
Abstract
Wildlife has been considered the main source of novel viruses causing emerging infectious diseases. Marmota himalayana is endemic to the Qinghai–Tibetan Plateau, China. Here, based on a high-throughput method using Illumina RNA sequencing, we studied the RNA virome of M. himalayana and discovered multiple novel viruses, especially picobirnaviruses (PBVs), which have a bi-segmented genome and belong to the family Picobirnaviridae. A total of 63% of the viral contigs corresponded to PBVs, comprising 274 segment 1 and 56 segment 2 sequences. Unexpectedly, four unsegmented PBV genomes were also detected and confirmed by PCR and resequencing. According to the phylogenetic analysis, the following nine PBV assortment types are proposed: C1:GI, C2:GIV, C4:GI, C4:GV, C5:GI, C7:GI, C8:GIV, C8:GV and C8:GII. We hypothesize a model of segmentation for the PBV genome, mediated by a 6-bp direct repeat sequence, GAAAGG. The model is supported by detection of the segmentation-associated sequence GAAAGG not only in the 5′ untranslated regions of segment 1 (221 in 289) and segment 2 (57 in 80) of bi-segmented PBVs but also in the 5′ untranslated regions and junction sequences between the capsid and RdRp genes of unsegmented PBVs. Therefore, with RNA sequencing, we found an unexpected biodiversity of PBVs in M. himalayana, indicating that M. himalayana is a special host for PBVs. We also proposed a putative model of how bi-segmented PBVs could be converted into unsegmented PBVs, which sheds new light on the processes of RNA virus genome evolution.
Xue-lian Luo and Shan Lu are contributed equally to this work.
Introduction
There have been frequent outbreaks of emerging infectious diseases caused by viruses of animal origin, such as the emergence of the Ebola virus in West Africa and the Middle East respiratory syndrome coronavirus in the Middle EastCitation1, Citation2. Investigations of the source of these novel viral infections have highlighted wildlife as a reservoir of zoonotic virusesCitation3, Citation4. Marmota himalayana is a species endemic to the Qinghai–Tibetan Plateau that belongs to the family Sciuridae in the order Rodentia. It is considered a major reservoir of Yersinia pestisCitation5. A novel hepatovirus and astrovirus have recently been detected in marmotCitation6, Citation7.
Segmented RNA viruses are widespread in nature and include important human, animal and plant pathogens, such as influenza virus and tick-borne Jingmen virus. Viruses with segmented and unsegmented genomes typically belong to different viral familiesCitation8. Unsegmented foot-and-mouth disease virus reportedly undergoes segmentation into two RNAs during prolonged cell cultureCitation9. Investigation of the recently discovered tick-borne Jingmen virus has revealed that a segmented RNA virus has a genome derived in part from unsegmented viral ancestorsCitation10. However, the mechanism by which an unsegmented RNA virus undergoes genome segmentation into a segmented virus is unclear.
Picobirnaviruses (PBVs) are small, non-enveloped, bi-segmented double-stranded RNA viruses of the family PicobirnavirdaeCitation11, Citation12. Segment 1 (2.2–2.7 kb) contains two open reading frames (ORF1 and ORF2) encoding a hydrophilic protein with unknown function and the capsid protein, respectively, whereas segment 2 (1.2–1.9 kb) has a single ORF and encodes the viral RNA-dependent RNA polymerase (RdRp)Citation13–Citation15. A recent study reported a fused RNA segment for PBV in horseCitation16. Based on the amino acid sequence of the RdRp, PBVs are classified into five genogroups: GI, GIICitation17, GIIICitation18, GIV and GVCitation16. The intra-genogroup and inter-genogroup sequence similarities of the RdRp nucleic acid fragment range from 49 to 100% and 28 to 37%, respectivelyCitation17. PBVs are frequently detected in fecal samples from mammals, birds, and wild animals, as well as environmental samples and in immunocompromised patients as an opportunistic diarrhea-causing pathogenCitation19, Citation20. In this study, using RNA sequencing (RNA-seq), we studied the RNA virome of M. himalayana and identified and characterized novel PBVs in this species, including bi-segmented and unsegmented genomes of PBVs. A putative model of PBV genome segmentation was also proposed.
