Abstract
Monoclonal antibodies (mAbs) are highly complex proteins that display a wide range of microheterogeneity that requires multiple analytical methods for full structure assessment and quality control. As a consequence, the characterization of mAbs on different levels is particularly product - and time - consuming. This work presents the characterization of trastuzumab sequence using sheathless capillary electrophoresis (referred as CESI) – tandem mass spectrometry (CESI-MS/MS). Using this bottom-up proteomic-like approach, CESI-MS/MS provided 100% sequence coverage for both heavy and light chain via peptide fragment fingerprinting (PFF) identification. The result was accomplished in a single shot, corresponding to the analysis of 100 fmoles of digest. The same analysis also enabled precise characterization of the post-translational hot spots of trastuzumab, used as a representative widely marketed therapeutic mAb, including the structural confirmation of the five major N-glycoforms.
Introduction
Since 1986 and the approbation of muromonab-CD3 by the US Food and Drug Administration (FDA), monoclonal antibodies (mAbs) have taken a major market share in the pharmaceutical industry and their development is constantly increasing.Citation1,Citation2 MAbs are highly complex glycoproteins potentially displaying many naturally-occurring molecular micro-heterogeneities and those that are introduced through imperfect processing, physico-chemical and enzymatic changes that occur during their production and long-term conservation. As a consequence, there is a continuous need for improvement of analytical methods to enable fast and accurate characterization. Mass spectrometry (MS) plays a major role in the characterization of therapeutic mAbs because it provides specificity, sensitivity and may also provide structural information. Characterization using MS has helped product and process developmentCitation3 and batch consistency assessment.Citation4,Citation5 Furthermore, recent raising importance of biosimilar and biobetter mAbs represents a major stake for characterization methodologies. Biosimilar mAbs are produced from different clones and processes.Citation6 The European Medicines Agency (EMA) and the FDA have published draft guidelines concerning the approval pathway of biosimilar mAbs. These documents discuss the physicochemical characterization steps required to assess biosimilarity. For example, EMA guidelines suggest that the biosimilar candidate should have the same amino acid sequence as the reference mAb and a comparable glycosylation profile.Citation7 The selection of a highly similar candidate to the innovator product is mandatory before performing pre-clinical and clinical biosimilarity studies. Obtaining suitable structural information rapidly, which allows quick elimination of candidates dissimilar to the reference protein, is therefore crucial. The methods used should cover a wide range of structural information, e.g., amino acid sequence, disulfide bonds, glycosylation, posttranslational modifications (PTM).Citation8 Additional to MS, mAbs characterization requires a panel of orthogonal methods to cover a full range of structural information,Citation9 including reverse phase liquid chromatography (RP-LC),Citation10-Citation12 ion exchange chromatography (IEC), size exclusion chromatography (SEC),Citation13 capillary isoelectric focusing (cIEF)Citation14 or micellar electrokinetic chromatography (MEKC).Citation15,Citation16
Peptide mapping of mAbs by MS is commonly performed using a bottom-up proteomic approach because it allows acquisition of information on the amino acid sequence and PTMs. In this approach, the protein undergoes enzymatic digestion, resulting in a peptide digest most often separated by liquid chromatography and detected by UV spectrophotometry and MS.
A novel sheathless CE-ESI-MS platform, referred as CESI, allowing hyphenation of capillary electrophoresis (CE) to electrospray ionization mass spectrometry (ESI-MS) has been developed by Beckman Coulter based on a design previously described by Moini.Citation17 This CESI prototype (also known as sheathless capillary electrophoresis) uses a bare fused silica capillary whose outlet has been etched using hydrofluoric acid, which makes it porous to the electrical transport of small ions without permitting significant matter transfer through the pores. The porous tip provides electrical contact via a second capillary. Detailed description of this interface has been given by Haselberg et al.Citation18 The sheathless interface has already been used to perform successful CE-ESI-MS experiments in proteomics,Citation19,Citation20 metabolomics,Citation21 intact proteinsCitation22,Citation23 demonstrating drastically increased sensitivity compared with sheath-liquid CE-MS due to operating flow rates of well below 100 nL/min. Whitmore and Gennaro recently used the CESI prototype to performed the peptide mass fingerprinting (PMF) of a mAb. They demonstrated the successful use of PMF to cover the amino sequence of a mAb, including the hydrophilic peptides that are eluted in the void volume in RP-LC-MS.Citation23
In this work, we used the CESI-MS system to develop a CE-MS/MS method that allowed the fast and precise characterization of a mAb digest. The mAb selected was trastuzumab which was approved by FDA and EMA, in 1998 and 2000 respectively. Trastuzumab has been intensively studied and can be considered as a highly representative therapeutic mAb.Citation24 It is a humanized immunoglobulin gamma 1 (HzIgG-1) directed against the HER2/neu receptor which is overexpressed in about 20–30% of invasive breast cancer patients.Citation25,Citation26 Trastuzumab is composed of two heavy chains (HC) having 449 amino acids and two light chains (LC) having 214 amino acids.Citation27 The HC has an N-glycosylation consensus site on its asparagine (Asn) in position 300, and the glycoprotein has a total molecular mass of about 148 kDa.Citation4 A tryptic digest of trastuzumab was prepared using a conventional in-solution digestion protocol.Citation28 The digest was then studied at different levels. First a peptide mapping of the mAb was performed using a CESI-MS/MS analysis. The MS/MS spectra were automatically analyzed to identify the peptides using a search engine widely used in proteomic studies in order to hasten peptide identifications. PTMs were also characterized using CESI-MS/MS, particularly those known as degradation hot spots. These PTMs were identified by Mascot search algorithm and manually confirmed. At a final level, glycosylation were also investigated using CESI-MS/MS. Although glycosylation is a type of PTM, they are treated in this article separately from amino acid PTMs because they are not degradation hot spots. The glycans could be located on the amino acid sequence and the structure of the major glycoforms could be deduced from MS/MS data. Remarkably, the characterization of the mAb over these different described levels was realized using only one CESI-MS/MS analysis of the digest.
