
Abstract
Wild aquatic birds are the major reservoir of influenza A virus. Cloacal swabs and feces samples (n = 6595) were collected from 62 bird species in Argentina from 2006 to 2016 and screened for influenza A virus. Full genome sequencing of 15 influenza isolates from 6 waterfowl species revealed subtypes combinations that were previously described in South America (H1N1, H4N2, H4N6 (n = 3), H5N3, H6N2 (n = 4), and H10N7 (n = 2)), and new ones not previously identified in the region (H4N8, H7N7 and H7N9). Notably, the internal gene segments of all 15 Argentine isolates belonged to the South American lineage, showing a divergent evolution of these viruses in the Southern Hemisphere. Time-scaled phylogenies indicated that South American gene segments diverged between ~ 30 and ~ 140 years ago from the most closely related influenza lineages, which include the avian North American (PB1, HA, NA, MP, and NS-B) and Eurasian lineage (PB2), and the equine H3N8 lineage (PA, NP, and NS-A). Phylogenetic analyses of the hemagglutinin and neuraminidase gene segments of the H4, H6, and N8 subtypes revealed recent introductions and reassortment between viruses from the Northern and Southern Hemispheres in the Americas. Remarkably and despite evidence of recent hemagglutinin and neuraminidase subtype introductions, the phylogenetic composition of internal gene constellation of these influenza A viruses has remained unchanged. Considering the extended time and the number of sampled species of the current study, and the paucity of previously available data, our results contribute to a better understanding of the ecology and evolution of influenza virus in South America.
Introduction
Influenza A Viruses (IAVs) belong to the family Orthomyxoviridae. The genome of IAVs is composed of 8 segments of negative-sense, single-stranded RNA. Wild aquatic birds of the order Anseriformes and Charadriiformes are considered the natural hosts of IAVs. Based on the antigenic characteristics of their surface glycoproteins, hemagglutinin (HA) and neuraminidase (NA), IAVs are divided into subtypes. To date, 16 HA subtypes (H1 to H16) and 9 NA subtypes (N1 to N9) have been recognized in wild birds. Due to the segmented nature of their genomes, IAVs frequently exchange gene segments, which has led to the identification of many subtype combinations in waterfowl speciesCitation1. Regardless of the subtype, IAV infections in wild aquatic birds occur mostly in the intestinal tract in an apparently asymptomatic fashion. The virus is excreted in large amounts in the feces, which allows for an efficient fecal-oral route mode of transmission among birds.
Many of the efforts to understand the ecology of IAVs in their natural reservoir have been driven by a desire to track the origin of pandemic strains; prompted in particular by realization that the pandemic IAV viruses of the 1957 Asian flu (H2N2) and of the 1968 Hong Kong flu (H3N2) carried gene segments (HA, NA, and PB1; and HA and PB1, respectively) similar to IAV strains from ducksCitation2. Renewed interest in understanding the ecology of IAVs in natural hosts resulted from the emergence of the HPAIVs of the H5 subtype in China with the ability to cause exacerbated and often lethal respiratory disease in humans. The HA gene segment of these viruses has continued to evolve and diversify into genetically distinct clades and sub-clades and have successfully spread across 4 different continentsCitation3,Citation4. Historically, long-term active IAV surveillance in wild birds has been circumscribed to the United States, Canada, Western Europe, Southeast Asia, and China. In other parts of the world, surveillance efforts have been mostly passive, in response to disease outbreaksCitation5,Citation6. As a result, only 0.1% of all available sequences from IAV isolated from wild birds originate from South AmericaCitation7. The first evidence of IAV in birds in South America was associated to an outbreak of highly pathogenic avian influenza virus (HPAIV) of the H7 subtype in commercial chickens and turkeys in Chile in 2002Citation8.
The evolution of IAVs in the natural reservoir is largely determined by the geographical separation and migratory patterns of the avian hosts. Thus, IAVs are broadly separated into lineages that correspond to vast geographic regions. IAVs of North American (NAm), Eurasian (EAs), and Australian lineages are well defined due to the significant amount of sequence information of IAV isolates available from those regionsCitation9–Citation11. More recently, additional lineages were proposed for South AmericaCitation12–Citation14 and AntarcticaCitation7 based on phylogenetic analyses of a limited number of IAV isolates obtained from wild birds.
Our group has been conducting active IAV surveillance in wild birds in Guatemala (Central America) and in Argentina (South America) as part of efforts to establish early warning systems for the potential introduction of IAVs from Asia into the Americas and/or from wild birds into commercial poultry. In the present study, we performed full genomic characterization and phylogenetic analyses of 15 low pathogenic avian influenza viruses (LPAIVs) isolated from 6 different avian species in Argentina between 2006 and 2016. We inferred time-scaled phylogenies using a previously described host specific local clock modelCitation15 using currently available sequences of IAVs from South America. Our studies provide further support for the existence of a South American (SAm) IAV lineage circulating in wild birds in Argentina. Interestingly, while most HA and NA subtypes obtained from the IAVs from Argentina follow unique evolutionary pathways within the SAm lineage, the HA and NA gene segments of three subtypes, H4, H6, and N8, appear to be recent introductions from IAVs of the NAm lineage. These analyses provide novel insights into the evolutionary patterns of these viruses. Taken together, our study highlights the importance of continuous surveillance of IAVs in South America as an integral part of worldwide efforts to better understand the ecology of these viruses and to better monitor the circulation of strains with zoonotic and/or pandemic concern.
Results
IAVs isolated from wild birds in Argentina with subtype combinations and from hosts not previously described in the region
In total, 6525 cloacal swabs and 70 fecal samples representing 62 different bird species were collected and tested for the presence of IAV (Fig. and STable 1). Birds of the order Anseriformes (n = 3258; 49.4%), Charadriiformes (n = 1973; 29.9%) and Sphenisciformes (n = 744; 11.3%) comprised the majority of the birds sampled. Of these samples, 55 tested positive for IAV by RT-qPCR (0.83%), in their majority from ducks (n = 53), one from gull (Larus dominicanus) and one from parrot (Amazona aestiva) (STable 2).
