
Abstract
Neurodegenerative diseases belong to a larger group of protein misfolding disorders, known as proteinopathies. There is increasing experimental evidence implicating prion-like mechanisms in many common neurodegenerative disorders, including Alzheimer disease, Parkinson disease, the tauopathies, and amyotrophic lateral sclerosis (ALS), all of which feature the aberrant misfolding and aggregation of specific proteins. The prion paradigm provides a mechanism by which a mutant or wild-type protein can dominate pathogenesis through the initiation of self-propagating protein misfolding. ALS, a lethal disease characterized by progressive degeneration of motor neurons is understood as a classical proteinopathy; the disease is typified by the formation of inclusions consisting of aggregated protein within and around motor neurons that can contribute to neurotoxicity. It is well established that misfolded/oxidized SOD1 protein is highly toxic to motor neurons and plays a prominent role in the pathology of ALS. Recent work has identified propagated protein misfolding properties in both mutant and wild-type SOD1, which may provide the molecular basis for the clinically observed contiguous spread of the disease through the neuroaxis. In this review we examine the current state of knowledge regarding the prion-like properties of SOD1 and comment on its proposed mechanisms of intercellular transmission.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neuromuscular condition characterized by degeneration of the upper and lower motor neurons causing progressive muscle paralysis and spasticity that affects mobility, speech, swallowing, and respiration.Citation1,Citation2 Half of affected individuals die within 3 years and less than 20% survive for more than 5 years.Citation3 The etiology of ALS is unknown; however, similar to other neurodegenerative diseases such as Alzheimer disease and Creutzfeldt-Jakob disease, 90–95% of cases are sporadic in which some predisposing gene mutations have been identified, such as those encoding ataxin-2 repeat expansions.Citation4 The remainder of cases are familial,Citation5 which are predominantly associated with Mendelian-inherited mutations in genes encoding Cu/Zn superoxide dismutase (SOD1), TAR-DNA binding protein 43 (TDP-43), fused in sarcoma/translocated in liposarcoma (FUS/TLS), and C9ORF72, but have been associated with other genes as well.Citation6-Citation9 Based on its pathobiology, ALS is considered a protein misfolding disorder, and as such is classified as a proteinopathy similar to other neurodegenerative diseases, including Alzheimer disease, Parkinson disease, other tauopathies, and the prion diseases. ALS patients typically feature abnormal accumulations of proteinaceous inclusions in motor neurons and neural accessory cells.
Mutations in SOD1, a ubiquitously-expressed gene encoding a free-radical scavenging enzyme, were the first genetic causes of ALS to be identifiedCitation10 and are implicated in ~20% of all familial ALS (FALS) cases; over 150 different disease-causing SOD1 mutations have been identified to date.Citation11-Citation13 Despite the intrinsic stability of the native SOD1 enzyme, the majority of these mutations induce misfolding and subsequent aggregation. Aggregate formation occurs through a mechanism by which the highly-stable native SOD1 homodimer is disrupted, thereby producing a misfolded monomeric intermediate that can be incorporated into higher-order oligomeric strucutres.Citation14-Citation16 However, genetic mutation is not the only way to destabilize, misfold, and aggregate SOD1. Aberrant oxidation or post-translational modification of wild-type (wt) SOD1 has been observed to mimic the aggregation-prone effects of mutant SOD1 in vitroCitation15,Citation17,Citation18 in a protein concentration-dependent manner.Citation15 SOD1-containing neural inclusions are typically detected in motor neurons from familial ALS patients,Citation19 and in transgenicCitation20 and tissue culture modelsCitation21 of the disease. There is increasing evidence that all types of ALS, including non-SOD1-linked familial and sporadic cases are associated with SOD1 misfolding, oxidation, and aggregation.Citation22,Citation23 Inclusions containing aggregated SOD1 have been detected in spinal cord tissues from both FALS and sporadic ALS (SALS) patientsCitation24-Citation26 in addition to biochemical, genetic and immunological evidence of misfolded SOD1 in cases of SOD1-excluded SALS.Citation27-Citation31 Misfolded SOD1 is therefore a prime candidate as a common molecular determinant for all forms of ALS and may play a key role in disease pathogenesis.
Native SOD1 scavenges cytotoxic superoxide radicals from the cytosol, converting them into less toxic hydrogen peroxide and oxygen.Citation32 Mutations in SOD1 often inhibit enzymatic function; however loss of superoxide dismutase function is not the cause of disease as SOD1 knockout mice do not develop an ALS-like motor neuron disease despite the build-up of superoxide radicals.Citation33 Instead, misfolding of SOD1 conferred by mutation, oxidation or other cell stresses is generally thought to acquire a toxic gain of function,Citation34-Citation36 although the precise nature of this toxicity and its specificity for motor neurons remains to be elucidated. Moreover, in its misfolded pathological form SOD1 can react non-specifically with a variety of substrates, causing it to become a net producer of reactive oxygen and nitrogen species,Citation37 as opposed to a free-radical scavenger, which can subsequently lead to intracellular damage of cellular protein, lipid, and nucleic acid.Citation38,Citation39 Toxicity of misfolded SOD1 has also been attributed to diverse deleterious effects including: cytoskeletal disruption, caspase activation, microglial activation, and proteasomal and autophagy pathway disruption.Citation40-Citation42
A significant clinical feature of ALS is its contiguous spread through the neuroaxis; pathology starts at a focal point of onset and spreads in a spatiotemporal fashion through adjacent neuroanatomical regions.Citation43,Citation44 Given the similarities in disease progression and anatomical spread between other neurodegenerative proteinopathies, including the classical prion diseases, a likely mechanism to account for this observation that has gained traction in recent years is the region to region prion-like spread of propagated protein misfolding. Indeed, the model of prion-like spread of misfolding fits the clinical pattern of ALS pathology and has previously been implicated in other relatively common neurodegenerative proteinopathies of aging, such as Parkinson diseaseCitation45 and Alzheimer disease.Citation46 The mechanism also accounts for how a mutant or even wild-type protein can dominate pathogenesis of a phenotypically diverse disease such as ALS, akin to when the normal cellular isoform of the prion protein (PrPC) converts to its pathological isoform PrPSc in prion disease. In this review, we will summarize the evidence for prion-like propagation of misfolded SOD1, the molecular mechanism by which this process can occur, and consider the pathways by which misfolded SOD1 transmits from cell-to-cell.