Materials and methods
Marmot sampling
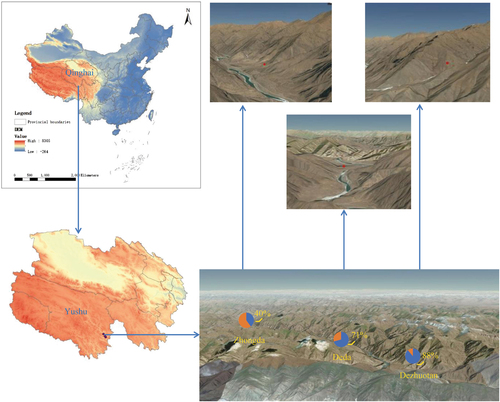
The 191 M. himalayana were sampled from 29 June to 8 August 2013 as part of the animal plague surveillance program conducted in Yushu Tibetan autonomous prefecture, Qinghai province, China. The locations of the sampling were the counties of Zhongda (with an altitude of 3599.6 m above sea level), Dezhuotan (3025 m above sea level) and Deda (3625.6 m above sea level) in Yushu Tibetan autonomous prefecture, Qinghai province, China (Fig. ). The marmots were captured live in cages in the field and sampled in the laboratory of the County’s Centre for Disease Control and Prevention. The sampling was performed in accordance with the protocol for the national plague surveillance program in animals. The intestinal contents were collected in 2 ml sterile tubes, which were kept at −80 °C until processing.
Red dots are the collection sites. Positive rates in three locations are indicated
Nucleic acid extraction
Each fecal sample was re-suspended (1:10, wt/vol) in PBS buffer and vortexed thoroughly. The suspension was clarified by centrifugation at 15,000×g for 10 min. The total viral nucleic acids were extracted with a QIAamp viral RNA mini kit (Qiagen, China) according to the manufacturer’s protocol. The concentration and quality of final RNA were examined using an ND-1000 UV spectrophotometer. These RNA were randomly merged into four pools for RNA-seq library construction and sequencing.
Illumina sequencing and data analysis
The library preparation and sequencing steps were performed by BGI Tech (Shenzhen, China). Briefly, the total RNA was subjected to an rRNA removal step using a Ribo-Zero Magnetic Gold Kit (Epicentre, Madison, WI). The remaining RNA was then fragmented, reverse-transcribed, ends repaired, dA-tailed, adapter ligated, purified, and quantified with an Agilent 2100 Bioanalyzer and ABI StepOnePlus Real-Time PCR System. Pair-end (approximately 100 bp) sequencing of the RNA library was performed on the HiSeq 2000 platform (Illumina, San Diego, CA). Raw reads were quality trimmed and assembled de novo into contigs using the Trinity programCitation21. The assembled contigs (>300 bp) were compared with the viral reference database and the GenBank non-redundant protein database using BLASTx search with an E-value of 10−5.
Quantification of relative transcript abundances
The relative abundance of each transcript is presented as transcripts per million and corrects for the total number of reads as well as for the transcript lengthCitation22. According to the procedure described, we first removed the rRNA reads from the data sets and then used the resulting rRNA contigs as a template for mapping using BOWTIE2Citation23. Finally, the remaining reads from each library were then mapped onto the assembled transcripts and analyzed with RSEMCitation24.
Sequence confirmation and complete genome sequencing
For the confirmation of high-throughput sequencing results, we used nested RT-PCR to examine each potential viral sequence by specific primers. To obtain longer sequences or the complete genome, we used genome walking (Takara, Japan) and 5′ and 3′ rapid amplification of cDNA ends (Roche, USA) according to the manufacturer’s protocol. All specific primers used are available on request.
Phylogenetic analysis
The viral amino acid sequences were predicted using the BioEdit program. Fifteen complete segment 1 sequences and 24 complete segment 2 sequences were downloaded from GenBank and used as reference sequences. Sequence alignment was performed using MAFFT version 7 with the E-INS-I algorithm. Phylogenetic trees were inferred using the maximum likelihood method implemented in PhyML version 3.0 with the WAG+Γ amino acid substitution model and a Subtree Pruning and Regrafting topology searching algorithmCitation25.