Species belong to orders Anseriformes (upper 14 species), Charadriiformes (middle 10 species) and Pscittaciformes (bottom species). Right panel: only species with more than 5 samples collected are shown. Left panel: number of isolates per species obtained from RT-qPCR positive samples
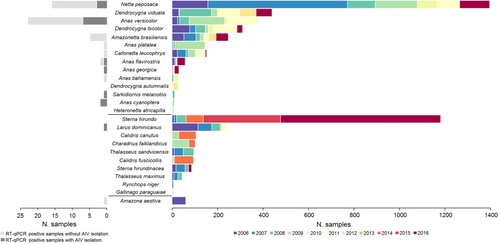
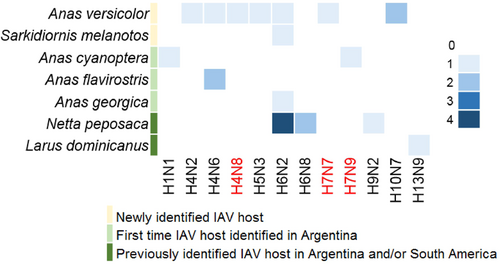
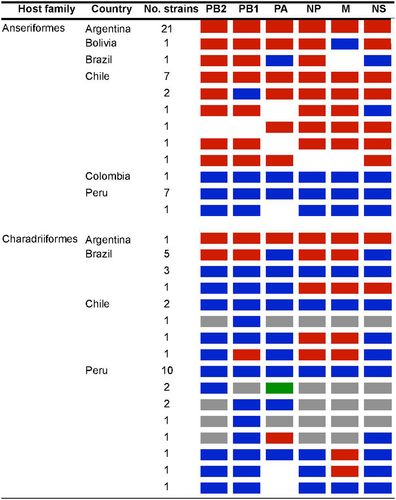
Viable IAVs were isolated from twenty-two RT-qPCR positive samples representing seven different bird species (Fig. ), including Anas versicolor (silver teal, n = 8), Netta peposaca (rosy-billed pochard, n = 7), Anas cyanoptera (cinnamon teal n = 2), Anas flavirostris (yellow-billed teal, n = 2), Anas georgica (yellow-billed pintail, n = 1), Sarkidiornis melanotos (comb duck, n = 1) and Larus dominicanus (kelp gull, n = 1). Of these isolates, 7 have been previously described (subtypes H6N2 (3), H6N8 (2), H9N2 and H13N9, STable 3)Citation12–Citation14. Full genome sequences of the additional 15 IAV isolates (HA subtypes H1, H4, H5, H6, H7, and H10, and NA subtypes N1, N2, N3, N6, N7, N8, and N9) were generated for further characterization (STable 3). The subtype combinations H4N8, H7N7 and H7N9 have not been reported previously in South America (shown in red in Fig. ). Previously unknown IAV hosts were also identified including Anas versicolor (silver teal) and Sarkidiornis melanotos (comb duck). Isolates from Anas cyanoptera (cinnamon teal), Anas flavirostris (yellow-billed teal), and Anas georgica (yellow-billed pintail) were obtained for the first time in Argentina (Fig. ).
The gradient in color intensity indicates the number of isolates obtained for each subtype combination. Novel IAV subtypes, reported for the first time in South America, are shown in red
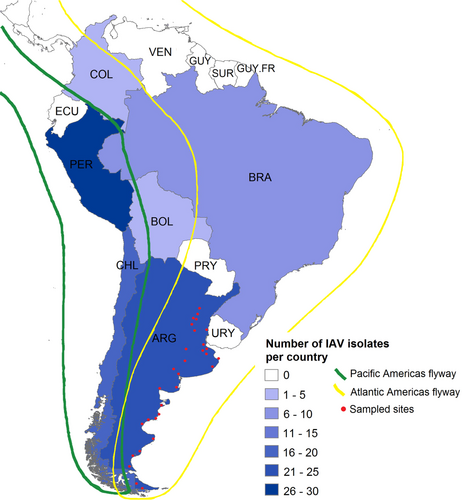
Together, the fifteen Argentinean IAVs account for nearly seventeen percent of IAVs obtained from wild birds throughout South America. With these data, Argentina is the country with the second-highest number of IAV isolates in the Southern Hemisphere (Fig. ).
Argentina (n = 23), Bolivia (n = 1), Brazil (n = 10), Chile (n = 20), Colombia (n = 2), and Peru (n = 30). Bird migration routes are indicated in green and yellow. Red dots represent all sites sampled for IAV in Argentina
Phylogenetic analyses of internal gene segment constellations
The major open reading frame (ORF) sequence of each internal gene segment was used for phylogenetic analyses. Given prior evidence of possible common ancestry, sequences derived from equine IAVs were included in our analysesCitation13,Citation16,Citation17. Prototypic nucleotide IAV sequences from other hosts (bats, swine, and human—except 2009 pandemic H1N1) were also included. The internal gene segments of the new 15 IAVs analyzed clustered together and are highly similar to other previously reported IAVs of avian origin from South America (SFig. 1–6). We used molecular clock analyses that incorporate host-specific evolutionary rates of IAV gene segmentsCitation15 to estimate the time of the most recent common ancestor (tMRCA) for the internal gene segments of the IAV isolates from Argentina (Fig. and STable 4), which indicated that these viruses diverged from other main IAV lineages between ~ 30 and ~ 140 years ago. A closer examination of each tree shows different topologies for each gene segment that indicate that the modern SAm lineage arose from multiple reassortment events, the exact timing of which remain unclear. The PB2 segment of the IAV strains from Argentina was introduced from an EAs-like IAV ancestor (Fig. and SFig. 1) in the late 1880’s (tMRCAPB2 ~ 1887), in agreement with the previous analysesCitation15. For this segment, the SAm avian clade diverged from the EAs lineage sometime between 1885 (tMRCA between EAs and SAm clades) and 1990 (tMRCA for SAm clade), implying a ~ 100-year gap of unknown diversity for the ancestors of the contemporary SAm clade. In contrast, the PB1 segment (Fig. and SFig. 2) diverged more recently from a NAm lineage-like ancestor (tMRCAPB1 ~ 1988). Interestingly, the PA (tMRCAPA ~ 1943, Fig. and SFig. 3) and NP (tMRCANP ~ 1951, Fig. and SFig. 4) segments diverged from the West equine-like (H3N8) lineage (Fig. and STable 4) and share common ancestry with other IAVs from South America, Antarctica, and other equine-origin IAVsCitation7,Citation18. Similar to the PB1, the M segment diverged more recently from a NAm lineage ancestor (tMRCAM ~ 1971, Fig. and SFig. 5) (STable 4).
Each dot represents the time of the most recent common ancestor, or node, between the SAm lineage and the most closely related lineage of each of 10,000 trees inferred for each segment. The identity of the most closely related lineage is shown by the colors. Mean tMRCA and 95% HPDs are represented by whiskers. Eurasian (EAs), North American (NAm)
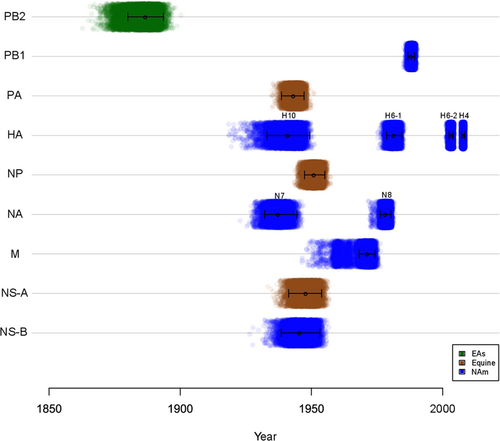
Phylogenetic analysis of the NS genes showed that 13 of the IAVs from Argentina contain the NS allele A (NS-A), whereas only two carry allele B (NS-B) (SFig. 6). Both of these alleles diverged from the main IAV lineages around the same time, between 1939 and 1954 (Fig. and STable 4). Like the PA and NP segments, the NS-A gene segments form a sister group with IAVs of equine originCitation13,Citation16,Citation17.