Evidence for the Molecular Conversion of SOD1
The requisite characteristic of prion-like activity on a molecular level is, at its core, defined by the ‘protein-only’ hypothesis that stipulates misfolded protein alone is necessary and sufficient to catalyze misfolding of native protein, without the requirement for any co-factors.Citation47 Although not yet as experimentally scrutinized as the prion protein, a considerable body of experimental evidence has accumulated in the literature indicating that misfolded SOD1 has the ability to transfer conformational information from one molecule to another, meeting some of the requirements of prion-like activity at the molecular level. SOD1, a small soluble ubiquitously-expressed 153-residue protein, exists as a dimer in its functional form. The native holo-enzyme contains an intramolecular disulfide bond within each monomer, which contributes to its high conformational stability and resistance to proteolytic digestion.Citation48 Despite this, SOD1 is highly prone to destabilization when mutated or aberrantly oxidized; over 150 pathological mutations have been identified within SOD1 affecting nearly every amino acid. In its mutated or modified state SOD1 has a high propensity to misfold and form oligomers and aggregates. Indeed, under denaturing conditions wild-type and mutant forms of SOD1 can spontaneously form aggregates and fibrils in vitro,Citation49 where the relative propensity for aggregation is dependent upon the variant of SOD1.Citation50 Indications of in vivo SOD1 fibril formation have been observed in transgenic mice expressing mutant SOD1Citation51,Citation52; however, the formation of highly-structured amyloid fibrils does not appear to be a consistent feature of SOD1 in human disease.Citation53
Intramolecular conversion of native wtSOD1 to a pathological form has been previously observed in a series of in vivo studies. Exacerbation of motor neuron disease in mutant SOD1 mouse models was observed upon co-expression of human wtSOD1, suggesting accelerated disease progression through molecular association with mutant SOD1,Citation54,Citation55 possibly through stabilization of mutant species through interaction with wtSOD1Citation56 or through the formation of non-native disulfide interactions that induce insoluble aggregate formation.Citation57 Coaggregation of mutant and wild-type SOD1 has also been detected in post-mortem tissue from familial ALS patients,Citation58 making intermolecular conversion in humans plausible and pathologically relevant. Direct evidence of intermolecular conversion of wtSOD1 to a misfolded isoform has been observed in human cell lines.Citation31,Citation59 Co-expression of misfolded human SOD1 mutants can confer a misfolded conformation on endogenous wtSOD1 that is revealed by conformation-specific antibodies whose epitopes are only accessible when the protein is misfolded; induced misfolding by mutant SOD1 results in enhanced protease sensitivity, an indicator of global loosening of the polypeptide backbone. Conformational conversion of wtSOD1 by a mutant conformer can also occur in a cell-free system, thus eliminating any unidentified tangential protein, lipid or nucleic acid factors from the process.Citation59 Interestingly, the intermolecular conversion of wtSOD1 to a misfolded form appears dependent upon the exposure of a single amino-acid residue, a tryptophan (W) at position 32,Citation59 indicating a physical point of contact between the converting and the converted species that is separate from the dimer interface region. W32 is the only tryptophan in the wild-type human protein sequence and has previously been identified as a site of oxidative modification and a potentiator of aggregation.Citation60 SOD1 constructs featuring ALS-causing mutations, but with an additional W32S substitution failed to induce wtSOD1 misfolding in human cell linesCitation59 indicating that sequence or structural requirements are required for efficient conversion to take place, a characteristic reminiscent of the ‘species barrier’ in prion diseases such as Chronic Wasting Disease, a transmissible spongiform encephalopathy that is thus far restricted to certain species of deer and elk.Citation61 Indeed, tryptophan is substituted for a serine at position-32 in mouse SOD1, which appears inert in the conversion process of human SOD1. Further evidence of the ‘species barrier’ is seen in vivo as wild-type human SOD1 does not accelerate motor neuron disease in mice expressing murine SOD1 with the G86R mutation.Citation62 Furthermore, murine SOD1 is not incorporated into aggregates of human mutant SOD1.Citation55,Citation57,Citation63 It is clear from this that SOD1 belongs to a distinct collection of proteins with intermolecular characteristics reminiscent of PrP.
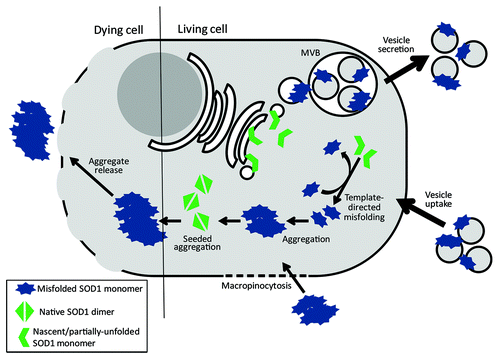
Figure 1. Intermolecular conversion and intercellular transmission of misfolded SOD1. Intermolecular conversion of wild-type SOD1 likely occurs via two possible mechanisms. Nascently-translated SOD1 that is presumably still partially unfolded are substrates for template-directed conversion by pathological forms of SOD1. Conversion of nascent SOD1 to a misfolded isoform provides template for further rounds of native SOD1 conversion. Additionally, homodimeric wild-type SOD1 (or soluble forms of misfolded SOD1) can be misfolded and incorporated into pre-existing insoluble aggregates of misfolded SOD1 to form larger complexes. Intercellular transmission of misfolded SOD1 isoforms also occurs via two possible mechanisms. Soluble forms of misfolded SOD1 (likely monomer and small oligomers) are packaged via the endocytic pathway into multivesicular bodies (MVBs); MVBs fuse with the plasma membrane thus releasing vesicles, such as exosomes, to the extracellular environment. Furthermore, exosomes, with misfolded SOD1 presented on the outer surface, are subsequently taken by neighboring cells. This process likely occurs between living cells in early stages of ALS. Conversely, SOD1 aggregate release likely occurs during neuronal cell death as plasma membrane integrity diminishes. Uptake of SOD1 aggregates occurs via macropinocytosis; however it remains unknown if this process is non-specific or is regulated by cell-surface receptors.
Self-Propagation of SOD1 Misfolding
A key aspect to the prion mechanism of misfolded SOD1 is the ability of the protein to impart its misfold onto wild-type protein, which in turn can provide new template for additional rounds of misfolding. Induced misfolding of SOD1 was shown to persist even in the absence of the misfolded ‘seed’ in cell culture,Citation59,Citation64 suggesting that newly misfolded polypeptide can act as template for subsequent cycles of SOD1 misfolding—an observation reminiscent of the definitive property of pathological prion protein. Two mechanisms have been proposed to account for this conversion process: nucleation- or seeded-polymerization, in which the misfolded monomeric species is intrinsically less stable as a monomer but becomes more stable than its native counterpart when recruited to a multimeric aggregate; and template-directed misfolding, in which the misfolded pathological conformer is more stable than the native form but is kinetically inaccessible without catalysis by interaction with the misfolded form.Citation65 Evidence for both mechanisms has been observed and likely suggests that propagated SOD1 misfolding of both mutant and wtSOD1 span a continuum between the two models of prion-like propagation (summarized in ). The work by Grad et al. suggests that the intracellular conversion of wtSOD1 to a misfolded conformer likely occurs in a template-directed fashion. Structural loosening of endogenous wtSOD1 is induced by contact with overexpressed wild-type misfolded template, but results in limited generation of non-native disulfide interactions and remains soluble, thus it is unlikely to be recruited to aggregates in human cell linesCitation59 demonstrating that soluble misfolded conformers of SOD1 can induce propagated protein misfolding in addition to insoluble aggregate seeds. Given the intrinsic stability of homodimeric native SOD1, and that co-expression of wtSOD1 can actually stabilize mutant misfolded SOD1,Citation66,Citation67 it is possible that nascent wtSOD1 polypeptide that has yet to be correctly folded likely represents the most thermodynamically favorable substrate for template-directed misfolding, although this remains to be experimentally verified.