Results
Peptide mapping of trastuzumab using CESI-MS/MS
Peptide mapping is commonly used to determine protein amino acid sequence and to locate or quantify PTMs. This methodology is quite important in early development of therapeutic antibodies as well as during long-term life cycle management of the biopharmaceutical products. As part of this work, one of the objective was to investigate the peptide mapping of trastuzumab by CESI-MS/MS. Tandem MS data interpretation and peptide identification was done automatically using Mascot to obtain fast and accurate data treatment.
A sample of trastuzumab was digested with trypsin enzyme. Tryptic digestion was performed using an in-solution digestion protocol applied on a routine basis in the laboratory. It includes reduction and alkylation of disulfide bonds. Prior to digestion, an unfolding step was performed using guanidine as a chaotropic agent. Prior to MS analysis, peptides were separated using the CESI prototype which allows direct coupling of CE with the nanospray source of the MS without using an additional liquid like in sheath-liquid CE-MS.Citation29
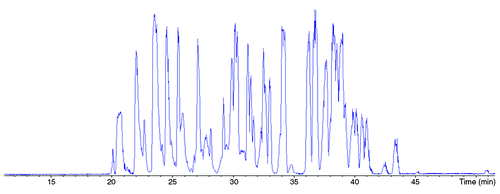
illustrates an example of the separation obtained for the tryptic digest of trastuzumab for 100 fmol of injected protein. The total duration of the separation was less than 50 min and the resulting electropherogram showed that all peptides were detected between 19 and 47 min. Separation was performed at a voltage of +20 kV to maintain a balance between peptide migration speed and separation efficiency.
Figure 1. Base Peak Electropherogram corresponding to the analysis by CESI-MS/MS of trastuzumab tryptic digest. Experimental conditions: bare fused silica capillary with porous tip, total length 95 cm (30 μm i.d., 150 μm o.d.); CE voltage +20 kV; BGE 10% acetic acid; sample trastuzumab tryptic digest 2.5 mg/mL in BGE (11nL injected). MS capillary voltage: -1.3 kV, m/z range: 50–3000.
Digested peptides were identified from MS/MS data acquired using a Mascot search algorithm, although 100% sequence coverage of a mAb has already been reported using the prototype CESI-MS platform.Citation23 In this work, peptide identification was automated and based on MS/MS data, meaning that identification was made on precursor ion mass measure and fragment identification. Results of Mascot search showed sequence coverage of 100% for both the HC and LC of the trastuzumab. The detailed sequence coverages for both chains are shown in . Some peptides were selected and fragmented several times during the analysis leading to the same identification. Similarly, missed cleavages could be promptly detected for several peptides reinforcing the confidence of the identification by enabling peptide overlapping.
Figure 2. Sequence coverage obtained by CESI-MS/MS for trastuzumab HC (left-hand side) and LC (right-hand side) based on the identified peptides. Experimental conditions: see materials and methods

Tryptic digestion produces a heterogeneous mixture of peptides, especially with regard to their number of amino acids. Results show the identification during the analysis of small peptides having 4–5 amino acids such as HT05 or LT09 () as well as a 63 amino acids peptide having a molecular mass of 6715.26 Da (HT21). During its elution, the ion corresponding to HT21 peptide was selected several time for fragmentation and resulted in the same identification, which reinforced the confidence of this important identification.
Figure 3. List of trastuzumab digested peptides identified by the CESI-MS/MS analysis. Experimental conditions: see material and methods

It should be noted that such heavy peptides usually can attain higher charge state; therefore, the search algorithm must consider high charge state as well. In the CESI-MS/MS analysis of the mAb, peptides having charge state up to 6+ could be detected. Fragmentation of the peptides enables an important part of the amino acid sequence to be retraced reinforcing the high confidence in peptide identification.
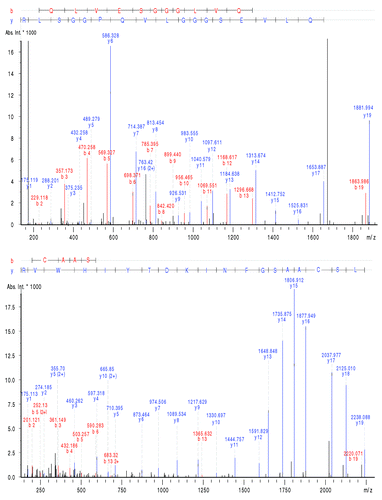
As seen in , the MS/MS spectra quality, has allowed the amino acids sequence composing the variable domain of the mAb HC for 109 of 120 amino acids to be retraced. Similarly 98 of the 107 amino acids sequence composing the variable domain of the LC could be retraced using the MS/MS data. Thus, in addition to allowing the identification of the digested peptides of the mAb, the MS/MS spectra enabled characterization, without ambiguity, of most amino acids composing such a sensitive part of the mAb, i.e., the variable domain.
Figure 4. MS/MS spectra of peptide HT01 and HT02 illustrating the amino acid sequence retracing in the variable domain. Experimental conditions: see material and methods.
To evaluate the robustness of the method, the CESI-MS/MS analysis of the mAb digest was repeated six times consecutively. Results of these analysis demonstrated 100% sequence coverage for each run and the same detected peptides in each analysis.