Analysis of the internal gene segment constellations for all Argentinean LPAIV strains and other viruses obtained from wild aquatic birds and poultry in South America revealed that all viruses from Argentina are positioned exclusively within the South American lineage. In contrast, viruses from Bolivia, Brazil, Chile, Colombia, and Peru include reassortants with IAVs of other genetic lineages (Fig. , STable 5 and SFigs. 1–6).
IAVs by host-order and country color-coded as South American lineage (red), North America lineage (blue), Eurasian lineage (green), other global avian lineages (gray), and no sequence data available (white)
Phylogenetic analyses of the surface gene segments
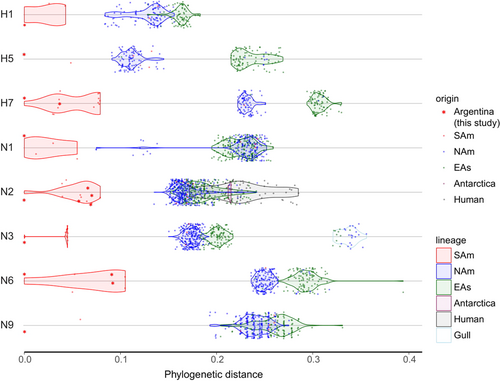
Overall the HA gene segments of Argentinean IAVs clustered with other South American IAVs (H1, H5, H7, H4, and a subset of H6, named H6-2), with the exception of the H10 and a subset of H6 (H6-1) gene segments that formed a cluster of IAVs exclusively from Argentina. We denoted all these clades as SAm lineage. Following is a description of the main findings for each HA subtype. The H1 and H5 isolates from Argentina grouped within the NAm lineage (Fig. and SFigs. 7 and 8). The H1 subtype SAm lineage viruses were recovered from resident wild birds (Anseriformes and Tinaniformes) from Argentina and Chile (Fig. and SFig. 7). The other H1 subtype IAVs identified in Peru (Charadriiformes and Gruiformes) belongs to the NAm lineage (Fig. and SFig. 7). The H5 subtype SAm lineage contains only two sequences from IAVs from resident duck species (silver teal and yellow-billed pintail) in Argentina and Chile. There were other two highly similar H5 sequences in the database (from Chile), but they were excluded from the analysis because they were only partial fragments of the H5. In addition, there are other H5 subtype IAVs isolated from different wild duck spp. in South America (Chile and Colombia), but they cluster in a different position within the NAm lineage and are less divergent compared to the Argentinean isolate (Fig. and SFig. 8). The H7 subtype from IAVs from the SAm lineage was basal to the main NAm lineage, showing two clades (SFig. 9). One clade contains the two H7 IAVs from Argentina and other IAVs from Chile (all IAV isolates from resident wild duck spp.). The second clade contains the HPAIV poultry isolates from the outbreak in Chile in 2002 and their closest wild birds LPAIV ancestor from Bolivia (cinnamon teal) (refs. Citation8,Citation17). Although all H7 from Argentina are LPAIV, they share a common ancestor with H7 HPAIVs from 2002. Similar to the H5 subtype, the other H7 subtype identified from a wild duck (unknown spp.) in Peru belongs to the NAm lineage (Fig. and SFig. 9).
Each dot represents a single sequence (virus) and it is colored by its source (geographic or host). Argentinean IAVs are shown with red asterisks. The continuous distribution of viruses within their main lineages is shown by the violin shapes for lineages with more than 2 observations: avian South American (SAm), North America (NAm), Eurasian (EAs). Argentinean isolates used as references and shown at zero distance: 432/H1N1 (H1 and N1), 1737/H5N3 (H5 and N3), 188/H7N7 (H7), 1174A/H6N2 (N2), 1227/H4N6 (N6), 1588/H7N9 (N9). Detailed phylogenies are provided in the supplementary material
The H10 subtype has been previously reported in Anseriformes and Charadriiformes in PeruCitation19. For this subtype, the two H10N7 IAV isolates from Argentina grouped together and formed a unique branch that is clearly separated from H10 gene segments from viruses from other geographic locations, and remarkably distant from the H10N7 viruses from Peru (SFig. 10, red branches). As this was the first isolation of a SAm H10 subtype, we inferred an MCC tree to estimate the time of divergence of these viruses. Both H10 Argentinean viruses, isolated from silver teal, have a common ancestor with H10 viruses from NAm lineage, with an estimated tMRCA between 1933 and 1949 (95% HPD) (Fig. and STable 4).
Phylogenetic analysis of H4 HA gene segment showed that the IAVs from Argentina (four isolates from 2011 and one from 2016) grouped within the NAm lineage and were closely related to other H4 viruses identified in Anseriformes in Chile in 2013 and 2015, suggesting dissemination of NAm H4 subtype IAVs in the region (SFig. 11). As this seemed to be a recent introduction to South America we estimated the time of introduction for this clade. Time-scaled phylogenetic analysis estimated that the introduction of these NAm H4 viruses in South America occurred between 2007–2009 (95% HPD) (Fig. and STable 4). Similarly, the MCC tree of H6 HA gene showed at least two separate introductions of H6 viruses in Argentina (identified as H6-1 and H6-2 in Fig. and SFig. 12). The first introduction (H6-1) includes two new H6 isolates (272/H6N2 and 1174A/H6N2) that clustered together with four H6 IAVs from Argentina published previouslyCitation13. In this case, we estimated that the SAm clade (as well as the most closely related NAm clade) has diverged from the EAs lineage sometime between 1978 and 1984 (95% HPD, Fig. and STable 4). The second most recent introduction (H6-2) includes other two new H6 isolates (49/H6N2 and 52/H6N2) that grouped together with one of our previous H6 subtype IAVs (1977/H6N2) and three IAVs from Brazil isolated in 2012 (SFig. 12). On this occasion, the SAm clade seems to have diverged from the NAm lineage sometime between 2002 and 2004 (95% HPD, Fig. and STable 4). The new Argentinean H6 IAVs were obtained from four wild duck spp. (comb duck, rosy-billed pochard, silver teal, and yellow-billed pintail), while Brazilian H6 IAVs were obtained from a small shorebird (white-rumped sandpiper, a Charadriiforme).