Seeded polymerization, also called fibrillar aggregation, is a phenomenon inherent to several proteinopathies, including the prion diseases,Citation68 Alzheimer diseaseCitation69,Citation70 and Parkinson disease.Citation71 Similar characteristics have been observed for SOD1 both in vitro and in cell culture, however the propensity to aggregate is dependent upon mutation or other structural modification,Citation50 and directly related to the relevant number of hydrophobic residues exposed due to the resulting conformational changes.Citation72 Aggregation propensity and structural stability of SOD1 mutants also plays a pivotal role in disease progression and patient survival.Citation73 Crystal structures from recombinant metal-deficient mutant SOD1 proteins reveal higher-order assemblies of aligned β-sheets forming amyloid-like filaments and water-filled nanotubes.Citation74 Recombinant apo-SOD1 without disulphide bonds, ALS mutant SOD1 protein or insoluble SOD1-containing aggregates isolated from transgenic mouse models of ALS expressing mutant SOD1 all have been shown to possess spontaneous fibrillization and seeding activity in vitro under various non-physiological conditions.Citation49,Citation52 Exogenously-applied aggregated mutant human SOD1 can induce subsequent aggregation of soluble transgenically-expressed mutant SOD1 in mouse neuroblastoma cells.Citation64 Similarly, exogenous fibrils of recombinant human mutant SOD1 can be taken up by mouse neuroblastoma cells expressing the same human SOD1 mutant and trigger intracellular aggregation.Citation75 Combined, these investigations clearly demonstrate behavior consistent with self-propagating misfolding and aggregation analogous to that which underlies the generation of pathological prions.
Intercellular Transmission of SOD1 Misfolding
It is clear that the intercellular spread of misfolding is a prevalent feature of neurodegenerative disease, with the distribution of misfolded protein seeds playing the critical role of expanding the pathology beyond the site of original involvement. A misfolded protein that cannot escape the local environment in which it was formed has no way to effectively transmit the misfold to other regions, therefore the misfolded protein must be transported in a way that allows it to spread from cell to cell and region to region. Indeed, intercellular spread of propagated protein misfolding and aggregation of pathogenic PrPSc is a hallmark of the prion disases. As described above, release and uptake of protein aggregates or proto-fibrils is one possible mechanism for intercellular spread in ALS, and likely occurs when dying neurons release their contents to the extracellular milieu. Exogenous mutant and wtSOD1 aggregates have been shown to efficeintly penetrate the cell membrane of neuron-like cells in a macropinocytosis-dependent mechanism,Citation64,Citation76 and become self-perpetuating in recruiting soluble SOD1 into insoluble aggregates. Aggregates of misfolded SOD1 likely represent a conformationally robust and thermodynamically stable formCitation77 that can better surive the relative hostilities of the extracellular evnironment than smaller soluble forms. Direct exposure of misfolded SOD1 to the extracellular space is an important characteristic as it allows for access of potential therapeutic molecules to recognize and bind directly to pathological protein, therby blocking its contact with native SOD1 substrate or targeting it for subsequent destruction. Indeed, intercellular propagated misfolding of SOD1 can be abrogated in the presence of misfolding-specific SOD1 anitbody.Citation76 Although intercellular seeded polymerization via large SOD1 aggregates and fibrils is an observable phenomenon, there is a question as to the pathological relevance of such a process in regards to ALS. In other neurodegenerative diseases, there is growing evidence pointing to soluble misfolded protein as the toxic species as opposed to large protein aggregates, inclusions or fibrils, which appear for the most part to be pathologically inert.Citation78 In Alzheimer disease, it is smaller oliogomeric forms of amyloid-β that impair synapic function,Citation79,Citation80 plasticity, and memory.Citation81 Likewise, in Parkinson disease there is growing evidence that pre-fibrillar oligomers of α-synuclein are responsible for disease progressionCitation82,Citation83 as opposed to the hallmark Lewy bodies, which may simply serve as benign reservoirs of aggregated protein. It is highly suggestive that a similar paradigm exists for SOD1 misfolding in ALS, which could explain why misfolded SOD1 is detectable in sporadic cases, but not typically associated with large protein inclusions usually observed in mutant SOD1-linked familial ALS.Citation29,Citation30,Citation76 Therefore, another mechanism is likely necessary for efficient cell-to-cell transport of soluble misfolded SOD1 species through the extracellular milieu.
Before neuronal cell death ensues in ALS or other neurodegenerative diseases, pathology can spread from cell to cell and region to region,Citation44 suggesting a cellular mechanism that may be more relevant to early stages of the disease. A highly plausible means by which this could occur is through small 30–80 nm-wide membrane-bounded vesicles, called exosomes, which normally contain and transport various proteins and nucleic acids between cells. Exosomes are formed within intracellular vesicles, or endosomes, by membrane invagination, resulting in the formation of multivesicular bodies (MVBs). MVBs are normally involved in the trafficking of proteins to the lysosomes for degradation. Alternatively, MVBs can fuse with the plasma membrane, leading to the release of its intraluminal vesicles into the extracellular environment; these are defined as exosomes.Citation84 The exosome secretion pathway is associated with numerous pathogen-associated proteins, often resulting in viral spread.Citation85-Citation87 In addition, exosomes have been increasingly implicated in the spread of pathogenic proteins involved in neurodegenerative diseases, such as Alzheimer disease,Citation88 Parkinson disease,Citation89,Citation90 and the prion diseases.Citation91,Citation92 The evidence supporting an analogous mechanism involved in pathogenic SOD1 transmission is ever-increasing. SOD1 itself is a well-known exosome resident protein from multiple cell types and species.Citation93 In SOD1-related ALS, the secretory pathway is thought to be compromised resulting in the secretion of mutant SOD1.Citation94,Citation95 Subsequent studies have identified exosomes as the secretion mechanism for both wild-type and mutant SOD1 in a motor neuron-like cell model,Citation96 potentially mediated by chromogranins, components of secretory vesicles that could serve as a secretory chaperone.Citation97 Further evidence suggests that mutant SOD1 oligomers accumulate in the endoplasmic reticulum-Golgi compartments of the endocytic pathway prior to their subsequent secretion.Citation98 More recent work has established that both mutant and wild-type misfolded SOD1 can be secreted from neuron-like cells via exosomesCitation99; these exosomes can then be subsequently taken up by fresh cells where the misfolded SOD1 cargo provides a template for subsequent induction of protein misfolding ().Citation76 The observation that wild-type SOD1 is capable of facilitating propagated SOD1 misfolding further implicates the protein as a common pathological target for ALS regardless of mutation within SOD1 itselfCitation30,Citation100 and may provide the means by which the disease contiguously spreads in cases where SOD1 is not mutated.Citation59 Interestingly, misfolded SOD1 is immunologically detectable on the outer leaflet of extracellular vesicle membranes,Citation76 providing accessibility to potential therapeutic molecules similar to non-vesicle associated aggregates of SOD1, although the reason for this is still unknown. Furthermore, the participation of the exosomal secretory pathway in intercellular transmission of misfolded SOD1 allows not only for the direct extracellular targeting of misfolded SOD1 that can effectively disrupt the physical contact between the converting species and its substrate, but also for the targeting of cellular processes that are involved in the packaging of misfolded SOD1 inside the cell, and its subsequent secretion via extracellular vesicles.