Characterization of amino acids PTMs hot-spots
Mabs are heterogeneous by nature because of structural modifications that the proteins may undergo during their lifetime. Those modifications may be Critical Quality Attributes (CQA) because they can modify the conformation of the protein and therefore influence the antibody activity. Posttranslational modifications and other physico-chemical modifications can be induced by intra or extra cellular processes, buffer, purification process, storage or stress conditions.Citation31,Citation32 Among those PTMs, several referred to as “hot spots” must be carefully monitored to determine the stability of the mAb during pharmaceutical development. Known degradation hot spots potentially modified on the HC are cyclization of N-terminal glutamic acid (Glu) into pyroglutamic acid and deamidation of Asn55 and Asn387 but also oxidation of Met255 and Met431. The main hot spot on the LC is the deamidation Asp30. For the characterization of trastuzumab PTMs, the same CESI-MS/MS data presented in the previous section to obtain the mAb peptide mapping was used. In this case again, the PTMs were automatically identified by the Mascot search algorithm. Mass spectra were further manually studied to confirm the presence of the modifications.
Results obtained showed glutamine cyclization of the N-terminal extremity of the HC. Also, the same peptide without the modification was identified, which suggests a partial modification of the protein. Use of the extracted ion electropherograms (XIE) for m/z ratio of 932.500 (2+) and 941.501 (2+) corresponding to the peptide HT01 with both N-terminal extremity forms allowed the determination of a migration time of 34.5 min for the intact form of HT01 and 45.6 min for the modified HT01. This charge difference influences the electrophoretic mobility of the modified peptide, leading to its separation.
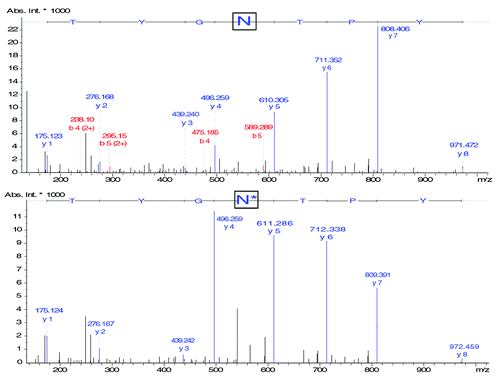
In the same manner, several deamidation hot spots were also identified. Results presented in show the correct identification of the deamidation of Asn55 (HT04). In this case again the parent peptide could be identified without the modification as well, and the fragmentation allowed confirmation of the deamidation of this specific amino acid. Another deamidation hot spot present on the HC on Asn387 (HT41) was also characterized with the method. In this case again, the MS/MS spectra obtained for HT41 allowed the identification of the deamidation on this specific amino acid which is crucial because the digested peptide is composed of several Asn. Other micro-variants, such as methionine (Met) oxidation hot spots, were also identified by the algorithm using the CESI-MS/MS data. Results allowed the identification of the oxidations of Met255 (HT25) and Met431 (HT48) as a confirmation of the trend of a partial modification/degradation; both peptides could also be observed without any oxidation. The peptide HT25 was detected at a time of 23.8 min for the intact form and 24.0 min for the oxidized HT25. A similar results could be observed for peptide HT48 as the intact peptide could be detected at 25.2 min and respectively at a time of 25.5 min for the oxidized HT25. Concerning the LC, the main deamidation hot spot located on Asn30 (LT03) was correctly identified by the algorithm. As for the other hot spots, the manual confirmation suggested a partial modification/degradation of the protein.
Figure 5. MS/MS spectra of peptide HT04 (IYPTNGYTR) (A) without modification and (B) with a deamidation on Asn55 illustrating the partial modification of the mAb and the characterization of both forms by CESI-MS/MS. Experimental conditions: see Materials and Methods.
These results unambiguously demonstrate the possibility of using CESI-MS to perform simultaneous MS/MS peptide mapping and amino acid PTM characterization. The separation of the digested peptide mixture allowed the MS to fragment a large number of ions, which enables the detection of PTMs. The mass spectra quality obtained using the CESI interface allowed the algorithm to successfully identify each studied modification.
Characterization of major N-glycoforms
For most IgGs, glycosylation represents only 2 to 3% of the total mass, but it can significantly influence the protein’s structure, immunogenicity and stability.Citation33,Citation34 Glycosylation is also a class of mAb PTMs: here their study was described separately from amino acid PTMs because they are not considered degradation hot spots. To perform an advanced characterization of trastuzumab, an estimation of the ratio of the main glycoforms, as well as a structural determination is necessary. Trastuzumab is N-glycosylated on the Asn300 in the CH2 region.Citation35,Citation36 G0F and G1F, the major glycoforms found on human and recombinant IgG produced in CHO, NS0 and SP2/0 cells, represent more than 80% of the glycoforms.Citation30,Citation37 Considering that Mascot search algorithm is only intended to identify the peptide sequences it was necessary to manually study the CESI-MS/MS data to identify the glycosylations and determine their structures. Interestingly, in this case as well, no extra experiments needed to be performed to focus on glycopeptides. The same data sets used for peptide mapping and hot spots characterization were also used here.
In conventional protocols, a mAb is commonly treated with PNGase F before analysis to release the glycans and allow study of them while not attached to the peptide backbone.Citation37 In this work, the mAb was digested by trypsin only, and therefore we expected to detect glycans still linked to the corresponding peptide backbone of the HC. The extracted ion electropherograms (XIE) of the m/z ratio matching to the peptide TKPREEQYN300STYR bearing respectively the glycosylation G0F, G1F, G0F-GlcNac, G2F and G0 are shown in −E, respectively. All the detected glycopeptides have a charge state of 3.
Figure 6. Extracted Ion Electropherogram of m/z ratios (A) 1039.80 (HT29 + G0F), (B) 1093.78 (HT29 + G1F), (C) 972.09 (HT29 + G0), (D) 1147.82 (HT29 + G2F), (E) 991.10 (HT29 + G0). Experimental conditions: see Material and Methods.