Nucleotide sequence accession numbers
Novel mammalian virus genome sequences in the study are at GenBank accession no. KY855432-KY855444. The RdRp and capsid sequences used in the study are at GenBank accession no. KY928683-KY928738 and KY928739-KY929012, respectively.
Results
Intestinal virome of M. himalayana
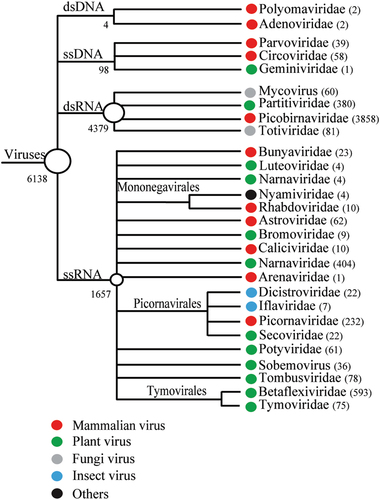
The intestinal contents of 191 M. himalayana individuals were pooled and sequenced using Illumina HiSeq 2000, which generated a total of ~120 Gb of 100-bp end reads. The 379,618 high-quality >300-bp contigs obtained were subjected to BLASTx searches against a viral reference database and the GenBank non-redundant protein database using an E-value of 10−5. A total of 6138 contigs matched the sequences of viruses, classified to 28 virus families. Among them, 11 were mammalian-related virus families, including Picobirnavirdae (n = 3858), Picornaviridae (n = 232), Astroviridae (n = 62), Bunyaviridae (n = 23), Caliciviridae (n = 10), Mononegavirales (n = 14), and Arenaviridae (n = 1). The DNA virus families included Circoviridae (n = 58), Parvoviridae (n = 39), Adenoviridae (n = 2) and Polyomaviridae (n = 2) (Fig. ). In addition, we obtained complete or nearly complete genome sequences for 13 new mammalian RNA viruses of three families, Picornaviridae, Astroviridae, and Caliciviridae (Supplementary Table S1). The polyproteins of Marmot sapalovirus HT5 and HT6 showed 54 and 56% amino acid sequence similarities to the genera Enterovirus and Sapelovirus, respectively, which are proposed new species of the Enterovirus/Sapelovirus supergroup (www.picornaviridae.com). The polyproteins of Marmot cardiovirus HT7 and Marmot mosavirus HT8 showed 59 and 46% amino acid sequence similarities to those of the viruses of the genera Cardiovirus and Mosavirus, respectively, which are considered new members of Cardiovirus and Mosavirus.
Numbers of virus contigs are shown in parentheses
Detection of unsegmented PBVs
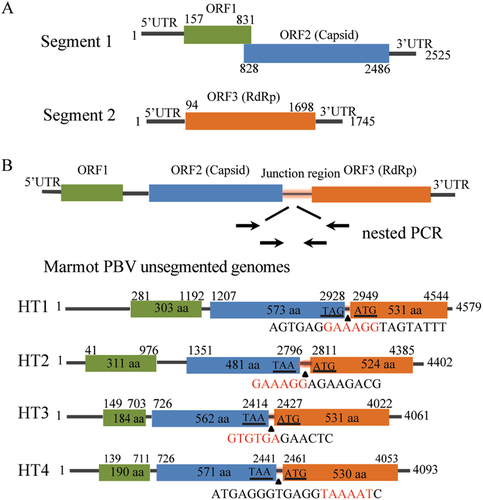
BLASTx analysis showed that of 6138 virus contigs 3858 (62.9%) belonged to PBVs and exhibited 25–74% amino acid similarities. A total of 274 segment 1 and 56 segment 2 sequences were obtained. Unlike the conventional two-segment genomes of PBVs (Fig. ), we found four unsegmented genomes (named Marmot PBV HT1-4) (Fig. , GenBank accession no. KY855428-KY855431). A nested polymerase chain reaction (PCR) bridging segment 1 and 2 was performed with two pairs of primers (Supplementary Table S2). The expected PCR product was sequenced, and the truth of the junction between both segments was confirmed. The four unsegmented genomes were also confirmed by re-sequencing. The junction sequences linking both segments of Marmot PBV HT1-4 were of 20, 14, 12, and 19-bp, respectively (Fig. ). Overall, 20 of 50 (40%), 17 of 24 (71%) and 104 of 117 (88%) intestinal samples from Zhongda, Deda, Dezhuotan, respectively, were positive for unsegmented PBVs (Fig. ).
