
Abstract
An electrochemical investigation of selenium species, using cyclic voltammetry, has been carried out for the purpose of determining the molar entropy of the Se(VI)/(IV) couple. To obtain the ion interaction coefficient between HSeO4− and Na+, ϵT(HSeO4−, Na+), the following reaction involving HSeO4− as an oxidant and the uncharged species (H2SeO3) as the reductant was used:
The Se(VI)/(IV) half-wave potentials were measured in acidic sodium nitrate solutions as a function of the molality of Na+ ranging from 0.500 to 2.00 mol kg−1. The temperatures for the measurement were 288, 298, 308 and 323 K. Specific ion interaction theory was used to calculate the HSeO4−/H2SeO3 standard redox potential, , and ϵT(HSeO4−, Na+) at each temperature. The following molar entropy was derived from the temperature dependence of
:
The value of ϵT(HSeO4−, Na+) at 298 K was determined to be 0.29 ± 0.03 kg mol−1. The following ∂ϵ/∂T was derived from the temperature dependence of ϵT(HSeO4−, Na+):
1. Introduction
Groundwater is the transport media for radionuclides relevant to the safety assessment of the geological disposal system. Selenium-79 (79Se) is a long-lived fission product with a half-life of 327,000 years [Citation1]. Se aqueous species in groundwater are present as anions. Sorption of Se aqueous species is relatively small, which results in only slight retardation of Se relative to groundwater movement. Therefore, Se is one of the key radionuclides for the safety assessment of the geological disposal system. The stability ranges of the predominant Se species in aqueous systems are summarized and displayed in a potential–pH diagram [Citation2] (). In neutral repository groundwaters under reducing conditions, the dominant aqueous Se species is HSe− [Citation3]. On the other hand, it is necessary to estimate the sensitivity of the system to perturbations in environmental conditions. Following a review of relevant international research and a consideration of characteristics specific to Japan, uplift, subsidence and erosion were considered to be potential natural phenomena related to the long-term stability of geological environments in Japan [Citation4]. Radiolysis of groundwater, due to radiation from the vitrified waste, generates oxidizing agents such as H2O2 [Citation4]. A decrease in the depth of repositories and the existence of oxidizing agents are of particular concern for the anticipated reducing conditions of repositories. The solubility and aqueous speciation of radionuclides can be altered due to the oxidation/reduction potentials of the geologic environments. The thermodynamic calculations indicate that the predominant Se oxidation state is Se(IV) in mildly reducing environments, and it is Se(VI) in oxidizing environments. If the environmental conditions of repositories are oxidized due to decrease in the depth of repositories and/or the presence of oxidizing agents, the oxidation state of Se is changed to Se(IV) and/or Se(VI). Increasing ionic strength decreased selenate ion (SeO2 −4) sorption on amorphous iron oxide (am-Fe(OH)3) and goethite (α-FeOOH), but did not affect selenite ion (SeO2 −3) sorption [Citation5]. Metallic iron and siderite (FeCO3) reduced and immobilized Se(IV) and Se(VI) as iron diselenide (FeSe2); the decrease in Se(IV) concentration was much greater than that in Se(VI) concentration under the same conditions [Citation6]. Because the extent of sorption and immobilization of Se(IV) is different from that of Se(VI), the accurate prediction of Se(VI)–Se(IV) ratios is crucial for understanding the migration behavior of Se in those cases where the repositories exhibit oxidizing environments. An engineered barrier system will be constructed up to a depth of 300 m or more. Because temperature is in the range of 313–323 K at a depth of 500 m and the geothermal gradient is 3 K/100 m [Citation4], the decrease in the depth of repositories occurs at a temperature over 298 K. Therefore, accurate prediction of Se(VI)–Se(IV) ratios is required for such a temperature.
Figure 1. Potential–pH diagram for Se based on the thermodynamic data reviewed and compiled by Olin et al. [Citation2]. The total concentration of Se is 9.3×10−4 mol kg−1. The line with arrow points indicates the scan region in this study (0.449↔1.439 V vs. SHE and pH = 1.13 ± 0.26).