For the NA gene segments, phylogenetic analysis of the N1, N2, N3, N6, and N9 gene segments grouped them into the unique SAm clades as previously observedCitation12–Citation14 (Fig. and SFigs. 13–17). The N1 subtype SAm lineage contains four similar sequences, three are paired with the H1 HA subtype from IAVs isolated in Argentina and Chile (from Tinaniformes and Anseriformes respectively), and one is paired with the H6 HA subtype from IAVs isolated from white-rumped sandpiper in Brazil (SFig. 13). These four viruses form a sister group with some IAVs isolated from shorebirds in Delaware Bay and New Jersey in a cluster that is basal to the NAm and EAs lineages. The N1 of other IAVs identified from Chardriiformes in Brazil and Peru belong to the NAm lineage. The N2 phylogenetic tree shows IAVs from SAm lineages share ancestry with avian IAVs from diverse origins that are basal to the main NAm and EAs lineages. The SAm viruses grouped in two clades (SFig. 14), one clade contains IAVs isolated exclusively from Anseriformes in Argentina and Chile between 2007 and 2014 (n = 9, four new sequences from this report). The second clade contains one N2 subtype from an IAV isolated in Argentina in 2016 (this report) and N2 sequences from IAVs from Chile (2015). The other N2 IAVs identified in Chile, Colombia, and Peru belong to the NAm lineage. The N3 subtype from IAVs from the SAm lineage also shows two clades (SFig. 15). One clade contains only two H5N3 IAVs isolated from wild ducks, one from Argentina (this report) and the other from Chile. There is a 6-year gap between these IAVs (2009 and 2015) and they were isolated from different species—silver teal and yellow-billed pintail, respectively. The second clade contains only H7N3 HPAIV isolate from poultry in Chile in 2002 (ref. Citation8) (SFig. 15). These two clades shared ancestry with non-contemporary NAm viruses, that are basal to NAm lineage. The N6 subtype SAm lineage, basal to the NAm lineage, contains the three newly identified N6 IAVs from Argentina and other sequences from Chile, all from resident wild bird spp. (SFig. 16). The only other N6 IAV identified in South America, in Peru, belongs to the NAm lineage and was isolated from a gull. Interestingly, our last N2 and N6 IAV isolates from 2016 (1174A/H6N2 and 1227/H4N6 respectively) are more distant from previous isolates from Argentina and more closely related to recent N2 and N6 segments from IAVs isolated in Chile (SFig. 14 and 16 respectively). With respect to the N9 gene segments, those obtained from IAVs in Argentina (n = 2) were basal to the main EAs and NAm lineages, whereas N9 subtype sequences from other IAV isolates from Brazil, Chile and Peru clearly belong to the NAm lineage (SFig. 17).
The N7 subtype phylogeny shows that sequences of those IAVs isolated in Argentina, all from silver teals, are unique and share no relationship with those obtained from wild bird isolates in ChileCitation20 or PeruCitation19 (SFig. 18, red branches). We estimated the time of divergence of these distinct viruses. The N7 subtype sequences from IAVs from Argentina have a NAm ancestor with a tMRCA between 1932–1945 (95% HPD), similar to the H10 HA subtype sequences (Fig. and STable 4).
Finally, three N8 IAVs from Argentina clustered together as a distinct clade within the NAm lineage. Time-scaled phylogenetic analysis estimated that the time of introduction of the N8 subtype in Argentina was between 1976–1980 (95% HPD) (Fig. and STable 4), which overlaps with the first introduction of H6 HA in SAm, suggesting common origins in a NAm ancestor. The N8 NA phylogenetic tree revealed that previously described sequences from IAVs from wild birds (not Anseriformes) in PeruCitation19 also belong to the NAm lineage but represent a more recent, independent introduction, different from Argentina (red branches SFig. 19).
Molecular analyses of virulence markers
The diversity of HA subtypes found in IAVs from Argentina confirm the circulation of group 1 (H1, H5, H6, H9, and H13) and group 2 (H4, H7, and H10) of HAs previously proposedCitation21. Previous studies have demonstrated the amino acid changes in the receptor binding site at positions 190, 225, 226, and 228 of the HA protein (H3 numbering) in a variety of subtype, including H1, H2, H3, H4, H5, H6, and H7Citation22–Citation25. Analysis of predicted HA amino acid sequences showed that all Argentinean viruses isolated from 2006 to 2016, regardless of subtype, contain amino acid residues consistent with binding to “avian-like” α2,3 sialic acid receptors (E190, G225, Q226, and G228, data not shown). In addition, 16 out of 22 Argentinean IAV strains shared potential glycosylation sites of asparagine (Asn) along the HA1 domain (H3 numbering) with IAVs previously isolated in other regions: Asn129Citation26 for 432/H1N1 strain; Asn6, Asn22, Asn165, Asn196, and Asn483Citation27 for all H4 strains (n = 5); Asn158Citation28 for 1737/H5N3 strain; and Asn21, Asn33, Asn170, Asn291, and Asn296Citation29 for all H6 strains (n = 9). Instead, the 188/H7N7 and 1588/H7N9 strains maintain Asn133 but have the mutation N158T that abolishes a potential glycosylation site.
The HA gene segments of the H1, H4 (n = 5), H5, H6 (n = 9), H9 and H13 subtypes, have cleavage site motifs typical of LPAIV (Table ). The HA gene segment of the H7 (n = 2) and H10 (n = 2) subtypes, have cleavage sites different from the consensus sequences described by Baron et al.Citation30 (Table ). In addition to the arginine at the P1 position, the H7 HA cleavage site contains basic amino acids (lysine) at the P3 and P5 positions, and the H10 HA cleavage site carries histidine at the P3 position. It remains to be determined if such cleavage site motifs modulate the virulence of these viruses in wild birds and/or poultry.
Cleavage site motifs of avian-origin influenza A viruses isolated in Argentina
No mammalian-associated virulence markers in NS1 protein (T92E)Citation31 or PB2 (E627K and D701N)Citation32–Citation34 were found in the viruses from Argentina (STable 6). None of the viruses contained mutations associated with drug resistance in the NA and M2 proteinsCitation35,Citation36, except for the 432/H1N1 virus, which carries the mutation NA H274Y predictive of oseltamivir resistanceCitation37. No mutations associated with enhanced transmission in mammals were found in the PB2 and PA proteinsCitation34,Citation38. In comparison to other lineages (NAm and EAs), none of the amino acid residues associated with drug resistance and virulence described here were unique to the Argentinean viruses or the SAm clade (STable 6). High degree of sequence homology at the protein level of the SAm lineage with other lineages was previously observedCitation13.