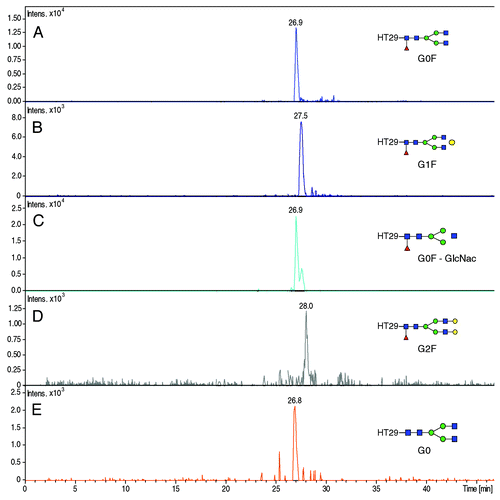
The glycopeptides were detected between 26.3 and 28.5 min. The electrophoretic resolution calculated between the peaks corresponding to the glycopeptides bearing G0F and G1F gave a value of 0.88, while the resolution between the peaks of glycopeptides G1F and G2F gave a value of 1.08. These results demonstrate the separation of the glycopeptides G0F, G1F and G2F glycan having only one galactose difference in each case. The glycopeptide bearing G0 could also be observed on its corresponding XIE; however, this glycopeptide co-migrated with glycopeptides G0F. Moreover, the sensitivity provided by the CESI prototype allows detection of these glycopeptides with significant intensities and the signal was sufficient to generate good quality MS/MS spectra of each glycopeptides. More generally, these results allowed us to accurately locate the position of the glycosylation on Asn300 because it is the only potential glycosylation site on this peptide. The nature of the glycosylation, however, had to be confirmed using MS/MS data.
MS/MS spectra () corresponding to the fragmentation of the different glycopeptides were subsequently extracted from the data. The precursor ion matches the mass of the glycopeptide being glycosylated by G0F with a precision of 8.66 ppm. As expected with low energy collision induced dissociation (CID), product ions observed in MS/MS mainly correspond to the fragmentation of the glycan moiety and the peptide backbone is usually preserved. Study of the fragmentation spectra allowed us to deduce the structure of the glycan G0F. In the same manner, the MS/MS spectra of the glycopeptides bearing G1F, G2F and G0 glycans were extracted from the data; in each case the mass measure precision on the precursor ion was inferior to 9.69 ppm. Similarly to the previous results, every ions corresponding to the different fragmentation of the glycan could be observed while the peptide backbone remained intact, allowing the structures of the studied glycans to be deduced.
Figure 7. MS/MS fragmentation spectra of (A) HT29 - G0F (precursor m/z 1039.78; charge state 3+), (B) HT29 - G1F (precursor ion 1093.80; charge state 3+), (C) HT29 - G2F (precursor ion 1147.82; charge state 3+), (D) HT29 - G0F-GlcNac (precursor ion 972.10; 3+), (E) HT29 - G0 (precursor ion 991.09; charge state 3+). Experimental conditions described in Materials and Methods section.
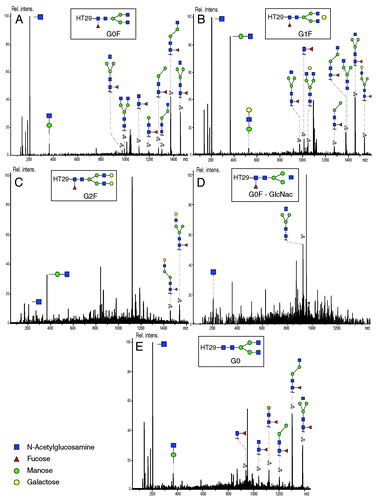
Results presented here demonstrate the possibility of using CE to separate and identify glycopeptides obtained from the trastuzumab enzymatic digestion mainly due to the baseline separation efficiency of the five major glycopeptides provided by the CE. As an additional benefit, the sensitivity provided by the CESI interface and the microTOFQ-II MS allowed us to obtain the complete fragmentation of the glycan and thereby deduce the structure of the five major glycans (G0F, G1F, G2F, G0F-GlcNac and G0). The same data was used for MS/MS peptide mapping and hot-spot characterization without requiring additional experiments.
Discussion
Peptide mapping is essential for the characterization of therapeutic mAbs, which are complex glycoproteins that contain a plethora of micro-heterogeneities due to the production process. In comparison to PMF, MS/MS peptide identification is not only based on digested peptide m/z ratio measure, but also on the fragmentation of those peptides, increasing the confidence of the identification. Capillary zone electrophoresis (CZE) separation is based on the migration of species present in a background electrolyte (BGE) under an electrical field. Separation is obtained by differences in electrophoretic mobility among analytes depending on size and charge of the molecule at the BGE pH.