![Figure 1. Potential–pH diagram for Se based on the thermodynamic data reviewed and compiled by Olin et al. [Citation2]. The total concentration of Se is 9.3×10−4 mol kg−1. The line with arrow points indicates the scan region in this study (0.449↔1.439 V vs. SHE and pH = 1.13 ± 0.26).](/cms/asset/4fa517e1-b253-4b6d-adc5-258b622cd84c/tnst_a_872059_f0001_oc.jpg)
Thermodynamic models are used to calculate solubility and speciation as a function of pH, redox potential and ligand concentrations. In the calculation of solubility and speciation, the reliability of the results is necessarily dependent on that of available thermodynamic data. The Se(VI)/(IV) standard redox potential, which determines Se(VI)–Se(IV) ratios at 298 K in thermodynamic models, was obtained using the following reaction [Citation7]:
(1) Because the molar entropy of the reaction, ΔrS0m, yields the temperature derivatives of the standard redox potential, ΔrS0m of the Se(VI)/(IV) couple is also required to predict Se(VI)–Se(IV) ratios at a temperature over 298 K. No existing experimental studies report the value for ΔrS0m of the Se(VI)/(IV) couple, which results in the poor prediction of Se(VI)–Se(IV) ratios. Therefore, the objective of this study is to obtain ΔrS0m of the Se(VI)/(IV) couple.
Conventionally, thermodynamic data are quoted for conditions for the standard state, i.e. zero ionic strength. The activities of all the species participating in reactions must be estimated in order to convert the thermodynamic data obtained at high ionic strengths to zero ionic strength. One method for estimating the activities is through the use of ion interaction parameters (ϵT(i, j)) in the specific ion interaction theory (SIT) model [Citation8] to calculate the ion activity coefficients (γi). SIT was used in the Nuclear Energy Agency chemical thermodynamics project sponsored by the Organization for Economic Cooperation and Development (OECD) [Citation8] and Japan Atomic Energy Agency Thermodynamic Data Base review [Citation9]. OECD has critically evaluated available thermodynamic data for Se [Citation2]. Because no experimental data were available at that time, these reviews estimated ϵT(i, j) for Se species using an analogy with sulfur (S). The uncertainty in ϵT(i, j) for Se species estimated by Olin et al. [Citation2] was the same as that in ϵT(i, j) for the similar S species. However, after that, Philippini et al. [Citation10] and Doi [Citation7] experimentally determined ϵT(i, j) for Se species and reported differences between ϵT(i, j) for Se species and S species. Based on these experimental investigations, the applicability of an analogy with S to estimate ϵT(i, j) for Se species is questionable. Therefore it is necessary to determine ϵT(i, j) for Se species by experimental investigations. Previous reports [Citation7,Citation11,Citation12] determined the stoichiometric sum of ϵT(i, j), ΔϵT(i, j), instead of ϵT(i, j). This was because ϵT(i, j) for both an oxidant and a reductant had to be considered when the half-wave potentials, E1/2(T, Im), determined experimentally at different ionic strengths were corrected by extrapolating to zero ionic strength to obtain the standard redox potential. If either an oxidant or a reductant is an uncharged species, then ϵT(i, j) for the other species can be determined because ϵT(i, j) for an uncharged species is zero [Citation2]. To obtain the value for ϵT(HSeO4−, Na+), the following reaction involving HSeO4− as the oxidant and the uncharged species (H2SeO3) as the reductant was used in this study:
(2) In spite of the fact that the pH of groundwater is anticipated to be 7 to 9 [Citation4], the above reaction occurring in an acidic solution was used. The standard thermodynamic data for the following protonation reactions are available [Citation2]:
(3)
(4)
(5) Because the Se speciation is calculated using a thermodynamic database including the standard thermodynamic data for these protonation reactions, it is possible to calculate the Se speciation at the anticipated pH of groundwater even though the standard thermodynamic data of the Se(VI)/(IV) couple included in a thermodynamic database are those of the reaction occurring in acidic conditions. Because this study assigned a priority to the determination of ϵT(i, j) for the Se species, the thermodynamic values for the reaction in Equation (2) were evaluated as functions of temperature and ionic strength.
2. Experimental
Cyclic voltammograms were obtained by using an electrochemical analyzer (BAS Co., Ltd., ALS Model 1100). A three-electrode system was employed. The working, the reference and the counter electrodes were a platinum disk (BAS Co., Ltd., 002013 PTE) with an exposed area of 2.0 mm2, Ag/AgCl with a saturated solution of KCl (BAS Co., Ltd., 002058 RE-1C) and a platinum wire (BAS Co., Ltd., 002233 VC-3), respectively. The working electrode was polished using a 0.05 μm alumina paste before each cyclic voltammetry experiment.