The IAVs from Argentina contain the typical PB1-F2 ORF of 90 amino acids in length commonly found in IAVs from ducksCitation39,Citation40. There are 45 amino acid differences among the PB1-F2 proteins in the IAVs from Argentina (SFig. 20). Interestingly, the PB1-F2 of Argentinean strains carry the N66S mutation implicated in increase virulence for mammalsCitation41, with the exception of the 52/H6N2 strain. In addition, the PB1-F2 protein of the 1227/H4N6 strain contains three amino acid markers, 51T, 56V, and 87E, associated with virulence in mallards of a highly pathogenic H5N1 avian influenza virusCitation42. Other Argentinean strains contain the 56V and 87E markers, the 1977/H6N2 contains only the 87E and the 1174A/H6N2 and 559/H9N2 strains contain only the 56V marker. Since the effects of PB1-F2 are virus- and host-dependent, further studies are needed to better understand the role of these PB1-F2 variants in the IAVs from ArgentinaCitation40,Citation41,Citation43.
The IAVs from Argentina also contain the predicted full-length PA-X ORF of 252 amino acids in length. There are 24 amino acid differences among the PA-X proteins in the IAVs from Argentina (SFig. 21). As is the case for PB1-F2, the effects of PA-X on virus replication and pathogenesis are strain- and host-dependentCitation44,Citation45, and further studies are necessary to determine the biological significance of these mutations in birds.
Discussion
Long-term IAV surveillance in wild waterfowl in Argentina between 2006 and 2016 resulted in the recovery of 22 isolates, 15 of which are newly reported here. Despite limited and opportunistic sampling across host species and locations within Argentina throughout the 10 years of the study (shown as red dots in Fig. ), we consistently observed that all internal segments of the Argentinean viruses were closely related to those from viruses previously isolated in Argentina, and exclusive to this geographical location in South America. Taken together, the results provide additional support for the existence of a unique SAm avian lineage with local independent evolution for all internal viral gene segmentsCitation12–Citation14,Citation17.
The phylogenetic analysis of the internal gene segments from the Argentinean IAVs showed shared ancestry with equine-origin IAVs (PA, NP, NS-A), and avian-origin IAVs of Eurasia (PB2), North America (PB1, M, NS-B) and Antarctica (PA, M), demonstrating that their genetic diversity has been shaped by multiple introductions and reassortment occurring as early as the end of the 19th centuryCitation15. Interestingly, the IAVs isolated from wild birds in Argentina have all internal gene segments of SAm origin, while many Chilean and Peruvian viruses have either all or most of their internal genes segments of NAm origin, and close to those from Central AmericaCitation19,Citation20,Citation46. More recently, H5N5 isolates from Antarctica were shown to carry gene constellations of NAm originCitation7,Citation18,Citation47. While reassortment of internal gene segments from IAV from different lineages do occur in parts of South America, the Argentinean IAVs circulate mostly in isolation with limited gene segment exchange with viruses from other latitudes, regardless of their subtype.
Phylogenies of the H1, H5, H7, and H10 HA subtypes and the N1, N2, N3, N6, N7, and N9 NA subtypes demonstrated that both genes follow unique evolutionary patterns in wild birds in Argentina. Although the primordial source of these viruses remains unknown, the results suggest that the Argentinean strains split from their ancestors, either Eurasian or North American, ≤ 30 years ago and continue an independent evolution pathway. It must be noted that our surveillance efforts in Argentina have covered limited areas of the country and a limited subset of avian species. Together with the scarce data available from IAVs in South America, it is difficult to confirm if the basal lineages of SAm viruses are ancestral to other main IAV lineages (for example NAm lineage) or whether they are a consequence of earlier virus introductions (from any lineage) that then diverged separately in this region. Expansion into broader geographic areas, animal species, and number of samples are indispensable as a first step to better understand the ecology and evolution of IAVs in wild birds in Argentina. Sequence analysis of the Argentinean IAVs are consistent with typical LPAIVs; however, there are remarkably divergent variants (e.g. H5, H7, H10) that warrant further experimental characterization in the laboratory to deepen our knowledge in the pathogenicity and host-range of these particular SAm IAVs.
In this study, we traced the origin of the Argentinean H4, H6, and N8 subtypes to North America, and detected at least four independent introduction events within the past 40 years. Particularly, the results of the H6 analyses further support the presence of two independent H6 subtype populations circulating in ArgentinaCitation13. In addition, these two Argentinean H6 gene clusters are different from the H6 subtype population that circulates in Charadriiforms and Pelecaniformes in Peru, indicating distinct ways of dissemination of NAm H6 subtype IAVs along the region. The IAV gene influx from North America into South America could be driven by the migration of different bird species that arrive to the Southern Hemisphere through the Atlantic and Pacific flyways (Fig. ). With the available data, however, the bird species involved in this process and the frequency of such events remains unclear.
At the level of internal gene constellations, the lack of reassortment between the viruses from Argentina and other latitudes is notable (Fig. and STable 5), but may be the result of sampling bias in geographical locations and/or host species. Several IAVs from Chile and Peru were obtained from gulls and shorebirds that perform long migrations that serve as important circuits for the dissemination of IAVCitation48,Citation49. In contrast, Argentinean isolates were obtained almost exclusively from resident wild duck species with distribution more restricted to South AmericaCitation50. Systematic surveillance of a larger number of IAV isolates obtained from South America will be necessary to trace the true origin of the SAm lineage, and further understanding of population densities and regional movement among migratory and resident birds in Argentina and other SAm countries is crucial to understand the dynamics of IAV transmission. Monitoring the movement of wild birds by geolocators, and identifying species subpopulations through analysis of stable isotopes and molecular genotypingCitation51–Citation53, have the potential to establish the possible points of contact in which wild birds from North and South America overlap to allow virus transmission and reassortment. Such studies would also serve to elucidate whether geographic barriers, such as the Andes Mountains (natural border between Argentina and Chile), restrict the movement of wild bird populations and spillover of viruses from other genetic lineages (e.g., N.Am) into resident species, explaining the absence of reassortant virus strains in Argentina.
Materials and methods
Sample collection
Trained biologists, veterinarians, and park rangers performed sampling between April–July (waterfowl and resident birds) and October–March (shorebirds and migratory species), from 2006 to 2016. Samples were obtained from a variety of seabirds (gulls, cormorants, penguins, terns, and shorebirds) captured along the Argentine Atlantic coast, and hunter-killed ducks in the Lower Paraná River Valley, as described previouslyCitation12,Citation13.
Cloacal swabs were collected using single-use sterile polyester swabs and then stored separately in single plastic cryovials, containing 2 ml of phosphate buffer solution (PBS) with 50% glycerol and penicillin 10,000 IU/ml, streptomycin 5 mg/ml, gentamicin Sulfate 1 mg/ml, kanamycin sulfate 700 µg/ml and amphotericin B 10 µg/ml (Sigma Chemical Co, St. Louis, MO, USA). Samples were frozen in liquid nitrogen and transported on dry ice. Once in the laboratory, all samples were stored at −80 °C until processing for molecular diagnosis and virus isolation.