(a) Schematic representation of the two segments of human picobirnavirus strain Hy005102 (GenBank accession no. NC_007026 and NC_007027). (B) The unsegmented genomes of picobirnaviruses detected in marmot. Arrows indicate the position and direction of primers for nested PCR. Stop codon of the capsid gene and the initiation codon of the RdRp are underlined. Junction sequences between segments 1 and 2 of bi-segmented picobirnaviruses are shown below the triangles. Segmentation-associated motifs are shown in red
Genome characterization of unsegmented PBVs
These unsegmented PBV sequences ranged from 4061 to 4579 bases in length with overall G + C contents of 39.9–45.4%. The 5′ untranslated regions (UTRs) (40–280 bases) were AU-rich (G + C contents of 28.2–32.6%). The 3′ UTRs (17–40 bases) contained G + C contents ranging from 37.1 to 64.7%. The genome of the unsegmented PBVs contained three ORFs, ORF1, ORF2 and ORF3 (Fig. ). ORF1 encoded a protein of 184–311 amino acids with unknown function. These proteins contained a different number of repetitions of the ExxRxNxxxE motif which was also observed in the corresponding protein in other known PBVsCitation26. There were six ExxRxNxxxE motifs in the ORF1-encoded protein of Marmot PBV HT1 but only two ExxRxNxxxE motifs in those of Marmot PBV HT2-4. ORF2 encoded the capsid protein of 481–573 amino acids. These capsid proteins shared low (23–29%) amino acid identities with those of other PBV strains, being most closely related to PBV Equ2 (GenBank accession no. AKN50623). ORF3 encoded the RdRp of 524–531 amino acids. These RdRp shared 52–68% amino acid identities with those of other PBV strains. The amino acid similarities of the capsid and RdRp proteins of Marmot PBV HT1-4 were 53 and 69%, respectively. The pairwise amino acid identities of the capsid and RdRp regions of Marmot PBV HT1-4 suggested a high level of diversity among the unsegmented PBVs (Supplementary Table S3).
A recent study had reported one fused PBV genome Equ4 (GenBank accession no. KR902502) in diseased horse feces. An alignment result revealed that the highest amino acid similarity of capsid and RdRp between Marmot PBV HT1-4 and PBV Equ4 was 14 and 39%, respectively. There were four conserved motifs in RdRp regions among unsegmented PBVs, such as MFP, HGM(L)G(A)SGS, GDD and RALG at 242, 366, 405 and 472 sits, respectively, whereas no conserved motifs were found among the capsid proteins. These results suggested that unsegmented PBVs exhibited a high diversity.
PBV assortment types
PBVs are classified into genogroups GI–GV. All five genogroups of RdRp were detected in marmot. In addition, an RdRp contig showed only 37% amino acid sequence similarity to those of the other genogroups. Thus, a new genogroup of RdRp named GVI was identified (Fig. ). The intra members of all six genogroups of RdRp shared similarity >42.41% at the amino acid level. The intergroup amino acid similarity for all RdRp groups ranged from 25.49–40.86% (Supplementary Figure S1). The RdRps of unsegmented Marmot PBV HT1-4 were grouped into GI, GII, and GIV, respectively (Fig. ).