Peptide mapping results showed the identification of wide variety of peptides regarding their chemical composition (). This result indicates clearly that CE separation driven by peptide migration phenomena is able to separate and transfer successfully to the MS every digested peptide regardless of its nature. In RP-LC, small peptides (from one to two amino acidsCitation23) and more generally hydrophilic peptides, are often not detected because they are not retained on the column and elute in the void volume.Citation19 The detection and identification of peptide such as LT09 or HT05 demonstrate the ability of CE to conserve and detect small peptides. The identification of the 63 amino acids peptide HT21 also demonstrates one of the advantages of using CE hyphenated to MS for peptide characterization. The presence of such large peptides is not surprising as trastuzumab HC does not have a tryptic cleavage site from amino acid 151 to 213. The elution and detection of such an imposing peptide may be difficult in RPLC-MS/MS because it can irreversibly interact with the stationary phase. Identification of very large peptides having no enzymatic digestion sites or corresponding to peptides or with several missed cleavages is also a very important asset of this method because 100% sequence coverage is obtainable in various cases. Indeed, some digestion sites in mAbs may not be accessible to trypsin due to the quaternary structure of the protein, which may cause missed cleavages during the digestion even when guanidine is present as a chaotropic reagent. In addition to the complete sequence coverage obtained of the HC and the LC, the sensitivity provided by the CESI prototype supported by the high resolution of the mass spectrometer allowed generation of high quality MS/MS spectra. This result is explained by a remarkable ionization efficiency of the peptides. The low BGE flow rate during the separation indeed enhances the ionization process and reduces ion suppression.Citation20,Citation38 The spectral quality is reflected by the possibility of retracing the major part of the amino acid sequence in the variable domain. As the CESI-MS system is able to transfer efficiently an important number of ions, MS/MS spectra were of an excellent quality in terms of resolution and intensities (). Therefore, a significant number of y/b fragments could be attributed, allowing retracing on the variable domain of the HC 109 of 120 amino acids and 98 of 107 on the LC. Understanding the variable domain of the mAb is crucial because it is directly involved in the antibody-antigen binding. It is noteworthy that such level of characterization could be attained with only one run of the CESI-MS/MS system.
Our results demonstrated the successful characterization of every single degradation hot spots on HC and LC and we observed in each case the partial modification of the protein because the modified and the intact form of the peptide could be identified (). In the case of the glutamine cyclization of the N-terminal extremity on the HC, the CE separation mechanism allowed the separation of the modified and intact forms of the peptide based on conformational changes induced by glutamine cyclization. Such separation is explained by the fact that even if the mass difference between the parent and the modified peptide is only 17.02 Da, Glu cyclization involves the loss of the amine function on its lateral chain; therefore, there is a difference of one charge between glutamic acid and pyroglutamic acid because the amine is protonated in 10% acetic acid (pH 2.2). The possibility of separate the peptides prior to MS analysis, allows the identification of both forms. Indeed as the number of precursor ions selected for fragmentation per cycle is fairly limited on the microTOF-Q II, and taking into account the complexity of the sample, it would have been difficult for the MS to automatically select the m/z ratio corresponding to both forms of this peptide during their detection window if they had co-migrated. In the case of the deamidation hot spots present on Asn55 and Asn387 on the HC and AsnCitation30 on the LC, the MS was able to fragment both the modified and the intact form for each peptide. Unlike in the glutamic acid cyclization, the XIE corresponding to those peptides (HT04, HT41 and LT03) showed in each case the complete co-migration between the intact peptide and its deamidated homolog. This is explained by the fact that the mass difference between asparagine and aspartic acid is only 0.98 Da. Regarding deamidation, the amide functional group located on asparagine lateral chain, is replaced by an alcohol function. Most importantly, there is thus no significant charge difference between the deamidated peptide and the intact peptide because those functions are protonated at BGE pH. In this case of oxidized Met255 (HT25) and Met431 (HT48), even if the detection times were not exactly the same for the intact peptide and its oxidized homologous, differences in electrophoretic mobilities could not lead to the baseline separation of those peptides. The sensitivity provided by the system allowed unambiguous characterization of each studied degradation hot spot, in addition to the low represented to PTMs and degradations present in small amounts.
Using the CESI-MS/MS data, the complete structural characterization of the glycosylation G0F, G1F, G2F, G0F-GlcNac and G0 could be achieved (). Because trypsin is not able to cleave glycosylation from the peptide, the glycan was observed on the corresponding digested peptide. The XIE corresponding to the glycopeptides bearing different glycans () demonstrated the separation of several glycopeptides in a reduced time. This result demonstrated the ability of the CE migration mechanism to completely separate glycopeptides having a difference of only one galactose (e.g., between G0F and G1F). Such levels of separation could be explained by the fact that one more galactose leads to a difference between the two glycopeptides in term of size (+ 162 Da). This difference affects their relative electrophoretic mobilities leading to their separation. The efficiency of CE in the glycopeptides separation has allowed the separation to be obtained in ~27 min. A difference of a fucose, however, did not lead to the separation of the glycopeptides G0F and G0, suggesting that the size of the glycopeptides is not significantly affected by the fucosylation at the BGE pH.
To evaluate the robustness of this method, the analysis was consecutively repeated six times using the same mAb digest sample. Each analysis resulted in 100% sequence coverage in term of MS/MS peptide mapping. Similarly, the degradation hot spots identified were the same in each case and the five same major N-glycans could also be structurally characterized in the six analyses. Intensity ratios between the different glycopeptides were calculated for each analysis; the results exhibited a standard deviations ranging from 6 to 13%. Based on this results, glycopeptide intensities appear to be related to glycan distribution. It is therefore it is possible to consider profiling the mAb glycan using this very same CESI-MS/MS method, which would enriching the amount of information obtain from only one analysis.
In conclusion, this work presents the development of a rapid and reliable method for performing a multi-level characterization of trastuzumab, which was used as reference therapeutic antibody in a single run. The mAb was digested using a conventional digestion protocol and the digest was analyzed by CESI hyphenated to a high resolution tandem MS. The CESI-MS/MS fragmentation obtained in only one acquisition allowed the complete amino acid sequence peptide map to be recorded. A Mascot search engine was used for peptide identification to provide a faster protocol. Also, the characterization of every PTM hot spot could be obtained within the same single run. Finally, separation of the major glycosylated peptides and their structural characterization could be obtained using the same CESI-MS/MS analysis data set. Such advanced characterization using only a single injection of 100 fmol of digested mAb peptide mixture in a 45 min analysis demonstrates the potential of CESI-MS as a fast and sensitive method in mAb characterization and more generally in bottom-up proteomic approach.