A solution of Se was prepared by the addition of Se standard (1003 ppm Se in 0.10 mol dm−3 HNO3, Kanto Kagaku Co., Ltd., 37808-1B) and sodium nitrate (NaNO3, Wako Jyunyaku Kogyo Co., Ltd., 195-02545) into 0.1000 mol dm−3 HNO3. The concentration of Se was 9.3×10−4 mol kg−1 in this solution. The molality of a sodium ion, mNa+, was adjusted to 0.500, 1.00, 1.50 and 2.00 mol kg−1. The tetravalent Se species was H2SeO3 because the values of −log10 aH+ were estimated to be 1.13 ± 0.26 as described in Section 3.1. The solution not containing Se (henceforth referred to as the “blank” solution) was prepared by dissolving NaNO3 into 0.1000 mol dm−3 HNO3. Aliquots of solution were transferred to the glass cell (BAS Co., Ltd., 001056). The temperature of the solution in the glass cell was maintained by the thermostat water bath (AS ONE Co., Ltd., Thermax TM-1). The temperatures for the measurements were selected to be 288, 298, 308 and 323 K because the values of the Debye–Hückel constants, A(T), are available for these temperatures [Citation8]. Before and after the cyclic voltammetry experiment, pH values at each temperature controlled by the thermostat water bath were measured using the combination glass pH electrode (TOA Co., Ltd., ION METER IM-55G). Because the reference electrode was immersed in the solution in the glass cell, it is reasonable to assume that the temperature of the internal solution of the reference electrode was same as that of the solution in the glass cell. All solutions were thoroughly degassed with nitrogen gas prior to recording cyclic voltammograms. For all the cyclic voltammetry experiments, the scan region was 0.250↔1.240 V vs. Ag/AgCl, which corresponded to 0.449↔1.439 V vs. SHE at 298 K, described as a line with arrow points in . The scan rate was 0.6 V s−1 except while investigating the dependence of the peak current and potential on the scan rate.
3. Data analysis
3.1. Dependence of the half-wave potential on the molality of a sodium ion
When the redox process is reversible or quasi-reversible and the diffusion coefficient of a reductant is equal to that of an oxidant, E1/2(T, Im) is the potential when the concentration of a reductant is equal to that of an oxidant [Citation11]. Nernst's equation for the redox reaction, , is
(6) The application of Nernst's equation to the HSeO4−/H2SeO3 couple and the Ag/AgCl couple yields E1/2(T, Im) and ERef(T, IRef), respectively, given by
(7)
(8) where
(9) J is constant. In this study, the value of 2log10 aCl− in Equation (8) is equal to the common logarithm of the equilibrium constant, log10 K1(T, 0), for the following reaction occurring in an internal solution of the Ag/AgCl reference electrode:
(10) SIT [Citation8] gives a good estimation of the activity coefficient in an ionic medium of up to 3.5 mol kg−1. SIT was used to estimate a value of log10
in Equation (7). Because the cations were Na+ and H+ in a solution of Se, log10
is described by
(11) where
(12) D(T, Im) is the Debye–Hückel term [Citation8]. The values of A(T) calculated from the static dielectric constant and the density of water as functions of temperature and pressure are listed in TDB-2 [Citation8]. The values of A(T) at a pressure of 1.00 bar for temperatures of 288, 298, 308 and 323 K were taken from TDB-2. By substituting Equation (11) for log10
in Equation (7), the following equations are finally obtained:
(13) where
(14)
(15) The values of Y are plotted against mNa+. As one would predict from Equation (13), the extrapolation of Y to
gives a straight line where the slope and intercept correspond to ϵT(HSeO4−, Na+) and Yintercept, respectively. Using Equation (15),
can be calculated from Yintercept.
Measurements of E1/2(T, Im) were made four times at each mNa+. Using Equation (14), Y was calculated from an average of E1/2(T, Im), E1/2ave(T, Im). The uncertainty 1.96σ of the four measurements, representing the 95% confidence level, was assigned to Eave1/2(T, Im).
In this experiment, the measured pH might be different from the value of −log10 aH+ because of the difference between the activity factor of the calibration buffer and high ionic strength solution. Therefore, the measured pH was not used to calculate Y. The value of is calculated by
(16) The H+ concentration is calculated from the amount of HNO3. During the cyclic voltammetry experiment, no oxygen gas bubble was found on the electrode surface. The pH value measured after the cyclic voltammetry experiment was the same as that measured before the cyclic voltammetry experiment. Therefore, it was considered that the cyclic voltammetry experiment made no changes in the H+ concentration. The value of log10 γH+ is calculated using SIT as follows:
(17) The value of
is 0.07 ± 0.01 kg mol−1 [Citation8]. The following relationship was proposed on the basis of a few experimental values for ΔϵT(i, j) [Citation13]:
(18) Using Equation (18) with ϵT(i, j) and
in place of ΔϵT(i, j) and
, the correction for ϵT0(NO3−, H+) was calculated to be less than 0.006 kg mol−1 for the temperature range of 288–323 K. Because this correction was smaller than the assigned uncertainty in ϵT0(NO3−, H+), ϵT(NO3−, H+) at 288, 308 and 323 K was equal to ϵT0(NO3−, H+) in this study.