a The phylogenetic trees of the full-sequence capsid (left) and RdRp (right) proteins. Sequences of picobirnaviruses obtained from marmot are shown in red. Sequences of picobirnaviruses reported previously are shown in black. Unsegmented picobirnaviruses from marmot are indicated by black triangles. Capsid and RdRp sequences of picobirnaviruses that underwent assortment are linked by lines. b Proposed assortment types of bi-segmented and unsegmented picobirnaviruses
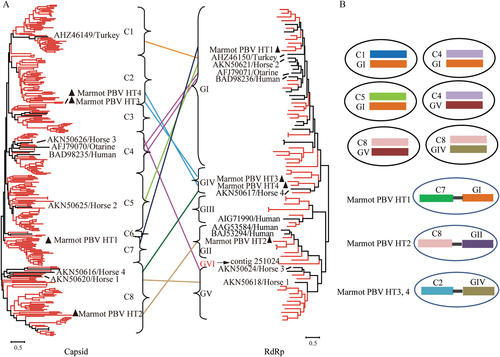
By phylogenetic analysis of 293 complete capsid sequences from marmot and other hosts, eight clusters of capsids (C1-8) were proposed (Fig. ). The capsid sequences of unsegmented Marmot PBV HT1-4 were classified into clusters C2, C7, and C8; Marmot PBV HT3 and four were in C2 (Fig. ).
Based on the genome sequences of isolated PBVs and the unsegmented genomes of Marmot PBVsCitation14, Citation15, Citation27–Citation29, we propose nine PBV assortment types (Fig. ): C1:GI, C2:GIV, C4:GI, C4:GV, C5:GI, C7:GI, C8:GIV, C8:GV, and C8:GII. Clearly, the genogroup G1 for RdRp could assort with clusters C1, C4, C5, and C7 for capsids (Fig. ). Cluster C8 for capsids could assort with the RdRp of genogroups GIV, GV, and GII (Fig. ). Unsegmented Marmot PBVs exhibited three assortment types—HT1 for C7:GI, HT2 for C8:GII, and HT3 and HT4 for C2:GIV (Fig. ); this was not observed in bi-segmented PBVs.
Direct repeat sequence segmentation model
Because both unsegmented and bi-segmented PBVs were identified in M. himalayana, we hypothesized that unsegmented PBVs underwent genome segmentation into segmented PBVs. First, the junction sequences linking the capsid and RdRp regions of unsegmented Marmot PBV HT1-4 were analyzed for possible segmentation-associated motifs. An alignment of these junction sequences and the 5′ UTR of the segment 2 showed three conserved motifs: TAAAAT, GAAAGG, and GTGTGA. Marmot PBV HT1 and HT2 have GAAAGG at the junction between the capsid and RdRp genes (Fig. ), whereas Marmot PBV HT3 and HT4 have GTGTGA or TAAAAT (Fig. ). These conserved motifs were also identified in 63 segment 2 of segmented PBVs in M. himalayana and other hosts (Supplementary Figure S4). However, an alignment of the junction sequences of unsegmented PBVs and the 3′ UTR of the segment 1 detected no conserved motifs. These findings indicated that the three conserved motifs might be involved in the segmentation of PBVs.
We next analyzed the UTR of the PBV sequences obtained in this study and those reported previously. Strikingly, most segment 1 and 2 sequences had the GAAAGG in 5′ UTRs. In particular, 57 segment 2 sequences (distributed among all six genotypes) had GAAAGG in the 5′ UTR (Fig. ). Of 289 segment 1 sequences analyzed, 221 (distributed among all eight clusters) had the GAAAGG motif in the 5′ UTR (Fig. ). Notably, 78 segment 1 sequences had two or three copies of GAAAGG in the 5′ UTR, and four segment 2 sequences had two copies of GAAAGG in their 5′ UTRs (Fig. ). Furthermore, Marmot PBV HT1 and HT2 had GAAAGG in their 5′ UTRs. Therefore, we propose a 6-bp direct repeat sequence GAAAGG-mediated segmentation model for Marmot unsegmented PBVs (Fig. ).