Materials and Methods
Materials
All chemicals used were of analytical grade or high purity grade. Acetic acid was provided by VWR. Bicarbonate ammonium, guanidine hydrochloride, iodoacetamide (IAM), dithiothreitol (DTT), trifluoroacetic acid, acetonitrile (ACN) and trypsin enzyme were purchased from Sigma-Aldrich. Water used to prepare buffers and sample solutions was obtained using an ELGA purelab UHQ PS water purification system. Desalting steps were performed using C8 spin tips purchased from Protea Biosciences.
Tryptic digestion procedure
Tryptic digests of antibodies were prepared using sequence-grade trypsin (1:10 w/w). Before digestion, sample was diluted to a concentration of 10 mg/mL. One mg of protein was mixed with 155 μL of denaturing reagent (guanidine HCl 6M in 50 mM ammonium bicarbonate) and 15 μL of DTT 100 mM were added. The mixture was incubated for 5 min at 95°C, then cooled to ambient temperature. Then, 30 μL of IAM 100 mM was added and the mixture was placed in the dark for 20 min. Ten μL of trypsin was added and the mixture was incubated at room temperature for 3 h; additional 10 μL of trypsin were added to the sample for overnight digestion at room temperature. Buffer exchange and concentration were performed using a C8 spin tips. Four hundred μL of mAb tryptic digest in BGE could be obtained. A volume of ten μL of the obtained digest was used to perform all CESI-MS/MS experiments.
Capillary electrophoresis
The CE experiments were performed with a PA 800 plus capillary electrophoresis (CE) system from Beckman Coulter equipped with a temperature controlled autosampler and a power supply able to deliver up to 30 kV. Hyphenation was realized using a CESI prototype made available by Beckman Coulter. Prototype of bare fused-silica capillaries (total length 100 cm; 30 µm i.d.) with characteristic porous tip on its final 3 cm supplied by Beckman Coulter, a second capillary (total length 80 cm; 50 μm i.d.) filled during experiments with BGE allows electric contact. New capillaries were flushed for 10 min at 75 psi (5.17 bar) with methanol, then 10 min with 0.1 M sodium hydroxide, followed 10 min with 0.1 M hydrochloric acid and water for 20 min also at 75 psi. Finally, the capillary was flushed 10 min at 75 psi with BGEn which was acetic acid 10%. Hydrodynamic injection (69 mbar for 1 min) corresponding to a total volume of 11 nL of sample injected was used. Separations were performed using a voltage of +20 kV.
Mass spectrometry
For antibody characterization, the CESI system was coupled to a microTOF-Q II mass spectrometer (Bruker Daltonics). The microTOF-Q II MS is equipped with a hybrid analyzer composed of a quadrupole followed by a time-of-flight (TOF) analyzer. Positive mode acquisition was used to detect precursor ions (MS) and fragmented product ions (MS/MS). Concerning the ESI source parameters, capillary voltage was set to -1.3 kV. Nebullizer gas was deactivated, the dry gas was set to 1.5 L/min and temperature of the source was set at 160°C. Spectra were collected at a data acquisition frequency of 2 Hz; for fragmentation spectra, collision energy ranged from 0 to 45 V depending on the m/z ratio and charge state of the precursor ion. For each MS scan, 3 precursor ions were selected for fragmentation, and total duty cycle was therefore 2 sec. Mass range was 50–3000 for MS as well as MS/MS scans.
MS/MS data analysis
Data obtained from CESI-MS/MS experiments were processed using Mascot search algorithm developed by Matrix Science. Tryptic cleavage rules were applied for both HC and LC sequences of the mAb. Carbamidomethylation of cysteine (+57.02 Da) was selected as a fixed modification, N-deamidation of aspartic/isoaspartic acid (+0.985 Da) or succinimide intermediate (-17.03 Da) were selected as a variable modifications. Methionine oxidation (+15.99 Da) and N-terminal glutamic acid cyclization (-17.02 Da) were also selected as variable modifications. The mass tolerance for precursor ions was set to ± 25 ppm and to ± 0.5 Da for fragments. A maximum of 3 missed cleavages was tolerated. MS/MS N-glycan identification and structural characterization were done manually.
| Abbreviations: | ||
| mAb | = | monoclonal antibody |
| HC | = | heavy chain |
| LC | = | light chain |
| HT | = | heavy chain tryptic peptide |
| LT | = | light chain tryptic peptide |
| PTM | = | posttranslational modification |
| RPLC | = | reverse phase liquid chromatography |
| MS | = | mass spectrometry |
| MS/MS | = | tandem mass spectrometry |
| CESI | = | sheathless capillary electrophoresis |
| ESI | = | electrospray ionization |
| XIE | = | extracted ion electropherogram |
| DTT | = | dithiothereitol |
| FA | = | formic acid |
Acknowledgments
Rabah Gahoual would like to thank the MRT for funding his Ph.D work. LSMIS would like to thank Beckman Coulter Inc. for lending a CESI prototype, Bruker Daltonics lending the microTOF-Q II and M. Anselme from Beckman Coulter Inc. for his help. The authors would also thanks Dr. E. Wagner-Rousset, Dr. D. Ayoub, MC. Janin-Bussat and O. Colas (Centre d’Immunologie Pierre Fabre, Saint-Julien en Genevois, France) for discussions around sample preparation and antibody LC-MS characterization.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Beck A, Wurch T, Bailly C, Corvaia N. Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol 2010; 10:345 - 52; http://dx.doi.org/10.1038/nri2747; PMID: 20414207
- Reichert JM. Marketed therapeutic antibodies compendium. MAbs 2012; 4:413 - 5; http://dx.doi.org/10.4161/mabs.19931; PMID: 22531442
- Ambrogelly A, Liu YH, Li H, Mengisen S, Yao B, Cannon-Carlson S, et al. Characterization of antibody variants during process development The tale of incomplete processing of N-terminal secretion peptide. MAbs 2012; 4:1 - 9; http://dx.doi.org/10.4161/mabs.21614; PMID: 22327425
- Damen CWN, Chen W, Chakraborty AB, van Oosterhout M, Mazzeo JR, Gebler JC, et al. Electrospray ionization quadrupole ion-mobility time-of-flight mass spectrometry as a tool to distinguish the lot-to-lot heterogeneity in N-glycosylation profile of the therapeutic monoclonal antibody trastuzumab. J Am Soc Mass Spectrom 2009; 20:2021 - 33; http://dx.doi.org/10.1016/j.jasms.2009.07.017; PMID: 19744865
- Committee for medicinal products for human use (CHMP), European Medicines Agency (EMA). Guideline on development, production, characterization and specifications for monoclonal antibodies and related products 2009; EMEA/CHMP/BWP/157653/2007.