Virus detection
Viral RNA was extracted from 140 µl of suspension from cloacal swabs using a QIAamp Viral RNA Mini Kit (Qiagen Inc., Valencia, CA, USA). RNA was eluted in a final volume of 60 µl and stored at −80 °C until further use. Viral cDNA was prepared using 15 µl of viral RNA and random hexamers in a final volume of 30 µl using a High Capacity cDNA Archive kit (Applied Biosystems, Foster City, CA, USA). The cDNA was tested for IAV by real-time reverse transcriptase PCR (RT-qPCR) using TaqMan Universal PCR Master Mix (Applied Biosystems) directed to the matrix (M) gene. Samples collected from 2006 to 2012 were tested using primers and probe previously described by Spackman et al.Citation54, and samples collected since 2013 were tested using primers (InfA Forward and InfA Reverse) and probe (InfA Probe) described in WHO/CDC protocol titled “CDC protocol of real time RTPCR for swine influenza A(H1N1)” and “28 April 2009 revision 1 (30 April 2009).” The PCR reaction was performed on an ABI Prism 7500 SDS (Applied Biosystems). Both methods were validated for sensitivity against avian influenza viruses and no differences were obtained with respect to IAV detection (data not shown).
Virus isolation
Swab samples that tested positive by RT-qPCR were inoculated into 9–11-day old specific-pathogen-free (SPF) embryonated chicken eggs. Briefly, 200 µl of PBS suspension from cloacal swab samples was injected into the allantoic cavity of the eggs, incubated for 72 h, and harvested in accordance with standardized protocols as suggested by the World Health Organization (http://www.wpro.who.int/emerging_diseases/documents/manual_on_animal_ai_diagnosis_and_surveillance/en/) and following Argentine regulations.
Full genome sequencing and assembly by Next Generation Sequencing
PCR was performed using specific primers for each gene segment as described previouslyCitation55. PCR products were purified with a QIAQuick PCR purification kit (Qiagen). Sequencing was performed using the BigDye Terminator v3.1 Cycle Sequencing Kit on an ABI PRISM 3700 DNA Analyzer (Applied Biosystems) following the manufacturer’s instructions. A subset of isolates (marked with “#” in STable 3) was re-sequenced by Next Generation Sequencing and nucleotide sequence identity from Sanger sequencing was verified. Briefly, products from a multisegment-RTPCR strategyCitation56 were cleaned by 0.45X of Agencourt AMPure XP Magnetic Beads (Beckman Coulter) according to manufacturer’s protocol. Eluate concentration was measured using the Qubit High Sensitivity dsDNA kit (Fisher Scientific) in the Qubit 3.0 fluorometer (ThermoFisher) and normalized to 0.2 ng/ul. Adaptors were added by tagmentation using the Nextera XT DNA library preparation kit (Illumina). Samples were purified using 0.7X of Agencourt AMPure XP Magnetic Beads and fragment size distributions were analyzed on a Bioanalyzer using the High Sensitivity DNA kit (Agilent). Next, samples were normalized to 4 nM and pooled. The final loading concentration of the pooled libraries was 15pM. Libraries were sequenced in a paired-end run using the MiSeq v2, 300 cycle reagent kit (Illumina).
Genome assembly was performed using a customized pipeline developed at the Icahn School of Medicine at Mount SinaiCitation56.
Phylogenetic and molecular analysis
The consensus amino acid and nucleotide sequences for all 8 gene segments of the viruses were generated using Megalign (DNASTAR, Madison, WI, USA). Phylogenetic analyses were performed using additional influenza virus sequence data available in the Influenza Research Database (https://www.fludb.org/brc/home.spg?decorator = influenza), and were run with Maximum clade credibility in BEAST or Maximum Likelihood (ML) in MEGA (see below).
Time-scaled phylogenetic analyses were performed in the program BEAST v.1.8.2. For the virus internal gene segments, and surface glycoproteins N7 and N8, previously published datasets were downloaded from the Dryad repositoryCitation15 with permission from the authors. Additional virus nucleotide sequences (2011–2016) were added to the original alignments, including all sequences from Argentina. Representative strains from South America were selected after inferring Maximum Likelihood phylogenetic trees for all internal gene segments available in IRD. The new alignments for PB2 (total number of sequences = 435), PB1 (n = 413), PA (n = 462), NP (n = 508), M (n = 519), NS (n = 442), N7 (n = 213) and N8 (n = 463) were performed with MUSCLE as implemented in the program MEGA 6.0. The MCC trees were inferred using a host-specific local clock model as previously describedCitation15, in which all host clades are permitted individual substitution rates. The parameters for the time-scaled phylogenetic analysis were set in the program Beauti, with manual edits for assignment of host-specific rates as specified in the original xml filesCitation15. We used the SRD06 model of nucleotide substitutionCitation57, and the coalescent tree prior GMRF Bayesian Skyride. For the H4, H6, and H10, the inferred NJ phylogenies were analyzed in the program Path-O-Gen to identify and remove outlier sequences. The final alignments of H4 (n = 264), H6 (n = 469) and H10 (n = 258) were used for phylogenetic analyses. The MCC trees were then inferred under the SRD06 model of nucleotide substitution, using the uncorrelated lognormal clock and a constant population size. For the mean rate, a prior of 0.001 was used in all cases. For each data set the MCMC chain was run for 20–100 million steps until convergence of parameters was observed in the program Tracer (v.1.6.0). For each chain, the first 10% states were removed (10% burn-in). Then the results of at least three independent runs were merged in the program LogCombiner. The MCC trees were summarized and annotated in TreeAnnotator, visualized and printed in FigTree (v.1.4.2). The times of divergence for the viruses from South America and Argentina were obtained from the node ages (and their corresponding 95% HPD) on the final MCC trees.
Maximum likelihood (ML) phylogenetic trees for H1 (n = 172), H5 (n = 183), H7 (n = 173), N1 (n = 339), N2 (n = 637), N3 (n = 380) and N6 (n = 264) and N9 (n = 297) gene segments were constructed by using MEGA 6.0 (https://www.megasoftware.net/), including the viruses from Argentina, other IAVs from South America and other background sequences representative of the LPAIV global diversity, available in Influenza Research Database (https://www.fludb.org/brc/home.spg?decorator=influenza). Estimates of the phylogenies were calculated by performing 1000 bootstrap replicates under a general reversible model with gamma distribution (GTR+G). The FigTree (v.1.4.2) program was used for visualization and printing of phylogenetic trees.
Nucleotide sequence accession numbers
The nucleotide sequences obtained in this study are available from GenBank under accession numbers MK070525 to MK070532 and MK071345 to MK071456.