Relative positions of the 6-bp direct repeat sequence GAAAGG in the 5′ UTR and junction between the capsid and RdRp genes of unsegmented Marmot picobirnaviruses are indicated by rectangles. A schematic of the three types of segment 1 harboring GAAAGG (S1-1-3) and two types of segment 2 harboring GAAAGG (S2-1-2) from bi-segmented picobirnaviruses. The proportions of S1-1-3 and S2-1-2 among the total sequences analyzed, together with their genotypes, are shown in the following tables. *The published segment 1 sequences (GenBank accession no. KJ495689; KF861768; JQ776551; AB186897; KR902506; KC692367; KR902502; KR902504; KF861770; KF861771; KF861772). The published segment 2 sequences (GenBank accession no. JQ776552; AAG53583; AHX00960; ACT64131; AFK81927; BAJ53294; AHZ46150; KR902507; KR902503)
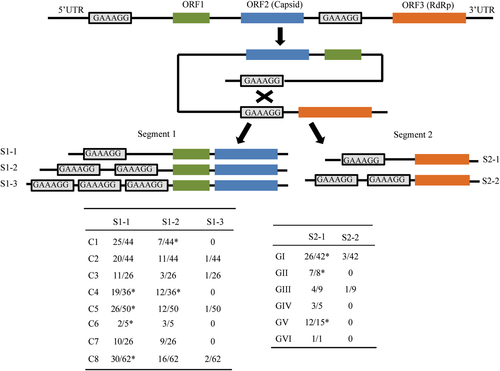
We also detected GTGTGA or TAAAAT in the 5′ UTR of segment 2 (Supplementary Figure S2). Ten segment 2 sequences had TAAAAT in their 5′ UTR while five had both the TAAAAT and the GAAAGG in their 5′ UTR. Three segment 2 sequences have GTGTGA in their 5′ UTRs. Neither the GTGTGA nor the TAAAAT motif was detected in the 5′ UTR of segment 1 or unsegmented PBVs from marmot.
Discussion
RNA-seq has been used to study the transcriptome in various cells and tissues based on high-throughput sequencing. Recently, this technique has been successfully used to discover a large number of novel RNA virusesCitation30, Citation31. In the present study, we used RNA-seq to study RNA virome in M. himalayana fecal samples and identified novel viruses belonging to different families, such as Picobirnaviridae, Picornaviridae, Astroviridae, and Caliciviridae (Fig. and Supplementary Table S1).
PBV sequences predominated in the intestine of M. himalayana. In addition, we also detected four unsegmented PBV genomes (named Marmot PBV HT1-4). Therefore, M. himalayana should be considered a special animal reservoir of PBVs. We failed to isolate unsegmented PBVs from the fecal samples of M. himalayana, hampering further biological research on unsegmented PBVs.
All of the PBV sequences obtained from M. himalayana, together with published reference PBV sequences, were divided into six genogroups for the RdRp genes and eight clusters for the capsid genes. A phylogenetic analysis (Fig. ) suggested recombination between the capsid and RdRp genes. This recombination was reflected not only in the different phylogenetic trees between the two genes but also in the marked diversity of capsid protein sequences. The four Marmot PBVs were dispersed within the segmented PBVs in phylogenetic trees (Fig. ), which suggests that multiple segmentation events might have occurred frequently.
The amino acid alignment analysis of RdRp sequences obtained in marmot with other RdRp sequences of PBVs showed three patterns: GI/GIV, GIII/GV/GVI, and GIII. The intra sequences of GI/GIV had higher conservation compared with that of GIII/GV/GVI, and GIII (Supplementary Figure S3). A motif GDD at the 472 site was conserved among all RdRp regions, whereas no conserved motifs were found among the capsid proteins. Of interest, the number of ExxRxNxxxE motifs was variable in ORF1-encoded proteins of marmot PBV sequences, ranging from 0 to 16. Half (~56%) of these proteins had three to six ExxRxNxxxE motifs, whereas ten of them did not possess this motif (Supplementary Figure S4).
Screening for1 segmentation-associated motifs revealed GAAAGG in the 5′ UTR and junction region between the capsid and RdRp genes of the unsegmented Marmot PBV HT1 and HT2 (Fig. ). Therefore, we proposed that the segmentation of unsegmented PBVs was mediated by GAAAGG (Fig. ). This is supported by the finding that 57 of 80 segment 2 sequences (covering all six genotypes of RdRp) had the GAAAGG motif in their 5′ UTRs (Fig. ). This motif was also detected in 221 of 289 segment 1 sequences (covering all eight clusters classified) (Fig. ). Direct repeat sequences have been identified in DNA viruses, RNA viruses or phagesCitation10, Citation32–Citation37. Direct repeat sequences evolved through multiple duplications, deletions, and mutations of a primordial sequence element in tick-borne flavivirusesCitation33. Interestingly, of 289 segment 1 sequences with the GAAAGG motif, 143, 73, and 5 had one, two, and three copies, respectively, in their 5′ UTR (Fig. ). Among the 80 segment 2 sequences with the GAAAGG motif, 55 and 4 had one and two copies, respectively, in their 5′ UTR (Fig. ). The specific mechanism by which GAAAGG-mediated genome segmentation occurs needs to be further studied.