- McCamish M, Woollett G. Worldwide experience with biosimilar development. MAbs 2011; 3:209 - 17; http://dx.doi.org/10.4161/mabs.3.2.15005; PMID: 21441787
- Reichert JM, Beck A, Iyer H. European Medicines Agency workshop on biosimilar monoclonal antibodies: July 2, 2009, London, UK. MAbs 2009; 1:394 - 416; http://dx.doi.org/10.4161/mabs.1.5.9630; PMID: 20065643
- Zhang Z, Pan H, Chen X. Mass spectrometry for structural characterization of therapeutic antibodies. Mass Spectrom Rev 2009; 28:147 - 76; http://dx.doi.org/10.1002/mas.20190; PMID: 18720354
- Harris RJ, Chin ET, Macchi F, Keck RG, Shyong BJ, Ling VT, et al. Analytical characterization of monoclonal antibodies: linking structure to function. Current Trends in Monoclonal Antibody Development and Manufacturing, Springer 2010; 193-205.
- Wagner-Rousset E, Bednarczyk A, Bussat MC, Colas O, Corvaïa N, Schaeffer C, et al. The way forward, enhanced characterization of therapeutic antibody glycosylation: comparison of three level mass spectrometry-based strategies. J Chromatogr B Analyt Technol Biomed Life Sci 2008; 872:23 - 37; http://dx.doi.org/10.1016/j.jchromb.2008.03.032; PMID: 18672411
- Beck A, Bussat MC, Zorn N, Robillard V, Klinguer-Hamour C, Chenu S, et al. Characterization by liquid chromatography combined with mass spectrometry of monoclonal anti-IGF-1 receptor antibodies produced in CHO and NS0 cells. J Chromatogr B Analyt Technol Biomed Life Sci 2005; 819:203 - 18; http://dx.doi.org/10.1016/j.jchromb.2004.06.052; PMID: 15833284
- Xie H, Chakraborty A, Ahn J, Yu YQ, Dakshinamoorthy DP, Gilar M, et al. Rapid comparison of a candidate biosimilar to an innovator monoclonal antibody with advanced liquid chromatography and mass spectrometry technologies. MAbs 2010; 2:379 - 94; PMID: 20458189
- Alvarez M, Tremintin G, Wang J, Eng M, Kao YH, Jeong J, et al. On-line characterization of monoclonal antibody variants by liquid chromatography-mass spectrometry operating in a two-dimensional format. Anal Biochem 2011; 419:17 - 25; http://dx.doi.org/10.1016/j.ab.2011.07.033; PMID: 21867674
- Michels DA, Tu AW, McElroy W, Voehringer D, Salas-Solano O. Charge heterogeneity of monoclonal antibodies by multiplexed imaged capillary isoelectric focusing immunoassay with chemiluminescence detection. Anal Chem 2012; 84:5380 - 6; http://dx.doi.org/10.1021/ac3008847; PMID: 22663182
- Kaschak T, Boyd D, Yan B. Characterization of glycation in an IgG1 by capillary electrophoresis sodium dodecyl sulfate and mass spectrometry. Anal Biochem 2011; 417:256 - 63; http://dx.doi.org/10.1016/j.ab.2011.06.024; PMID: 21756870
- Rustandi RR, Wang Y. Use of CE-SDS gel for characterization of monoclonal antibody hinge region clipping due to copper and high pH stress. Electrophoresis 2011; 32:3078 - 84; http://dx.doi.org/10.1002/elps.201100186; PMID: 22145164
- Moini M. Simplifying CE-MS operation. 2. Interfacing low-flow separation techniques to mass spectrometry using a porous tip. Anal Chem 2007; 79:4241 - 6; http://dx.doi.org/10.1021/ac0704560; PMID: 17447730
- Haselberg R, Ratnayake CK, de Jong GJ, Somsen GW. Performance of a sheathless porous tip sprayer for capillary electrophoresis-electrospray ionization-mass spectrometry of intact proteins. J Chromatogr A 2010; 1217:7605 - 11; http://dx.doi.org/10.1016/j.chroma.2010.10.006; PMID: 20970804
- Faserl K, Sarg B, Kremser L, Lindner H. Optimization and evaluation of a sheathless capillary electrophoresis-electrospray ionization mass spectrometry platform for peptide analysis: comparison to liquid chromatography-electrospray ionization mass spectrometry. Anal Chem 2011; 83:7297 - 305; http://dx.doi.org/10.1021/ac2010372; PMID: 21848273
- Busnel JM, Schoenmaker B, Ramautar R, Carrasco-Pancorbo A, Ratnayake C, Feitelson JS, et al. High capacity capillary electrophoresis-electrospray ionization mass spectrometry: coupling a porous sheathless interface with transient-isotachophoresis. Anal Chem 2010; 82:9476 - 83; http://dx.doi.org/10.1021/ac102159d; PMID: 21028888
