs. Irreversible TKIs bind with high affinity to receptors carrying the T790M mutation. MET or IGF activation induces activation of PI3K/Akt pathway independent of EGFR activation. In these cases MET-specific inhibitor or HGF-inhibitor, inhibition of parallel pathway is a feasible strategy.
Case Report
A 64-y-old non-smoker female was diagnosed adenocarcinoma in the middle right lobe (T1N0M0) in November 2005.The patient underwent right middle lobectomy with lymphnode dissection. In November 2007, we found metastasis in the vertebrae and multiple metastases in the lung. At that time, the patient didn’t take EGFR gene mutation analysis. Because the patient refused to use pemetrexedfor some economic reasons, he was treated with chemotherapy including cisplatin (40 mg/days 1–3) and gemcitabine (1600 mg/days 1 and 8) every 3 weeks up to 6 cycles and concurrent radiotherapy (30 Gy/10 fr). The patient was classified as having a stable disease (SD) according to the Response Evaluation Criteria in Solid Tumors (RECIST1.0).Citation7
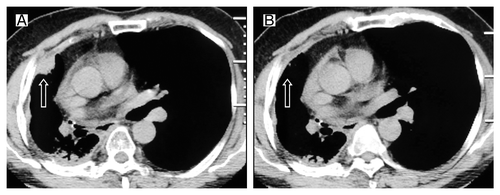
In July 2009, the patient felt right chest pain; right pleura metastasis was showed in CT (). The EGFR exon 19 deletion mutation was identified through the analyses of exons 18 through 21 performed by the polymerase chain reaction (PCR) method in the primary tumor tissues obtained when the diagnosis was first made. Erlotinib was then administered at the standard dose of 150 mg once daily. A grade 1 skin rash was observed. Significant shrinkage was showed at 2 mo () while progression of disease at 11 mo after initiation of erlotinib treatment (). The erlotinib was discontinued. We had to give up using docetaxel due to insufferable allergic reactions, then the patient was given gemcitabine (1600 mg, days 1 and 8) every 3 weeks up to 2 cycles. A reduction in CT (CT) of tumor volume was evident after 2 cycles of chemotherapy (). The tumor was shown to progress 6 mo later (). Erlotinib was again administered as a standard dose. After 3 mo of treatment with erlotinib, a CT scan again showed response (). However, the erlotinib was discontinued because of the disease progression 8 mo later. The patient was given gemcitabine (1600 mg, days 1 and 8, 1 cycle/3 weeks) again. Further chemotherapy was refused by the patient because of insufferable side effects. The disease progressed again one month later (). Erlotinib was administered at the standard dose and surprisingly the tumor regressed after 2 mo (). What’s more, the patient’s condition was stable until now. Unfortunately, we failed to gain the condition of EGFR gene mutation during the second and third erlotinib treatments because of the patient’s refusal.
Figure 2. Tumor volume changes seen on CT (A) before the first erlotinib treatment; (B) 2 mo after the first erlotinib treatment.
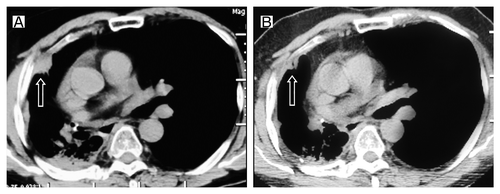
Figure 3. Tumor volume changes seen on CT (A) before the gemcitabine treatment; (B) 2 cycles after gemcitabine retreatment.
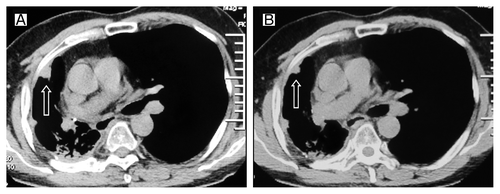
Figure 4. Tumor volume changes seen on CT (A) before the second erlotinib treatment; (B) 3 mo after the second erlotinib treatment.
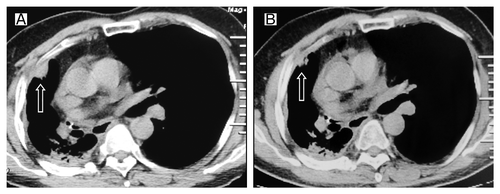
Figure 5. Tumor volume changes seen on CT (A) before the third erlotinib treatment; (B) 2 mo after the third time erlotinib treatment.