The values of log10 aH2O are calculated by
(19) where φ is the osmotic coefficient of the mixture and the summation extends over all solute species k with molality mk present in the solution [Citation2]. The following equations yield the osmotic coefficient for a mixed electrolyte [Citation14]:
(20)
(21)
(22)
(23) where f φ is the Debye–Hückel term extended to include osmotic effects and c is an index covering all cations, while a covers all anions. The parameters of β(0)ca and β(1)ca define the second virial coefficient, and Cφca defines the third virial coefficient [Citation15].
The EAg/AgCl(T, 0) values are calculated using the following equation, which is applicable for any temperature from 273 to 368 K [Citation16]:
(24) The standard molar Gibbs energy of formation of K+ and Cl− [Citation2] and that of KCl [Citation17] were used to calculate log10 K1(T0, 0). Equation (25) can be used to calculate equilibrium constant for any temperature from 273 to 473 K [Citation18]:
(25) Equation (25) was used to calculate log10 K1(T, 0) at 288, 308 and 323 K. The standard molar enthalpy of formation of KCl, K+ and Cl− [Citation17] were used to calculate ΔrH0m (T0) of Equation (10). Following the simplification that the heat capacity for any temperature is equal to that at 298 K for the temperature range between 273 and 423 K [Citation18], the standard molar heat capacity of KCl, K+ and Cl− [Citation17] were used to calculate ΔrC0p, m of Equation (10).
3.2. Temperature dependence of the standard redox potential
The Gibbs–Helmholtz equation [Citation12] is represented by
(26) On the other hand,
(27) Equation (28) is obtained by substituting Equation (27) into Equation (26) [Citation12]:
(28) Equation (28) indicates that the determination of EOx/Red(T, 0) as a function of temperature allows us to determine ΔrS0m.
4. Results and discussion
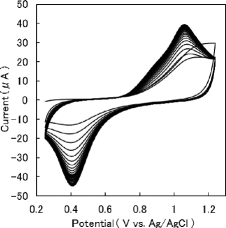
shows the cyclic voltammogram of a solution containing H2SeO3 after repetitive potential cycling. Peaks were assigned as arising from Se species present in solution. This was because no voltammetric peaks associated with the oxidation–reduction reaction of NaNO3 and HNO3 were observed in a voltammogram of the blank solution. With increasing number of cycles, two peaks appear clearly, consistent with electrochemical investigations of Se species, using cyclic voltammetry conducted with the working platinum electrode [Citation7,Citation19]. As described in the previous work [Citation7], this voltammetric behavior arises from a passivating layer covering the electrode surface. The drop of the anodic current at the upper reversal potential, observed during the course of the second cycle, suggests a decrease in the active electrode area available for the electron-transfer process owing to the formation of a passivating layer. The growth of the redox wave is indicative of the increase in the active electrode area owing to the removal of this layer by repetitive potential cycling. The cyclic voltammogram became steady by the 35th cycle in this study. The complete removal of this passivating layer resulted in a steady voltammogram. The E1/2(T, Im) value was obtained from the 40th cyclic voltammogram in this study.
Figure 2. Cyclic voltammogram of a solution containing H2SeO3 after repetitive potential cycling .
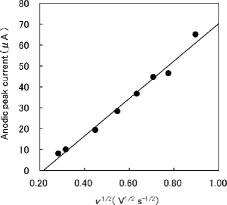
The anodic peak current of the 40th cyclic voltammogram after repetitive potential cycling is plotted as a function of v1/2 in . The linear dependence of the anodic peak current on v1/2 is indicative of the diffusion-controlled process [Citation20,21]. The peak at about 1.0 V vs. Ag/AgCl and that at about 0.4 V vs. Ag/AgCl are attributed to the oxidation of H2SeO3 to HSeO4− and to the corresponding reduction of HSeO4− to H2SeO3, respectively.
Figure 3. Dependence of the anodic peak current on v1/2. The eight filled circles corresponding to each scan rate in the figure are obtained from the 40th cyclic voltammogram after repetitive potential cycling measured in a solution containing H2SeO3 . The solid line is the linear least-squares fit of data. The linear dependence of the anodic peak current on v1/2 is indicative of the diffusion-controlled process.