The evidence supporting an exosome-mediated mechanism facilitating intercellular transmission of propagated SOD1 misfolding continues to build; however, the link between this mechanism and ALS pathology is more tenuous. To address this, more physiologically-relevant models have been utilized in order to relate exosome-mediated misfolded SOD1 transmission back to those cell-types directly affected by ALS, namely motor neurons and their accessory cells. Secreted mutant SOD1 is observed to have deleterious effects as demonstrated by induced microgliosis and increased motor neuron death in co-cultures.Citation97 More recently, mouse astrocyte-derived exosomes were observed to efficiently transfer mutant SOD1 to spinal neurons and subsequently cause selective motor neuron death,Citation99 a further exemplar of the non-cell autonomous nature of ALS and evidence that transmitted misfolded SOD1 can be pathogenic. Similarly, astrocytes derived from familial and sporadic ALS patients were toxic to motor neurons derived from mouse embryonic stem cells.Citation101 Interestingly, suppression of SOD1 expression in both FALS and SALS-derived astrocytes attenuates motor neuron toxicity, highly suggestive of SOD1-mediated neurotoxicity. However, the results of the study conflict with a prion-like mode of action as the affected motor neurons were derived from wild-type non-transgenic mice; mouse SOD1 has been shown to be resistant to propagated protein misfolding when induced by misfolded human SOD1.Citation59 Therefore the observations by Haidet-Phillips et al. imply an additional SOD1-associated factor may be responsible for neurotoxicity in their cultured neuronal cell system; however, there remains to be explained the self-propagating region to region spread of neurotoxicity that perpetuates once an initial pathological event has occurred. Needless to say, future work is needed to solidify the connection between propagated SOD1 misfolding, neurotoxicity and the contiguous spread of pathology within the neuroaxis of ALS patients.
Perspectives: Is ALS a Prion Disease?
The intercellular transmission of self-perpetuating protein misfolding now appears to be a prevalent mechanism in neurodegenerative diseases of aging, and one that explains the relatively rare frequency of ALS within the population and the progressive nature of its disease biology. The contiguous spatiotemporal spread of pathology and the demonstrated intercellular passage of misfolded SOD1 argue that the event precipitating disease is endogenous spontaneous misfolding arising from a rare conformational change of a susceptible protein. However, can ALS, or any of the other non-prion protein neurodegenerative diseases mentioned in this review, be truly considered an authentic prion disease? One would argue that despite fulfilling certain required characteristics of pathological prion proteins, such as evidence of protein recruitment and misfolding conversion, intercellular transmission, and tissue migration, SOD1 and other analogous “prion-like” proteins involved in neurodegenerative proteinopathies do not share other key traits synonymous with the classical prion diseases, namely transmission between organismsCitation77 and the evidence of strains that vary in their infectivity and toxicity.Citation102 Furthermore, there is little evidence in the literature thus far that firmly cements the “protein-only” hypothesis in any of the other non-classical prion diseases under physiological conditions; the lone exception may be for SOD1 as template-directed misfolding was observed in a completely cell-free environment using purified recombinant proteinCitation59; however, the reaction was dependent upon a buffer composition that only mimicked the reducing and metal cation-buffering capacity of the cytosol, which may not replicate the intracellular milieu exactly.
When it comes to infectivity, no other propagated misfolded protein comes close to the robustness of PrPSc in prion disease. To date, there is no published experimental evidence demonstrating that any neurodegenerative proteinopathy, other than the classical prion diseases, can spread from individual to individual in humans or experimental model systems via natural infection pathways such as oral or intravenous inoculation. In fact, a recent study firmly states that person-to-person transmissibility in Alzheimer and Parkinson diseases is highly unlikely to occur.Citation103 However, one cannot ignore the more demonstrative observations of facilitated trans-organism disease transmission when intraperitoneal inoculation of misfolded amyloid-β-enriched brain extract from Alzheimer patients into transgenic mice induces amyloidosis in the brain, a region significantly distal to the site of injection.Citation104 It therefore seems that the point of entry of other “prion-like” agents is crucial for disease propagation, indicating a relatively weak robustness of transmission compared with PrPSc, the prototypical highly-infectious prion. Sensitivity to the extracellular environment may explain the lack of evidence thus far for organism-to-organism spread of ALS, at least via misfolded SOD1. Unlike PrPSc, which is a highly-stable and protease-resistant isoform compared with normal PrPC and represents the epitome of a transmissible pathological protein-only agent, many pathological species of misfolded SOD1 become increasingly protease-sensitive and unstable.Citation48,Citation59 This would make a misfolded SOD1-mediated “infectious particle” less likely to stand up to the environmental rigours of person-to-person transmission, although the presence of native wild-type SOD1 has been shown to increase the stability of mutant misfolded SOD1.Citation105 Other factors that may confound interorganism “infection” of SOD1 misfolding include misfolding dynamics, i.e. it simply takes too long for suitable levels of misfolded template to replicate to levels that facilitate infection, and host attributes that may simply be incompatible for misfolded SOD1 transmission between individuals. However, SOD1 does appear to meet the intramolecular, intermolecular, and intercellular requirements associated with prions. Taken together, the literature would suggest that the pathological agents of neurodegenerative proteinopathies could all be considered “prions” with transmissibility and toxicity characteristics falling within a wide spectrum, with PrPSc representing the one end (highly transmissible, highly toxic) and proteins associated with more relatively common neurodegenerative diseases, such as SOD1, amyloid-β or α-synuclein, on the other end representing less robust infectious agents, but with the ability to induce protein misfolding and perpetuate it from cell to cell.
There is no doubt that further in vivo characterization of misfolded SOD1 transmissibility is required to determine the risks, if any, for interorganism infection, and how much of a role misfolded SOD1 propagation plays in disease progression and pathology. ALS itself features other misfolded/aggregated proteins associated with pathology, such as TDP-43, FUS, and C9ORF72, that may also play a role in disease spread and transmission. Recently, TDP-43, a nuclear-expressed RNA-binding protein found in large cytosolic inclusions in the brains of patients with ALS and frontotemporal dementia, was also found to induce seed-dependent aggregation in living cells.Citation106,Citation107 This recent data extends the prion paradigm of ALS to new levels and compounds the difficulty of elucidating the precise molecular mechanisms governing the pathogenesis and spread of the disease.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
NRC is the Canada Research Chair in Neurodegeneration and Protein Misfolding Diseases at the University of British Columbia, and is supported by donations from the Webster Foundation, the Allen T. Lambert Neural Research Fund and the Temerty Family Foundation, and also by grants from PrioNet Canada, the Canadian Institutes of Health Research (CIHR), and Biogen-Idec Corp.
References
- Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci 2001; 2:806 - 19; http://dx.doi.org/10.1038/35097565; PMID: 11715057
- Bradley WG. Updates on amyotrophic lateral sclerosis: improving patient care. Ann Neurol 2009; 65:Suppl 1 S1 - 2; http://dx.doi.org/10.1002/ana.21546; PMID: 19191303
- Strong M, Rosenfeld J. Amyotrophic lateral sclerosis: a review of current concepts. Amyotroph Lateral Scler Other Motor Neuron Disord 2003; 4:136 - 43; http://dx.doi.org/10.1080/14660820310011250; PMID: 13129799
- Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010; 466:1069 - 75; http://dx.doi.org/10.1038/nature09320; PMID: 20740007
- Haverkamp LJ, Appel V, Appel SH. Natural history of amyotrophic lateral sclerosis in a database population. Validation of a scoring system and a model for survival prediction. Brain 1995; 118:707 - 19; http://dx.doi.org/10.1093/brain/118.3.707; PMID: 7600088
- Mutation Database ALS. http://reseq.biosciencedbc.jp/resequence/SearchDisease.do?targetId=1. The University of Tokyo, 2007.
- Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, Shi Y, Zhai H, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011; 477:211 - 5; http://dx.doi.org/10.1038/nature10353; PMID: 21857683
- Wu CH, Fallini C, Ticozzi N, Keagle PJ, Sapp PC, Piotrowska K, Lowe P, Koppers M, McKenna-Yasek D, Baron DM, et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 2012; 488:499 - 503; http://dx.doi.org/10.1038/nature11280; PMID: 22801503
- Stewart H, Rutherford NJ, Briemberg H, Krieger C, Cashman N, Fabros M, Baker M, Fok A, DeJesus-Hernandez M, Eisen A, et al. Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol 2012; 123:409 - 17; http://dx.doi.org/10.1007/s00401-011-0937-5; PMID: 22228244
- Rosen DR. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993; 364:362; http://dx.doi.org/10.1038/364362c0; PMID: 8332197
- Andersen PM. Genetic factors in the early diagnosis of ALS. Amyotroph Lateral Scler Other Motor Neuron Disord 2000; 1:Suppl 1 S31 - 42; http://dx.doi.org/10.1080/14660820052415899; PMID: 11464924
- Robberecht W. Genetics of amyotrophic lateral sclerosis. J Neurol 2000; 247:2 - 6; http://dx.doi.org/10.1007/PL00007785; PMID: 11200702
- Andersen PM, Sims KB, Xin WW, Kiely R, O’Neill G, Ravits J, Pioro E, Harati Y, Brower RD, Levine JS, et al. Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: a decade of discoveries, defects and disputes. Amyotroph Lateral Scler Other Motor Neuron Disord 2003; 4:62 - 73; http://dx.doi.org/10.1080/14660820310011700; PMID: 14506936
- Cashman NR, Chakrabartty A, Rakhit R, Ostermann JB. EP 2495327 A3. Methods and compositions to treat and detect misfolded SOD1 mediated diseases 2007.
- Rakhit R, Crow JP, Lepock JR, Kondejewski LH, Cashman NR, Chakrabartty A. Monomeric Cu,Zn-superoxide dismutase is a common misfolding intermediate in the oxidation models of sporadic and familial amyotrophic lateral sclerosis. J Biol Chem 2004; 279:15499 - 504; http://dx.doi.org/10.1074/jbc.M313295200; PMID: 14734542
- Rakhit R, Robertson J, Vande Velde C, Horne P, Ruth DM, Griffin J, Cleveland DW, Cashman NR, Chakrabartty A. An immunological epitope selective for pathological monomer-misfolded SOD1 in ALS. Nat Med 2007; 13:754 - 9; http://dx.doi.org/10.1038/nm1559; PMID: 17486090
- Rakhit R, Cunningham P, Furtos-Matei A, Dahan S, Qi XF, Crow JP, Cashman NR, Kondejewski LH, Chakrabartty A. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J Biol Chem 2002; 277:47551 - 6; http://dx.doi.org/10.1074/jbc.M207356200; PMID: 12356748
- Casoni F, Basso M, Massignan T, Gianazza E, Cheroni C, Salmona M, Bendotti C, Bonetto V. Protein nitration in a mouse model of familial amyotrophic lateral sclerosis: possible multifunctional role in the pathogenesis. J Biol Chem 2005; 280:16295 - 304; http://dx.doi.org/10.1074/jbc.M413111200; PMID: 15699043
- Kato S, Takikawa M, Nakashima K, Hirano A, Cleveland DW, Kusaka H, Shibata N, Kato M, Nakano I, Ohama E. New consensus research on neuropathological aspects of familial amyotrophic lateral sclerosis with superoxide dismutase 1 (SOD1) gene mutations: inclusions containing SOD1 in neurons and astrocytes. Amyotroph Lateral Scler Other Motor Neuron Disord 2000; 1:163 - 84; http://dx.doi.org/10.1080/14660820050515160; PMID: 11464950
- Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 1997; 18:327 - 38; http://dx.doi.org/10.1016/S0896-6273(00)80272-X; PMID: 9052802
- Durham HD, Roy J, Dong L, Figlewicz DA. Aggregation of mutant Cu/Zn superoxide dismutase proteins in a culture model of ALS. J Neuropathol Exp Neurol 1997; 56:523 - 30; http://dx.doi.org/10.1097/00005072-199705000-00008; PMID: 9143265
- Matias-Guiu J, Galan L, Garcia-Ramos R, Barcia JA. Superoxide dismutase: the cause of all amyotrophic lateral sclerosis?. Ann Neurol 2008; 64:356 - 7, author reply 358; http://dx.doi.org/10.1002/ana.21373; PMID: 18350588
- Synofzik M, Ronchi D, Keskin I, Basak AN, Wilhelm C, Gobbi C, Birve A, Biskup S, Zecca C, Fernández-Santiago R, et al. Mutant superoxide dismutase-1 indistinguishable from wild-type causes ALS. Hum Mol Genet 2012; 21:3568 - 74; http://dx.doi.org/10.1093/hmg/dds188; PMID: 22595972
- Shibata N, Hirano A, Kobayashi M, Sasaki S, Kato T, Matsumoto S, Shiozawa Z, Komori T, Ikemoto A, Umahara T, et al. Cu/Zn superoxide dismutase-like immunoreactivity in Lewy body-like inclusions of sporadic amyotrophic lateral sclerosis. Neurosci Lett 1994; 179:149 - 52; http://dx.doi.org/10.1016/0304-3940(94)90956-3; PMID: 7845611
- Chou SM, Wang HS, Komai K. Colocalization of NOS and SOD1 in neurofilament accumulation within motor neurons of amyotrophic lateral sclerosis: an immunohistochemical study. J Chem Neuroanat 1996; 10:249 - 58; http://dx.doi.org/10.1016/0891-0618(96)00137-8; PMID: 8811414
- Chou SM, Wang HS, Taniguchi A. Role of SOD-1 and nitric oxide/cyclic GMP cascade on neurofilament aggregation in ALS/MND. J Neurol Sci 1996; 139:Suppl 16 - 26; http://dx.doi.org/10.1016/0022-510X(96)00090-1; PMID: 8899653
- Gruzman A, Wood WL, Alpert E, Prasad MD, Miller RG, Rothstein JD, Bowser R, Hamilton R, Wood TD, Cleveland DW, et al. Common molecular signature in SOD1 for both sporadic and familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 2007; 104:12524 - 9; http://dx.doi.org/10.1073/pnas.0705044104; PMID: 17636119
- Broom WJ, Greenway M, Sadri-Vakili G, Russ C, Auwarter KE, Glajch KE, Dupre N, Swingler RJ, Purcell S, Hayward C, et al. 50bp deletion in the promoter for superoxide dismutase 1 (SOD1) reduces SOD1 expression in vitro and may correlate with increased age of onset of sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2008; 9:229 - 37; http://dx.doi.org/10.1080/17482960802103107; PMID: 18608091
- Forsberg K, Jonsson PA, Andersen PM, Bergemalm D, Graffmo KS, Hultdin M, Jacobsson J, Rosquist R, Marklund SL, Brännström T. Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS One 2010; 5:e11552; http://dx.doi.org/10.1371/journal.pone.0011552; PMID: 20644736
- Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, Goolsby H, Fontaine BA, Lemay N, McKenna-Yasek D, et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci 2010; 13:1396 - 403; http://dx.doi.org/10.1038/nn.2660; PMID: 20953194
- Pokrishevsky E, Grad LI, Yousefi M, Wang J, Mackenzie IR, Cashman NR. Aberrant localization of FUS and TDP43 is associated with misfolding of SOD1 in amyotrophic lateral sclerosis. PLoS One 2012; 7:e35050; http://dx.doi.org/10.1371/journal.pone.0035050; PMID: 22493728
- Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol 1996; 271:C1424 - 37; PMID: 8944624
- Shefner JM, Reaume AG, Flood DG, Scott RW, Kowall NW, Ferrante RJ, Siwek DF, Upton-Rice M, Brown RH Jr.. Mice lacking cytosolic copper/zinc superoxide dismutase display a distinctive motor axonopathy. Neurology 1999; 53:1239 - 46; http://dx.doi.org/10.1212/WNL.53.6.1239; PMID: 10522879
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994; 264:1772 - 5; http://dx.doi.org/10.1126/science.8209258; PMID: 8209258
- Andersen PM, Nilsson P, Ala-Hurula V, Keränen ML, Tarvainen I, Haltia T, Nilsson L, Binzer M, Forsgren L, Marklund SL. Amyotrophic lateral sclerosis associated with homozygosity for an Asp90Ala mutation in CuZn-superoxide dismutase. Nat Genet 1995; 10:61 - 6; http://dx.doi.org/10.1038/ng0595-61; PMID: 7647793
- Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH Jr., et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet 1996; 13:43 - 7; http://dx.doi.org/10.1038/ng0596-43; PMID: 8673102
- Said Ahmed M, Hung WY, Zu JS, Hockberger P, Siddique T. Increased reactive oxygen species in familial amyotrophic lateral sclerosis with mutations in SOD1. J Neurol Sci 2000; 176:88 - 94; http://dx.doi.org/10.1016/S0022-510X(00)00317-8; PMID: 10930589
- Beckman JS, Carson M, Smith CD, Koppenol WH. ALS, SOD and peroxynitrite. Nature 1993; 364:584; http://dx.doi.org/10.1038/364584a0; PMID: 8350919
- Beckman JS, Estévez AG, Crow JP, Barbeito L. Superoxide dismutase and the death of motoneurons in ALS. Trends Neurosci 2001; 24:Suppl S15 - 20; http://dx.doi.org/10.1016/S0166-2236(00)01981-0; PMID: 11881740
- Cleveland DW, Liu J. Oxidation versus aggregation - how do SOD1 mutants cause ALS?. Nat Med 2000; 6:1320 - 1; http://dx.doi.org/10.1038/82122; PMID: 11100110
- Bendotti C, Marino M, Cheroni C, Fontana E, Crippa V, Poletti A, De Biasi S. Dysfunction of constitutive and inducible ubiquitin-proteasome system in amyotrophic lateral sclerosis: implication for protein aggregation and immune response. Prog Neurobiol 2012; 97:101 - 26; http://dx.doi.org/10.1016/j.pneurobio.2011.10.001; PMID: 22033150
- Bard F, Fox M, Friedrich S, Seubert P, Schenk D, Kinney GG, Yednock T. Sustained levels of antibodies against Aβ in amyloid-rich regions of the CNS following intravenous dosing in human APP transgenic mice. Exp Neurol 2012; 238:38 - 43; http://dx.doi.org/10.1016/j.expneurol.2012.07.022; PMID: 22892246
- Ravits J, Paul P, Jorg C. Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology 2007; 68:1571 - 5; http://dx.doi.org/10.1212/01.wnl.0000260965.20021.47; PMID: 17485643
- Ravits JM, La Spada AR. ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology 2009; 73:805 - 11; http://dx.doi.org/10.1212/WNL.0b013e3181b6bbbd; PMID: 19738176
- Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012; 338:949 - 53; http://dx.doi.org/10.1126/science.1227157; PMID: 23161999
- Eisele YS, Bolmont T, Heikenwalder M, Langer F, Jacobson LH, Yan ZX, Roth K, Aguzzi A, Staufenbiel M, Walker LC, et al. Induction of cerebral beta-amyloidosis: intracerebral versus systemic Abeta inoculation. Proc Natl Acad Sci U S A 2009; 106:12926 - 31; http://dx.doi.org/10.1073/pnas.0903200106; PMID: 19622727
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 44; http://dx.doi.org/10.1126/science.6801762; PMID: 6801762
- Ratovitski T, Corson LB, Strain J, Wong P, Cleveland DW, Culotta VC, Borchelt DR. Variation in the biochemical/biophysical properties of mutant superoxide dismutase 1 enzymes and the rate of disease progression in familial amyotrophic lateral sclerosis kindreds. Hum Mol Genet 1999; 8:1451 - 60; http://dx.doi.org/10.1093/hmg/8.8.1451; PMID: 10400992
- Chia R, Tattum MH, Jones S, Collinge J, Fisher EM, Jackson GS. Superoxide dismutase 1 and tgSOD1 mouse spinal cord seed fibrils, suggesting a propagative cell death mechanism in amyotrophic lateral sclerosis. PLoS One 2010; 5:e10627; http://dx.doi.org/10.1371/journal.pone.0010627; PMID: 20498711
- Prudencio M, Hart PJ, Borchelt DR, Andersen PM. Variation in aggregation propensities among ALS-associated variants of SOD1: correlation to human disease. Hum Mol Genet 2009; 18:3217 - 26; http://dx.doi.org/10.1093/hmg/ddp260; PMID: 19483195
- Wang J, Xu G, Gonzales V, Coonfield M, Fromholt D, Copeland NG, Jenkins NA, Borchelt DR. Fibrillar inclusions and motor neuron degeneration in transgenic mice expressing superoxide dismutase 1 with a disrupted copper-binding site. Neurobiol Dis 2002; 10:128 - 38; http://dx.doi.org/10.1006/nbdi.2002.0498; PMID: 12127151
- Furukawa Y, Kaneko K, Yamanaka K, O’Halloran TV, Nukina N. Complete loss of post-translational modifications triggers fibrillar aggregation of SOD1 in the familial form of amyotrophic lateral sclerosis. J Biol Chem 2008; 283:24167 - 76; http://dx.doi.org/10.1074/jbc.M802083200; PMID: 18552350
- Kerman A, Liu HN, Croul S, Bilbao J, Rogaeva E, Zinman L, Robertson J, Chakrabartty A. Amyotrophic lateral sclerosis is a non-amyloid disease in which extensive misfolding of SOD1 is unique to the familial form. Acta Neuropathol 2010; 119:335 - 44; http://dx.doi.org/10.1007/s00401-010-0646-5; PMID: 20111867
- Jaarsma D, Haasdijk ED, Grashorn JA, Hawkins R, van Duijn W, Verspaget HW, London J, Holstege JC. Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiol Dis 2000; 7:6 Pt B 623 - 43; http://dx.doi.org/10.1006/nbdi.2000.0299; PMID: 11114261
- Wang L, Deng HX, Grisotti G, Zhai H, Siddique T, Roos RP. Wild-type SOD1 overexpression accelerates disease onset of a G85R SOD1 mouse. Hum Mol Genet 2009; 18:1642 - 51; http://dx.doi.org/10.1093/hmg/ddp085; PMID: 19233858