- Ramautar R, Busnel JM, Deelder AM, Mayboroda OA. Enhancing the coverage of the urinary metabolome by sheathless capillary electrophoresis-mass spectrometry. Anal Chem 2012; 84:885 - 92; http://dx.doi.org/10.1021/ac202407v; PMID: 22148170
- Haselberg R, Harmsen S, Dolman MEM, de Jong GJ, Kok RJ, Somsen GW. Characterization of drug-lysozyme conjugates by sheathless capillary electrophoresis-time-of-flight mass spectrometry. Anal Chim Acta 2011; 698:77 - 83; http://dx.doi.org/10.1016/j.aca.2011.04.050; PMID: 21645662
- Whitmore CD, Gennaro LA. Capillary electrophoresis-mass spectrometry methods for tryptic peptide mapping of therapeutic antibodies. Electrophoresis 2012; 33:1550 - 6; http://dx.doi.org/10.1002/elps.201200066; PMID: 22736356
- Demeule B, Palais C, Machaidze G, Gurny R, Arvinte T. New methods allowing the detection of protein aggregates: a case study on trastuzumab. MAbs 2009; 1:142 - 50; http://dx.doi.org/10.4161/mabs.1.2.7632; PMID: 20061815
- Hudis CA. Trastuzumab--mechanism of action and use in clinical practice. N Engl J Med 2007; 357:39 - 51; http://dx.doi.org/10.1056/NEJMra043186; PMID: 17611206
- Carter P, Presta L, Gorman CM, Ridgway JBB, Henner D, Wong WL, et al. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci U S A 1992; 89:4285 - 9; http://dx.doi.org/10.1073/pnas.89.10.4285; PMID: 1350088
- Harris RJ, Kabakoff B, Macchi FD, Shen FJ, Kwong M, Andya JD, et al. Identification of multiple sources of charge heterogeneity in a recombinant antibody. J Chromatogr B Biomed Sci Appl 2001; 752:233 - 45; http://dx.doi.org/10.1016/S0378-4347(00)00548-X; PMID: 11270864
- Vlasak J, Bussat MC, Wang S, Wagner-Rousset E, Schaefer M, Klinguer-Hamour C, et al. Identification and characterization of asparagine deamidation in the light chain CDR1 of a humanized IgG1 antibody. Anal Biochem 2009; 392:145 - 54; http://dx.doi.org/10.1016/j.ab.2009.05.043; PMID: 19497295
- Gennaro LA, Salas-Solano O, Ma S. Capillary electrophoresis-mass spectrometry as a characterization tool for therapeutic proteins. Anal Biochem 2006; 355:249 - 58; http://dx.doi.org/10.1016/j.ab.2006.04.002; PMID: 16712766
- Beck A, Sanglier-Cianférani S, Van Dorsselaer A. Biosimilar, biobetter, and next generation antibody characterization by mass spectrometry. Anal Chem 2012; 84:4637 - 46; http://dx.doi.org/10.1021/ac3002885; PMID: 22510259
- Houde D, Peng Y, Berkowitz SA, Engen JR. Post-translational modifications differentially affect IgG1 conformation and receptor binding. Mol Cell Proteomics 2010; 9:1716 - 28; http://dx.doi.org/10.1074/mcp.M900540-MCP200; PMID: 20103567
- Liu H, Gaza-Bulseco G, Faldu D, Chumsae C, Sun J. Heterogeneity of monoclonal antibodies. J Pharm Sci 2008; 97:2426 - 47; http://dx.doi.org/10.1002/jps.21180; PMID: 17828757
- Jefferis R. Glycosylation of recombinant antibody therapeutics. Biotechnol Prog 2005; 21:11 - 6; http://dx.doi.org/10.1021/bp040016j; PMID: 15903235
- Beck A, Reichert JM. Marketing approval of mogamulizumab: a triumph for glyco-engineering. MAbs 2012; 4:1 - 7; http://dx.doi.org/10.4161/mabs.20996; PMID: 22327425
- Beck A, Wagner-Rousset E, Bussat MC, Lokteff M, Klinguer-Hamour C, Haeuw JF, et al. Trends in glycosylation, glycoanalysis and glycoengineering of therapeutic antibodies and Fc-fusion proteins. Curr Pharm Biotechnol 2008; 9:482 - 501; http://dx.doi.org/10.2174/138920108786786411; PMID: 19075687
- Beck A, Cochet O, Wurch T. GlycoFi’s technology to control the glycosylation of recombinant therapeutic proteins. Expert Opin Drug Discov 2010; 5:95 - 111; http://dx.doi.org/10.1517/17460440903413504; PMID: 22823974
- Damen CWN, Chen W, Chakraborty AB, van Oosterhout M, Mazzeo JR, Gebler JC, et al. Electrospray ionization quadrupole ion-mobility time-of-flight mass spectrometry as a tool to distinguish the lot-to-lot heterogeneity in N-glycosylation profile of the therapeutic monoclonal antibody trastuzumab. J Am Soc Mass Spectrom 2009; 20:2021 - 33; http://dx.doi.org/10.1016/j.jasms.2009.07.017; PMID: 19744865
- Wilm MS, Mann M. Electrospray and Taylor-Cone theory, Dole’s beam of macromolecules at last?. Int J Mass Spectrom Ion Process 1994; 136:167 - 80; http://dx.doi.org/10.1016/0168-1176(94)04024-9