Discussion
There are substantial literatures on the concept of retreatment of EGFR-TKI with previously given conventional chemotherapy after the initial treatment. Unlike other studies, the survival after the initial treatment of erlotinib is more than 5 years in our report. The acquired resistance of erlotinib was reversed twice, and had good improvement of outcomes each time, which is crucial for the patients who acquired resistance after retreatment of EGFR-TKI. The third time use of EGFR-TKI may be a choice selectively. We reviewed the literatures for erlotinib retreatment and we found that there are some preconditions for valid response to EGFR-TKI retreatment: (1) the character of the patient would better to be female, non-smoker, and with adenocarcinoma histology; (2) initial treatment shows a favorable clinical response (e.g., partial response [PR] or long SD better more than 4–5 mo); (3) a long period of time since the termination of initial EGFR-TKI treatment. On the contrary, the dose of erlotinib and the pattern of EGFR mutation are small related to the retreatment response. And the disease control may be achieved more effectively in the patients with systemic progression disease (PD) than local PD to previous TKI treatment.Citation8
Even after the progression, some of the patients can still be sensitive to TKIs after a “drug holiday” with chemotherapy or radiotherapy. There are some possible reasons for this phenomenon.
Tumor heterogeneity
Despite identification of EGFR exon 19 deletion mutation in the biopsy tumor tissue and an initial PR to erlotinib in this case, it may be that only some clones are sensitive to erlotinib. Cytotoxic chemotherapy or radiotherapy administered after a resistance to the initial EGFR-TKI may modify the heterogeneous tissue distribution in sensitive or resistant cells. T790M, the most common resistant mutation, which enhanced oncogenicity, while T790M-harboring tumors in patients can display surprisingly slow rates of growth.Citation9 Oxnard et al. observed a distinct growth disadvantage in T790M-containing cells vs. their TKI sensitive parental counterparts, this may be partly responsible for the re-response phenomenon, and they allow us to predict that resistant tumors are likely a mixed population of TKI-sensitive and TKI-resistant cells. Upon withdrawal of the selective pressure of TKI, previously arrested TKI-sensitive cells can now repopulate more quickly than resistant cells, and tumors may regain sensitivity to TKI. Transformation to small cell carcinoma is another possible mechanism of TKI resistance. Recently, it has been reported that even in a tumor that shows transformation to small cell carcinoma, there potentially remains a subpopulation of cells that are still sensitive to TKI.Citation6 Switching to chemotherapy completely might allow these TKI-sensitive cells to regrow, likely causing the severe “flare” that has been described in 23% of patients a median of 8 d after stopping TKI.Citation10 For these reasons, some clinicians prefer to combine TKI with chemotherapy to simultaneously treat TKI-resistant and TKI-sensitive cells. This strategy is feasible given that chemotherapy plus TKI has been found to be only minimally more toxic than chemotherapy alone.Citation11The optimal sequential schedule of erlotinib with chemotherapy remains to be confirmed.
Gene mutation changes
The patient was treated with gemcitabine between the initial and retreatment of erlotinib. Treatment with cytotoxic chemotherapy or the “drug holiday” might produce genetic changes in EGFR or other associated genes that regulate resistance to erlotinib. It is reported that patients who develop recurrence after stopping TKI are unlikely to harbor a detectable T790M mutation and can have better responses to TKI retreatment than those recurred while receiving TKI.Citation12 Withdrawal of TKI from tumor cells in patients with acquired resistance leads to gradual loss of the T790M mutation, which might be responsible for this phenomenon,Citation9,Citation13 such that sensitivity to EGFR-TKI is reacquired. Indolent growth of T790M-mutant cells, leading to overgrowth of parental cells harboring only the EGFR-sensitizing mutation (as mentioned above), or due to lost of EGFR alleles lying in extra-chromosomal double-minutes from cells without appropriate selection pressure (TKI).Citation14
Chemotherapy attacks tumor stem cell
EGFR-TKI therapy may increase the population of stem-like cells and induce acquired resistance to EGFR-TKIs.Citation15 It is strongly suggested that cancer stem-like cells are more resistant for cancer therapy than other cells with accumulating evidence. The treatment with EGFR-TKIs may cause more cancer stem-like cells survive than other cells. It is not easy for them to survive in such a severe condition due to chemotherapy or radiotherapy. This might be the reason for the response to erlotinib retreatment in our case, where chemotherapy reduced cancer stem-like cells. While in order to overcome the resistance for the EGFR-TKIs, more evidences of cancer stem-like cells existence are necessary, and it would be very important to clarify the molecular mechanisms and biology of cancer stem-like cells.
Minakata et al. reported that hypoxia causes gefitinib resistance in NSCLC with both EGFR-Mut+ and EGFR-Mut− through the activation of wild-type EGFR mediated by the upregulation of tumor growth factor α (TGFα).Citation16 Chemotherapy or radiotherapy might improve the hypoxic microenvironment, which reduce the resistance to TKI. Shingo Kagawa et al. proved that inhibition of Akt/mammalian target of rapamycin (mTOR) signal activation recovers sensitivity to gemcitabine in pancreatic cancer cells, combination of mTOR-inhibitor and gemcitabine is effective to gemcitabine-resistant pancreatic cancer.Citation17Akt/mTOR pathway is involved in the mechanism of gemcitabine resistance by annexin II, erlotinib administered between gemcitabine inhibits Akt/mTOR pathway, which might be the reason why gemcitabine was effective after failing first time in our case. What’s more, the resistance to TKI may naturally change over time.