In conclusion, we have identified a number of previously unknown RNA viruses in the intestine of M. himalayana on the Qinghai–Tibetan Plateau, China, especially novel PBVs. Discovering and characterizing both bi-segmented and unsegmented PBVs in M. himalayana encourages us to rethink the evolution and classification of PBVs. In addition, the putative model of PBV segmentation provides a new perspective on genome segmentation in RNA viruses.
Supplementary Figure S1
Download MS Word (866.2 KB)Supplementary Figure S2
Download MS Word (1.4 MB)Supplementary Figure S3
Download MS Word (1.5 MB)Supplementary Figure S4
Download MS Word (776.6 KB)Supplementary Table S1
Download MS Word (15.1 KB)Supplementary Table S2
Download MS Word (12.8 KB)Supplementary Table S3
Download MS Word (14.2 KB)Acknowledgements
This work was supported by the National Key R&D Program of China (2016YFC1201903 and 2016YFC1202700) and National Natural Science Foundation of China (81290340 and 81290345).
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41426-018-0020-6).
References
- JonesKEPatelNGLevyMAGlobal trends in emerging infectious diseasesNature2008451 990 99310.1038/nature06536
- KooriyamaTOkamotoMYoshidaTEpidemiological study of zoonoses derived from humans in captive chimpanzeesPrimates201354899810.1007/s10329-012-0320-8
- KhalafallaAIMERS-CoV in upper respiratory tract and lungs of dromedary camels, Saudi Arabia, 2013-2014Emerg. Infect. Dis.2015211153115810.3201/eid2107.1500704480395
- WacharapluesadeeSOlivalKJKanchanasakaBSurveillance for Ebola virus in wildlife, ThailandEmerg. Infect. Dis.2015212271227310.3201/eid2112.1508604672430
- GePXiJDingJPrimary case of human pneumonic plague occurring in a Himalayan marmot natural focus area Gansu Province, ChinaInt. J. Infect. Dis.201533677010.1016/j.ijid.2014.12.044
- AoYYuJLiLDiverse novel astroviruses identified in wild Himalayan marmotsJ. Gen. Virol.20179861262310.1099/jgv.0.000709
- YuJMLiLLZhangCYA novel hepatovirus identified in wild woodchuck Marmota himalayanaSci. Rep.2016610.1038/srep223614770319
- McDonaldSMNelsonMITurnerPEPattonJTReassortment in segmented RNA viruses: mechanisms and outcomesNat. Rev. Microbiol.20161444846010.1038/nrmicro.2016.465119462
- MorenoEExploration of sequence space as the basis of viral RNA genome segmentationProc. Natl. Acad. Sci. USA20141116678668310.1073/pnas.13231361114020086
- QinXCShiMTianJHA tick-borne segmented RNA virus contains genome segments derived from unsegmented viral ancestorsProc. Natl. Acad. Sci. USA20141116744674910.1073/pnas.13241941114020047
- CarstensEBRatification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2009)Arch. Virol.201015513314610.1007/s00705-009-0547-x
- CarstensEBBallLARatification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2008)Arch. Virol.20091541181118810.1007/s00705-009-0400-2
- GaneshBMasachessiGMladenovaZAnimal picobirnavirusVirusdisease20142522323810.1007/s13337-014-0207-y4188182
- BanyaiKPotgieterCGellertAGenome sequencing identifies genetic and antigenic divergence of porcine picobirnavirusesJ. Gen. Virol.2014952233223910.1099/vir.0.057984-0
- WakudaMPongsuwannaYTaniguchiKComplete nucleotide sequences of two RNA segments of human picobirnavirusJ. Virol. Methods200512616516910.1016/j.jviromet.2005.02.010
- LiLGiannittiFLowJExploring the virome of diseased horsesJ. Gen. Virol.2015962721273310.1099/vir.0.0001994635498