- Fukada K, Nagano S, Satoh M, Tohyama C, Nakanishi T, Shimizu A, Yanagihara T, Sakoda S. Stabilization of mutant Cu/Zn superoxide dismutase (SOD1) protein by coexpressed wild SOD1 protein accelerates the disease progression in familial amyotrophic lateral sclerosis mice. Eur J Neurosci 2001; 14:2032 - 6; http://dx.doi.org/10.1046/j.0953-816x.2001.01828.x; PMID: 11860498
- Deng HX, Shi Y, Furukawa Y, Zhai H, Fu R, Liu E, Gorrie GH, Khan MS, Hung WY, Bigio EH, et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci U S A 2006; 103:7142 - 7; http://dx.doi.org/10.1073/pnas.0602046103; PMID: 16636275
- Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 1998; 281:1851 - 4; http://dx.doi.org/10.1126/science.281.5384.1851; PMID: 9743498
- Grad LI, Guest WC, Yanai A, Pokrishevsky E, O’Neill MA, Gibbs E, Semenchenko V, Yousefi M, Wishart DS, Plotkin SS, et al. Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc Natl Acad Sci U S A 2011; 108:16398 - 403; http://dx.doi.org/10.1073/pnas.1102645108; PMID: 21930926
- Taylor DM, Gibbs BF, Kabashi E, Minotti S, Durham HD, Agar JN. Tryptophan 32 potentiates aggregation and cytotoxicity of a copper/zinc superoxide dismutase mutant associated with familial amyotrophic lateral sclerosis. J Biol Chem 2007; 282:16329 - 35; http://dx.doi.org/10.1074/jbc.M610119200; PMID: 17389599
- Raymond GJ, Bossers A, Raymond LD, O’Rourke KI, McHolland LE, Bryant PK 3rd, Miller MW, Williams ES, Smits M, Caughey B. Evidence of a molecular barrier limiting susceptibility of humans, cattle and sheep to chronic wasting disease. EMBO J 2000; 19:4425 - 30; http://dx.doi.org/10.1093/emboj/19.17.4425; PMID: 10970836
- Audet JN, Gowing G, Julien JP. Wild-type human SOD1 overexpression does not accelerate motor neuron disease in mice expressing murine Sod1 G86R. Neurobiol Dis 2010; 40:245 - 50; http://dx.doi.org/10.1016/j.nbd.2010.05.031; PMID: 20573565
- Prudencio M, Durazo A, Whitelegge JP, Borchelt DR. Modulation of mutant superoxide dismutase 1 aggregation by co-expression of wild-type enzyme. J Neurochem 2009; 108:1009 - 18; http://dx.doi.org/10.1111/j.1471-4159.2008.05839.x; PMID: 19077113
- Münch C, O’Brien J, Bertolotti A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc Natl Acad Sci U S A 2011; 108:3548 - 53; http://dx.doi.org/10.1073/pnas.1017275108; PMID: 21321227
- Horwich AL, Weissman JS. Deadly conformations--protein misfolding in prion disease. Cell 1997; 89:499 - 510; http://dx.doi.org/10.1016/S0092-8674(00)80232-9; PMID: 9160742
- Witan H, Gorlovoy P, Kaya AM, Koziollek-Drechsler I, Neumann H, Behl C, Clement AM. Wild-type Cu/Zn superoxide dismutase (SOD1) does not facilitate, but impedes the formation of protein aggregates of amyotrophic lateral sclerosis causing mutant SOD1. Neurobiol Dis 2009; 36:331 - 42; http://dx.doi.org/10.1016/j.nbd.2009.07.024; PMID: 19660548
- Prudencio M, Durazo A, Whitelegge JP, Borchelt DR. An examination of wild-type SOD1 in modulating the toxicity and aggregation of ALS-associated mutant SOD1. Hum Mol Genet 2010; 19:4774 - 89; http://dx.doi.org/10.1093/hmg/ddq408; PMID: 20871097
- Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 2009; 64:783 - 90; http://dx.doi.org/10.1016/j.neuron.2009.12.016; PMID: 20064386
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 2002; 297:353 - 6; http://dx.doi.org/10.1126/science.1072994; PMID: 12130773
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 2006; 313:1781 - 4; http://dx.doi.org/10.1126/science.1131864; PMID: 16990547
- Luk KC, Song C, O’Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A 2009; 106:20051 - 6; http://dx.doi.org/10.1073/pnas.0908005106; PMID: 19892735
- Münch C, Bertolotti A. Exposure of hydrophobic surfaces initiates aggregation of diverse ALS-causing superoxide dismutase-1 mutants. J Mol Biol 2010; 399:512 - 25; http://dx.doi.org/10.1016/j.jmb.2010.04.019; PMID: 20399791
- Wang Q, Johnson JL, Agar NY, Agar JN. Protein aggregation and protein instability govern familial amyotrophic lateral sclerosis patient survival. PLoS Biol 2008; 6:e170; http://dx.doi.org/10.1371/journal.pbio.0060170; PMID: 18666828
- Elam JS, Taylor AB, Strange R, Antonyuk S, Doucette PA, Rodriguez JA, Hasnain SS, Hayward LJ, Valentine JS, Yeates TO, et al. Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat Struct Biol 2003; 10:461 - 7; http://dx.doi.org/10.1038/nsb935; PMID: 12754496
- Furukawa Y, Kaneko K, Watanabe S, Yamanaka K, Nukina N. Intracellular seeded aggregation of mutant Cu,Zn-superoxide dismutase associated with amyotrophic lateral sclerosis. FEBS Lett 2013; 587:2500 - 5; http://dx.doi.org/10.1016/j.febslet.2013.06.046; PMID: 23831581
- Grad LI, Yerbury JJ, Turner BJ, Guest WC, Pokrishevsky E, O’Neill MA, Yanai A, Silverman JM, Zeineddine R, Corcoran L, et al. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc Natl Acad Sci U S A 2014; http://dx.doi.org/10.1073/pnas.1312245111; PMID: 24550511
- Guest WC, Plotkin SS, Cashman NR. Toward a mechanism of prion misfolding and structural models of PrP(Sc): current knowledge and future directions. J Toxicol Environ Health A 2011; 74:154 - 60; http://dx.doi.org/10.1080/15287394.2011.529065; PMID: 21218344
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol 2007; 8:101 - 12; http://dx.doi.org/10.1038/nrm2101; PMID: 17245412
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003; 300:486 - 9; http://dx.doi.org/10.1126/science.1079469; PMID: 12702875
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006; 440:352 - 7; http://dx.doi.org/10.1038/nature04533; PMID: 16541076
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 2008; 14:837 - 42; http://dx.doi.org/10.1038/nm1782; PMID: 18568035
- El-Agnaf OM, Salem SA, Paleologou KE, Curran MD, Gibson MJ, Court JA, Schlossmacher MG, Allsop D. Detection of oligomeric forms of alpha-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J 2006; 20:419 - 25; http://dx.doi.org/10.1096/fj.03-1449com; PMID: 16507759
- Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T, Lansbury PT Jr.. Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol 2002; 322:1089 - 102; http://dx.doi.org/10.1016/S0022-2836(02)00735-0; PMID: 12367530