Supplementary Figure Legends 1-21
Download PDF (33.1 KB)Supplementary Table 1
Download MS Word (139 KB)Supplementary Table 2
Download MS Word (63.5 KB)Supplementary Table 3
Download MS Word (79 KB)Supplementary Table 4
Download MS Word (64.5 KB)Supplementary Table 5
Download MS Word (206.5 KB)Supplementary Table 6
Download MS Word (125 KB)Supplementary Figure 1
Download PDF (735.2 KB)Supplementary Figure 2
Download PDF (394.6 KB)Supplementary Figure 3
Download PDF (762.4 KB)Supplementary Figure 4
Download PDF (450.9 KB)Supplementary Figure 5
Download PDF (462.9 KB)Supplementary Figure 6
Download PDF (417.3 KB)Supplementary Figure 7
Download PDF (317.4 KB)Supplementary Figure 8
Download PDF (290.1 KB)Supplementary Figure 9
Download PDF (295.8 KB)Supplementary Figure 10
Download PDF (286.6 KB)Supplementary Figure 11
Download PDF (298 KB)Supplementary Figure 12
Download PDF (424.7 KB)Supplementary Figure 13
Download PDF (469.4 KB)Supplementary Figure 14
Download PDF (764.6 KB)Supplementary Figure 15
Download PDF (490.3 KB)Supplementary Figure 16
Download PDF (416 KB)Supplementary Figure 17
Download PDF (414.2 KB)Supplementary Figure 18
Download PDF (258.9 KB)Supplementary Figure 19
Download PDF (285.8 KB)Supplementary Figure 20
Download PDF (79.1 KB)Supplementary Figure 21
Download PDF (96.8 KB)Acknowledgements
The National Institute of Allergy and Infectious Diseases (NIAID) Center for Research on Influenza Pathogenesis (CRIP) (contracts HHSN266200700010C and HHSN272201400008C) and the Instituto Nacional de Tecnología Agropecuaria (INTA) (PNSA 1115052 and PNSA 1115056) funded this study. This study was supported in part by resources and technical expertize from the Georgia Advanced Computing Resource Center, a partnership between the University of Georgia’s Office of the Vice President for Research and Office of the Vice President for Information Technology. This work was also supported by PICT 2008 – 0922 from MINCyT and CONICET, through a fellowship gave to Agustina Rimondi during part of her work. The content is solely the responsibility of the authors and does not represent official views of the National Institutes of Health.
Author contributions
Author contributions were as follows: Study design: A.R., A.P. and D.R.P. Samples and data collection: A.R., V.S.O., J.D., G.C. and M.R. Virus detection and virus sequencing: A.R., V.S.O., A.S.G., A.P, L.F, G.G. and H.B. Phylogenetic analyses: A.R. (maximum likelihood phylogenetic inference) and A.S.G. (time-scaled Bayesian phylogenetic analyses). Data analysis and interpretation: A.R., A.S.G. and M.N. Writing of the manuscript: A.R., A.S.G. and D.R.P. All authors approved the manuscript before submission.
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41426-018-0190-2).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related Research Data
References
- FouchierRAMMunsterVJEpidemiology of low pathogenic avian influenza viruses in wild birdsRev. Sci. Tech.200928 49 5810.20506/rst.28.1.1863
- KawaokaYKraussSWebsterRGAvian-to-Human Transmission of the PB1 Gene of Influenza A Viruses in the 1957 and 1968 PandemicsJ. Virol.19896346034608251093
- WHO/OIE/FAO H5N1 Evolution Working Group.Toward a unified nomenclature system for highly pathogenic avian influenza virus (H5N1)Emerg. Infect. Dis.200814e110.3201/eid1407.071681
- LeeDHet al.Intercontinental Spread of Asian-Origin H5N8 to North America through Beringia by Migratory BirdsJ. Virol.2015896521652410.1128/JVI.00728-15
- VergneTet al.Avian influenza vaccination of poultry and passive case reporting, EgyptEmerg. Infect. Dis.2012182076207810.3201/eid1812.120616
- WaziriNEet al.Evaluating a surveillance system: live-bird market surveillance for highly pathogenic avian influenza, a case studyPan Afr. Med. J.201418114199346
- HurtACet al.Detection of evolutionarily distinct avian influenza A viruses in AntarcticamBio20145e01098e0111410.1128/mBio.01098-14
- SuarezDLet al.Recombination resulting in virulence shift in avian influenza outbreak, ChileEmerg. Infect. Dis.20041069369910.3201/eid1004.030396
- ObenauerJCet al.Large-Scale Sequence Analysis of Avian Influenza IsolatesScience20063111576158010.1126/science.1121586
- BulachDet al.Molecular analysis of H7 avian influenza viruses from Australia and New Zealand: genetic diversity and relationships from 1976 to 2007J. Virol.2010849957996610.1128/JVI.00930-10
- WebsterRGBeanWJGormanOTChambersTMKawaokaYEvolution and ecology of influenza A virusesMicrobiol. Rev.199256152179372859
- PeredaAJet al.Avian influenza virus isolated in wild waterfowl in Argentina: evidence of a potentially unique phylogenetic lineage in South AmericaVirology200837836337010.1016/j.virol.2008.06.010
- RimondiAet al.Phylogenetic analysis of H6 influenza viruses isolated from rosy-billed pochards (Netta peposaca) in argentina reveals the presence of different HA gene clustersJ. Virol.201185133541336210.1128/JVI.05946-11
- XuKet al.Isolation and characterization of an H9N2 influenza virus isolated in ArgentinaVirus Res.2012168414710.1016/j.virusres.2012.06.010
- WorobeyMHanGZRambautAA synchronized global sweep of the internal genes of modern avian influenza virusNature201450825425710.1038/nature13016
- SpackmanEMcCrackenKGWinkerKSwayneDEAn avian influenza virus from waterfowl in South America contains genes from North American avian and equine lineagesAvian Dis.20075127327410.1637/7529-032106R.1
- SpackmanEMcCrackenKGWinkerKSwayneDEH7N3 avian influenza virus found in a South American wild duck is related to the Chilean 2002 poultry outbreak, contains genes from equine and North American wild bird lineages, and is adapted to domestic turkeysJ. Virol.2006807760776410.1128/JVI.00445-06
- HurtACet al.Evidence for the introduction, reassortment, and persistence of diverse influenza A viruses in AntarcticaJ. Virol.2016909674968210.1128/JVI.01404-16
- NelsonMIet al.The genetic diversity of influenza A viruses in wild birds in PeruPLoS One201611e014605910.1371/journal.pone.0146059
- Jiménez-BluhmPet al.Wild birds in Chile Harbor diverse avian influenza A viruses articleEmerg. Microbes Infect.201874710.1038/s41426-018-0046-9