There are still some disadvantages in our report. First, erlotinib showed modest effects, almost no PR, on the CT images themselves. Nevertheless, targeted therapy is designed to interfere with specific aberrant biological pathways involved in oncogenesis and angiogenesis, which is in contrast to the generalized cytotoxic effects of standard chemotherapy. The effects of the new targeted therapy, such as angiogenesis inhibitors and antivascular therapies, are more complex. Necrosis and cavitation without a change in size are frequently observed over a short period of time. With these newer treatments, lack of progression may be associated with a good improvement in outcome, even in the absence of major shrinkage of tumors as evidenced by PR or complete response (CR). Therefore, the effect of targeted therapy is often underestimated by RECIST criteria based on tumor size. Considering these above limitations of CT on monitoring targeted therapy by RECIST, molecular imaging, which shows specific molecules and cellular processes, such as glycose metabolism and cell proliferation, may be more valid. Unfortunately, positron emission tomography (PET)/CT scanning was refused by the patient because of the unaffordable costs of this examination.
Second, unfortunately, tumor biopsy specimens failed to be obtained after both resistance to erlotinib and response to erlotinib retreatment due to the following reasons: (1) the patient refused to take percutaneous needle lung biopsy (PNLB) repeatedly because it is invasive; (2) considering the patient’s weakness, it was too risky to take biopsies again. The change of gene mutation or microenvironment in tumor was not clear along with the therapy. Besides, from this, we found biopsies are invasive and not frequently repeated; in addition, the predictive power may be limited due to intratumoral expression heterogeneity. Considering these limitations of tissue specimens, some new methods for monitoring the therapeutic efficacy of drugs during the whole treatment course are needed, such as elevated plasma levels or serum cell free DNA levels or molecular imaging, which needs further researches.
Third, because the patient refused to choose any other chemotherapy regimens except gemcitabine, we had to give him gemcitabine in the second-line and third-line chemotherapy after resistance to erlotinib, which doesn’t comply with standard treatment.
Conclusion
Erlotinib readministration after a certain interval should be considered as one of the therapeutic options for selected patients who showed a good response to initial erlotinib treatment. Considering the repeated reversion of erlotinib resistance, TKI plus chemotherapy may be a feasible choice to avoid TKI resistance. Sequential or concurrent therapy needs more study. Besides, the patients must be selected, and the appropriate dose, the predictive biomarker, the timing of TKI therapy, which all require further more researches.
| Abbreviations: | ||
| EGFR | = | epidermal growth factor receptor |
| NSCLC | = | non-small cell lung cancer |
| Mut− | = | non-mutant |
| EGFR-TKIs | = | EGFR tyrosine kinase inhibitors |
| EGFR-Mut+ | = | EGFR-mutant |
| T790M | = | threonine-methionine substitution at position 790 |
| MET | = | mesenchymal–epithelial transition factor |
| OS | = | overall survival |
| HGF | = | hepatocyte growth factor |
| IGF | = | insulin-like growth factor |
| TGFα | = | tumor growth factor alpha |
| CR | = | complete response |
| PR | = | partial response |
| PCR | = | polymerase chain reaction |
| SD | = | stable disease |
| PD | = | progression disease |
| CT | = | computed tomography |
| PFS | = | progression-free survival |
| mTOR | = | mammalian target of rapamycin |
| PET | = | positron emission tomography |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by The National Natural Science Foundation, 81201827.