- Keller S, Sanderson MP, Stoeck A, Altevogt P. Exosomes: from biogenesis and secretion to biological function. Immunol Lett 2006; 107:102 - 8; http://dx.doi.org/10.1016/j.imlet.2006.09.005; PMID: 17067686
- Flanagan J, Middeldorp J, Sculley T. Localization of the Epstein-Barr virus protein LMP 1 to exosomes. J Gen Virol 2003; 84:1871 - 9; http://dx.doi.org/10.1099/vir.0.18944-0; PMID: 12810882
- Gould SJ, Booth AM, Hildreth JE. The Trojan exosome hypothesis. Proc Natl Acad Sci U S A 2003; 100:10592 - 7; http://dx.doi.org/10.1073/pnas.1831413100; PMID: 12947040
- Nguyen DG, Booth A, Gould SJ, Hildreth JE. Evidence that HIV budding in primary macrophages occurs through the exosome release pathway. J Biol Chem 2003; 278:52347 - 54; http://dx.doi.org/10.1074/jbc.M309009200; PMID: 14561735
- Perez-Gonzalez R, Gauthier SA, Kumar A, Levy E. The exosome secretory pathway transports amyloid precursor protein carboxyl-terminal fragments from the cell into the brain extracellular space. J Biol Chem 2012; 287:43108 - 15; http://dx.doi.org/10.1074/jbc.M112.404467; PMID: 23129776
- Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci 2010; 30:6838 - 51; http://dx.doi.org/10.1523/JNEUROSCI.5699-09.2010; PMID: 20484626
- Danzer KM, Kranich LR, Ruf WP, Cagsal-Getkin O, Winslow AR, Zhu L, Vanderburg CR, McLean PJ. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener 2012; 7:42; http://dx.doi.org/10.1186/1750-1326-7-42; PMID: 22920859
- Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, Laude H, Raposo G. Cells release prions in association with exosomes. Proc Natl Acad Sci U S A 2004; 101:9683 - 8; http://dx.doi.org/10.1073/pnas.0308413101; PMID: 15210972
- Coleman BM, Hanssen E, Lawson VA, Hill AF. Prion-infected cells regulate the release of exosomes with distinct ultrastructural features. FASEB J 2012; 26:4160 - 73; http://dx.doi.org/10.1096/fj.11-202077; PMID: 22767229
- Kim DK, Kang B, Kim OY, Choi DS, Lee J, Kim SR, Go G, Yoon YJ, Kim JH, Jang SC, et al. EVpedia: an integrated database of high-throughput data for systemic analyses of extracellular vesicles. J Extracell Vesicles 2013; 2.
- Turner BJ, Atkin JD, Farg MA, Zang DW, Rembach A, Lopes EC, Patch JD, Hill AF, Cheema SS. Impaired extracellular secretion of mutant superoxide dismutase 1 associates with neurotoxicity in familial amyotrophic lateral sclerosis. J Neurosci 2005; 25:108 - 17; http://dx.doi.org/10.1523/JNEUROSCI.4253-04.2005; PMID: 15634772
- Gomes C, Palma AS, Almeida R, Regalla M, McCluskey LF, Trojanowski JQ, Costa J. Establishment of a cell model of ALS disease: Golgi apparatus disruption occurs independently from apoptosis. Biotechnol Lett 2008; 30:603 - 10; http://dx.doi.org/10.1007/s10529-007-9595-z; PMID: 18004513
- Gomes C, Keller S, Altevogt P, Costa J. Evidence for secretion of Cu,Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis. Neurosci Lett 2007; 428:43 - 6; http://dx.doi.org/10.1016/j.neulet.2007.09.024; PMID: 17942226
- Urushitani M, Sik A, Sakurai T, Nukina N, Takahashi R, Julien JP. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci 2006; 9:108 - 18; http://dx.doi.org/10.1038/nn1603; PMID: 16369483
- Urushitani M, Ezzi SA, Matsuo A, Tooyama I, Julien JP. The endoplasmic reticulum-Golgi pathway is a target for translocation and aggregation of mutant superoxide dismutase linked to ALS. FASEB J 2008; 22:2476 - 87; http://dx.doi.org/10.1096/fj.07-092783; PMID: 18337461
- Basso M, Pozzi S, Tortarolo M, Fiordaliso F, Bisighini C, Pasetto L, Spaltro G, Lidonnici D, Gensano F, Battaglia E, et al. Mutant copper-zinc superoxide dismutase (SOD1) induces protein secretion pathway alterations and exosome release in astrocytes: implications for disease spreading and motor neuron pathology in amyotrophic lateral sclerosis. J Biol Chem 2013; 288:15699 - 711; http://dx.doi.org/10.1074/jbc.M112.425066; PMID: 23592792
- Kabashi E, Valdmanis PN, Dion P, Rouleau GA. Oxidized/misfolded superoxide dismutase-1: the cause of all amyotrophic lateral sclerosis?. Ann Neurol 2007; 62:553 - 9; http://dx.doi.org/10.1002/ana.21319; PMID: 18074357
- Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, Song S, Likhite S, Murtha MJ, Foust KD, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol 2011; 29:824 - 8; http://dx.doi.org/10.1038/nbt.1957; PMID: 21832997
- Legname G, Nguyen HO, Baskakov IV, Cohen FE, Dearmond SJ, Prusiner SB. Strain-specified characteristics of mouse synthetic prions. Proc Natl Acad Sci U S A 2005; 102:2168 - 73; http://dx.doi.org/10.1073/pnas.0409079102; PMID: 15671162
- Irwin DJ, Abrams JY, Schonberger LB, Leschek EW, Mills JL, Lee VM, Trojanowski JQ. Evaluation of potential infectivity of Alzheimer and Parkinson disease proteins in recipients of cadaver-derived human growth hormone. JAMA Neurol 2013; 70:462 - 8; http://dx.doi.org/10.1001/jamaneurol.2013.1933; PMID: 23380910
- Eisele YS, Obermüller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H, Walker LC, Staufenbiel M, Heikenwalder M, Jucker M. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science 2010; 330:980 - 2; http://dx.doi.org/10.1126/science.1194516; PMID: 20966215
- Weichert A, Besemer AS, Liebl M, Hellmann N, Koziollek-Drechsler I, Ip P, Decker H, Robertson J, Chakrabartty A, Behl C, et al. Wild-type Cu/Zn superoxide dismutase stabilizes mutant variants by heterodimerization. Neurobiol Dis 2014; 62:479 - 88; http://dx.doi.org/10.1016/j.nbd.2013.10.027; PMID: 24200866
- Furukawa Y, Kaneko K, Watanabe S, Yamanaka K, Nukina N. A seeding reaction recapitulates intracellular formation of Sarkosyl-insoluble transactivation response element (TAR) DNA-binding protein-43 inclusions. J Biol Chem 2011; 286:18664 - 72; http://dx.doi.org/10.1074/jbc.M111.231209; PMID: 21454603
- Nonaka T, Masuda-Suzukake M, Arai T, Hasegawa Y, Akatsu H, Obi T, Yoshida M, Murayama S, Mann DM, Akiyama H, et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep 2013; 4:124 - 34; http://dx.doi.org/10.1016/j.celrep.2013.06.007; PMID: 23831027