- RosenBIFangZYGlassRIMonroeSSCloning of human picobirnavirus genomic segments and development of an RT-PCR detection assayVirology200027731632910.1006/viro.2000.0594
- SmitsSLSchapendonkCMvan BeekJNew viruses in idiopathic human diarrhea cases, the NetherlandsEmerg. Infect. Dis.2014201218122210.3201/eid2007.1401904073879
- GiordanoMOMartinezLCRinaldiDDiarrhea and enteric emerging viruses in HIV-infected patientsAIDS Res. Hum. Retrovir.1999151427143210.1089/088922299309937
- ValleMCMartinezLCFerreyraLJViral agents related to diarrheic syndrome in kidney transplanted patientsMed. (B. Aires).200161179182
- GrabherrMGHaasBJYassourMFull-length transcriptome assembly from RNA-Seq data without a reference genomeNat. Biotechnol.20112964465210.1038/nbt.18833571712
- LangmeadBSalzbergSLFast gapped-read alignment with Bowtie 2Nat. Methods2012935735910.1038/nmeth.19233322381
- LiBRuottiVStewartRMThomsonJADeweyCNRNA-Seq gene expression estimation with read mapping uncertaintyBioinformatics20102649350010.1093/bioinformatics/btp692
- KatohKStandleyDMMAFFT multiple sequence alignment software version 7: improvements in performance and usabilityMol. Biol. Evol.20133077278010.1093/molbev/mst0103603318
- GuindonSGascuelOA simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihoodSyst. Biol.20035269670410.1080/10635150390235520
- Da CostaBDuquerroySTarusBDelmasBPicobirnaviruses encode a protein with repeats of the ExxRxNxxxE motifVirus Res.201115825125610.1016/j.virusres.2011.02.018
- GhoshSKobayashiNNagashimaSNaikTNMolecular characterization of full-length genomic segment 2 of a bovine picobirnavirus (PBV) strain: evidence for high genetic diversity with genogroup I PBVsJ. Gen. Virol.2009902519252410.1099/vir.0.013987-0
- NgTFVegaEKondovNODivergent picobirnaviruses in human fecesGenome Announc20142e004151410.1128/genomeA.00415-144022810
- VermaHMorSKErberJGoyalSMPrevalence and complete genome characterization of turkey picobirnavirusesInfect. Genet. Evol.20153013413910.1016/j.meegid.2014.12.014
- ShiMLinXDVasilakisNDivergent viruses discovered in arthropods and vertebrates revise the evolutionary history of the Flaviviridae and related virusesJ. Virol.20159065966910.1128/JVI.02036-154702705
- LiCShiMTianJHUnprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA virusesElife20154e053784384744
- DambaughTRKieffEIdentification and nucleotide sequences of two similar tandem direct repeats in Epstein-Barr virus DNAJ. Virol.198244823833256339
- GritsunTSGouldEAThe 3’ untranslated region of tick-borne flaviviruses originated by the duplication of long repeat sequences within the open reading frameVirology200635026927510.1016/j.virol.2006.03.002
- VolozhantsevNVMyakininaVPPopovaAVComplete genome sequence of novel T7-like virus vB_KpnP_KpV289 with lytic activity against Klebsiella pneumoniaeArch. Virol.201616149950110.1007/s00705-015-2680-z
- WangYTaoQ[Function of direct repeat sequences in DNA replication of hepatitis B virus]Zhonghua. Yi. Xue. Za. Zhi.199070604607
- WohlrabFMcLeanMJWellsRDThe segment inversion site of herpes simplex virus type 1 adopts a novel DNA structureJ. Biol. Chem.198726264076416
- YalamanchiliRRO’CallaghanDJSequence and organization of the genomic termini of equine herpesvirus type 1Virus Res.19901514916110.1016/0168-1702(90)90005-V