References
- Moran T, Sequist LV. Timing of epidermal growth factor receptor tyrosine kinase inhibitor therapy in patients with lung cancer with EGFR mutations. J ClinOncol 2012; 30:3330 - 6; http://dx.doi.org/10.1200/JCO.2012.43.1858; PMID: 22753908
- Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009; 361:947 - 57; http://dx.doi.org/10.1056/NEJMoa0810699; PMID: 19692680
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005; 2:e73; http://dx.doi.org/10.1371/journal.pmed.0020073; PMID: 15737014
- Miller VA, Hirsh V, Cadranel J, Chen YM, Park K, Kim SW, Zhou C, Su WC, Wang M, Sun Y, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol 2012; 13:528 - 38; http://dx.doi.org/10.1016/S1470-2045(12)70087-6; PMID: 22452896
- Brugger W, Thomas M. EGFR-TKI resistant non-small cell lung cancer (NSCLC): new developments and implications for future treatment. Lung Cancer 2012; 77:2 - 8; http://dx.doi.org/10.1016/j.lungcan.2011.12.014; PMID: 22281074
- Oxnard GR. Strategies for overcoming acquired resistance to epidermal growth factor receptor: targeted therapies in lung cancer. Arch Pathol Lab Med 2012; 136:1205 - 9; http://dx.doi.org/10.5858/arpa.2012-0254-RA; PMID: 23020725
- Nishino M, Jackman DM, Hatabu H, Yeap BY, Cioffredi LA, Yap JT, Jänne PA, Johnson BE, Van den Abbeele AD. New Response Evaluation Criteria in Solid Tumors (RECIST) guidelines for advanced non-small cell lung cancer: comparison with original RECIST and impact on assessment of tumor response to targeted therapy. AJR Am J Roentgenol 2010; 195:W221-8; http://dx.doi.org/10.2214/AJR.09.3928; PMID: 20729419
- Oh IJ, Ban HJ, Kim KS, Kim YC. Retreatment of gefitinib in patients with non-small-cell lung cancer who previously controlled to gefitinib: a single-arm, open-label, phase II study. Lung Cancer 2012; 77:121 - 7; http://dx.doi.org/10.1016/j.lungcan.2012.01.012; PMID: 22333554
- Chmielecki J, Foo J, Oxnard GR, Hutchinson K, Ohashi K, Somwar R, Wang L, Amato KR, Arcila M, Sos ML, et al. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. SciTransl Med 2011; 3:90ra59; http://dx.doi.org/10.1126/scitranslmed.3002356; PMID: 21734175
- Chaft JE, Oxnard GR, Sima CS, Kris MG, Miller VA, Riely GJ. Disease flare after tyrosine kinase inhibitor discontinuation in patients with EGFR-mutant lung cancer and acquired resistance to erlotinib or gefitinib: implications for clinical trial design. Clin Cancer Res 2011; 17:6298 - 303; http://dx.doi.org/10.1158/1078-0432.CCR-11-1468; PMID: 21856766
- Herbst RS, Prager D, Hermann R, Fehrenbacher L, Johnson BE, Sandler A, Kris MG, Tran HT, Klein P, Li X, et al, TRIBUTE Investigator Group. TRIBUTE: a phase III trial of erlotinib hydrochloride (OSI-774) combined with carboplatin and paclitaxel chemotherapy in advanced non-small-cell lung cancer. J ClinOncol 2005; 23:5892 - 9; http://dx.doi.org/10.1200/JCO.2005.02.840; PMID: 16043829
- Oxnard GR, Janjigian YY, Arcila ME, Sima CS, Kass SL, Riely GJ, Pao W, Kris MG, Ladanyi M, Azzoli CG, et al. Maintained sensitivity to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer recurring after adjuvant erlotinib or gefitinib. Clin Cancer Res 2011; 17:6322 - 8; http://dx.doi.org/10.1158/1078-0432.CCR-11-1080; PMID: 21831955
- Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. SciTransl Med 2011; 3:75ra26; http://dx.doi.org/10.1126/scitranslmed.3002003; PMID: 21430269
- Ercan D, Zejnullahu K, Yonesaka K, Xiao Y, Capelletti M, Rogers A, Lifshits E, Brown A, Lee C, Christensen JG, et al. Amplification of EGFR T790M causes resistance to an irreversible EGFR inhibitor. Oncogene 2010; 29:2346 - 56; http://dx.doi.org/10.1038/onc.2009.526; PMID: 20118985
- Nakata A, Gotoh N. Recent understanding of the molecular mechanisms for the efficacy and resistance of EGF receptor-specific tyrosine kinase inhibitors in non-small cell lung cancer. Expert OpinTher Targets 2012; 16:771 - 81; http://dx.doi.org/10.1517/14728222.2012.697155; PMID: 22762482
- Minakata K, Takahashi F, Nara T, Hashimoto M, Tajima K, Murakami A, Nurwidya F, Yae S, Koizumi F, Moriyama H, et al. Hypoxia induces gefitinib resistance in non-small-cell lung cancer with both mutant and wild-type epidermal growth factor receptors. Cancer Sci 2012; 103:1946 - 54; http://dx.doi.org/10.1111/j.1349-7006.2012.02408.x; PMID: 22863020
- Kagawa S, Takano S, Yoshitomi H, Kimura F, Satoh M, Shimizu H, Yoshidome H, Ohtsuka M, Kato A, Furukawa K, et al. Akt/mTOR signaling pathway is crucial for gemcitabine resistance induced by Annexin II in pancreatic cancer cells. J Surg Res 2012; 178:758 - 67; http://dx.doi.org/10.1016/j.jss.2012.05.065; PMID: 22726648