- NobusawaEet al.Comparison of complete amino acid sequences and receptor-binding properties among 13 serotypes of hemagglutinins of influenza A virusesVirology199118247548510.1016/0042-6822(91)90588-3
- ShiYWuYZhangWQiJGaoGFEnabling the “host jump”: structural determinants of receptor-binding specificity in influenza A virusesNat. Rev. Microbiol.20141282283110.1038/nrmicro3362
- WangFet al.Adaptation of avian influenza A (H6N1) virus from avian to human receptor-binding preferenceEMBO J.2015341661167310.15252/embj.201590960
- TzarumNet al.Structure and receptor binding of the hemagglutinin from a human H6N1 influenza virusCell. Host. Microbe20151736937610.1016/j.chom.2015.02.005
- SongHet al.Avian-to-human receptor-binding adaptation by influenza A virus hemagglutinin H4Cell Rep.2017201201121410.1016/j.celrep.2017.07.028
- DeomCMCatonAJSchulzeITHost cell-mediated selection of a mutant influenza A virus that has lost a complex oligosaccharide from the tip of the hemagglutininProc. Natl. Acad. Sci. U. S. A.1986833771377510.1073/pnas.83.11.3771
- LiangLet al.Genetics, receptor binding, replication, and mammalian transmission of H4 Avian influenza viruses isolated from live poultry markets in ChinaJ. Virol.2016901455146910.1128/JVI.02692-15
- WangWet al.Glycosylation at 158N of the hemagglutinin protein and receptor binding specificity synergistically affect the antigenicity and immunogenicity of a live attenuated H5N1 A/Vietnam/1203/2004 vaccine virus in ferretsJ. Virol.2010846570657710.1128/JVI.00221-10
- WangGet al.H6 influenza viruses pose a potential threat to human healthJ. Virol.2014883953396410.1128/JVI.03292-13
- BaronJet al.Matriptase, HAT, and TMPRSS2 activate the hemagglutinin of H9N2 influenza A virusesJ. Virol.2013871811182010.1128/JVI.02320-12
- HeuiSSHoffmannEWebsterRGLethal H5N1 influenza viruses escape host anti-viral cytokine responsesNat. Med.2002895095410.1038/nm757
- SubbaraoEKLondonWMurphyBRA single amino acid in the PB2 gene of influenza A virus is a determinant of host rangeJ. Virol.19936717611764240216
- HattaMGaoPHalfmannPKawaokaYMolecular basis for high virulence of Hong Kong H5N1 influenza A virusesScience20012931840184210.1126/science.1062882
- ShinyaKet al.PB2 amino acid at position 627 affects replicative efficiency, but not cell tropism, of Hong Kong H5N1 influenza A viruses in miceVirology200432025826610.1016/j.virol.2003.11.030
- McKimm-BreschkinJLResistance of influenza viruses to neuraminidase inhibitors–a reviewAntivir. Res.20004711710.1016/S0166-3542(00)00103-0
- AbedYGoyetteNBoivinGGeneration and characterization of recombinant influenza A (H1N1) viruses harboring amantadine resistance mutationsAntimicrob. Agents Chemother.20054955655910.1128/AAC.49.2.556-559.2005
- BazMAbedYSimonPHamelinMBoivinGEffect of the neuraminidase mutation H274Y conferring resistance to oseltamivir on the replicative capacity and virulence of old and recent human influenza A(H1N1) virusesJ. Infect. Dis.201020174074510.1086/650464
- GabrielGet al.The viral polymerase mediates adaptation of an avian influenza virus to a mammalian hostProc. Natl Acad. Sci. USA.2005102185901859510.1073/pnas.0507415102
- KrumbholzAet al.Current knowledge on PB1-F2 of influenza A virusesMed. Microbiol. Immunol.2011200697510.1007/s00430-010-0176-8
- KošíkIHollýJRussGPB1-F2 expedition from the whole protein through the domain to aa residue functionActa Virol.20135713814810.4149/av_2013_02_138
- ConenelloGMZamarinDPerroneLATumpeyTPalesePA single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulencePLoS. Pathog.200731414142110.1371/journal.ppat.0030141
- MarjukiHet al.Three amino acid changes in PB1-F2 of highly pathogenic H5N1 avian influenza virus affect pathogenicity in mallard ducksArch. Virol.201015592593410.1007/s00705-010-0666-4
- ZamarinDOrtigozaMBPalesePInfluenza A virus PB1-F2 protein contributes to viral pathogenesis in miceJ. Virol.2006807976798310.1128/JVI.00415-06
- JaggerBWet al.An overlapping protein-coding region in influenza A virus segment 3 modulates the host responseScience201233719920410.1126/science.1222213
- GaoHet al.The contribution of PA-X to the virulence of pandemic 2009 H1N1 and highly pathogenic H5N1 avian influenza virusesSci. Rep.2015510.1038/srep08262
- Gonzalez-ReicheASet al.Evidence of intercontinental spread and uncommon variants of low-pathogenicity avian influenza viruses in ducks overwintering in GuatemalamSphere20172e003621610.1128/mSphere.00362-16
- BarrigaGPet al.Avian influenza virus H5 strain with north American and Eurasian lineage genes in an Antarctic penguinEmerg. Infect. Dis.2016222221222310.3201/eid2212.161076
- KraussSet al.Coincident ruddy turnstone migration and horseshoe crab spawning creates an ecological “hot spot” for influenza virusesProc. Biol. Sci.20102773373337910.1098/rspb.2010.1090
- De AraujoJet al.Avian Influenza Virus (H11N9) in Migratory Shorebirds Wintering in the Amazon Region, BrazilPLoS One20149e11014110.1371/journal.pone.0110141
- Kear, J. (eds). Ducks, geese, and swans (Oxford University Press, 2005).
- HobsonKAStable isotopes and the determination of avian migratory connectivity and seasonal interactionsAm. Ornithol. Soc.200512210371048
- Ruegg, K. C. et al. Identifying migrant origins using genetics, isotopes, and habitat suitability. bioRxiv10.1101/085456 (2013).
- BridgeESet al.Bird migration and avian influenza: A comparison of hydrogen stable isotopes and satellite tracking methodsEcol. Indic.20144526627310.1016/j.ecolind.2014.04.027
- SpackmanEet al.Development of a real-time reverse transcriptase PCR assay for type A influenza virus and the avian H5 and H7 hemagglutinin subtypesJ. Clin. Microbiol.2002403256326010.1128/JCM.40.9.3256-3260.2002
- HoffmannEStechJGuanYWebsterRGPerezDRUniversal primer set for the full-length amplification of all influenza A virusesArch. Virol.20011462275228910.1007/s007050170002
- MenaIet al.Origins of the 2009 H1N1 influenza pandemic in swine in MexicoeLife20165e1677710.7554/eLife.16777
- ShapiroBRambautADrummondAJ Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequencesMol. Biol. Evol.2005237910.1093/molbev/msj021