Compared with the cathodic wave, an extra increase in the anodic current occurs in the potential range 0.87 ± 0.08 V vs. Ag/AgCl, where the stripping peak was observed in the cyclic voltammogram of a solution containing 1×10−3 mol dm−3 of SeO2 [Citation22]. Similar behavior was observed in the cyclic voltammogram of a solution containing Se species [Citation7,Citation19]. The stripping peak is associated with the removal of a previously deposited film of Se [Citation23] and can be considered independent of the oxidation of H2SeO3 to HSeO4−. The anodic current is the sum of the current associated with the removal of the Se film and that associated with the oxidation of H2SeO3 to HSeO4−. The cathodic wave is associated only with the corresponding reduction of HSeO4− to H2SeO3. The similarity in the peak shape between the anodic wave and the cathodic wave indicates that the contribution of the removal of the Se film to the anodic current is not significant. Without subtracting this contribution from the total anodic current, evidence of a diffusion-controlled process was obtained as described earlier. These voltammetric features were seen from the voltammogram of a solution containing SeO32− [Citation7]. Therefore, as was the case of the cyclic voltammogram of a solution containing SeO32−, the peak potential and current of the anodic wave were determined without any correction in this study.
Ep(Ox), Ep(Red) and E1/2(T, Im) of the 40th cyclic voltammogram after repetitive potential cycling, measured at different scan rates are listed in . As confirmed by the data in , E1/2(T, Im) is independent of the scan rate. This feature corresponds to the quasi-reversible reaction [Citation11,12]. Assuming that the diffusion coefficient of H2SeO3 is equal to that of HSeO4−, E1/2(T, Im) is the potential when the concentration of H2SeO3 is equal to that of HSeO4−. Based on this assumption, Equation (7) was used to determine at each temperature in this study.
Table 1. Ep(Ox), Ep(Red) and E1/2(T, Im) of the 40th cyclic voltammogram after repetitive potential cycling, measured at different scan rates in a solution containing H2SeO3 .
shows a summary of the cyclic voltammetry experiments to measure the half-wave potentials for the HSeO4−/H2SeO3 couple in acidic sodium nitrate solutions as a function of mNa+. A plot of the Y values vs. mNa+ is shown in . The values of Yintercept and ϵT(HSeO4−, Na+) are obtained by using the extrapolations of weighted linear regression [Citation24] through Y determined at various mNa+, which correspond to the lines in . Using Equation (15), were calculated from Y intercept, EAg/AgCl(T, 0) and log10 K1(T, 0). The values of Y intercept, ϵT(HSeO4−, Na+), EAg/AgCl(T, 0), log10 K1(T, 0) and
are listed in . A plot of the
values vs. temperature is shown in , where the solid line represents a weighted linear regression of
. The slope of this line corresponds to ΔrS0m/2F given by
Table 2. Summary of the cyclic voltammetry experiments to measure the half-wave potentials for the HSeO4−/H2SeO3 couple in acidic sodium nitrate solutions as a function of mNa+.
Table 3. Values of Yintercept, ϵT(HSeO4−, Na+), EAg/AgCl(T, 0), log10 K1(T, 0) and . The intercept and slope of each line in correspond to Y intercept and ϵT(HSeO4−, Na+), respectively, at each temperature.
Figure 4. Determination of the standard redox potential for the HSeO4−/H2SeO3 couple using SIT. Experimental data as a function of are shown as the solid bars with a vertical bar showing the standard deviation in each data point. The solid line is a weighted linear regression [Citation24] of data. The slope and intercept of each line correspond to ϵT(HSeO4−, Na+) and Yintercept, respectively, at each temperature.
, where J = RT/2F log10 e. Yintercept = [
– EAg/AgCl(T, 0)]/J + log10 K1(T, 0).
![Figure 4. Determination of the standard redox potential for the HSeO4−/H2SeO3 couple using SIT. Experimental data as a function of are shown as the solid bars with a vertical bar showing the standard deviation in each data point. The solid line is a weighted linear regression [Citation24] of data. The slope and intercept of each line correspond to ϵT(HSeO4−, Na+) and Yintercept, respectively, at each temperature. , where J = RT/2F log10 e. Yintercept = [ – EAg/AgCl(T, 0)]/J + log10 K1(T, 0).](/cms/asset/04d0bb24-e61c-4314-99d0-e520c2f4fbdb/tnst_a_872059_f0004_b.gif)
Figure 5. Determination of the molar entropy for the HSeO4−/H2SeO3 couple. The HSeO4−/H2SeO3 standard redox potential as a function of temperature is shown as the filled circle with vertical bar showing the standard deviation in each data point. The solid line is a weighted linear regression [Citation24] of data. The slope of this line corresponds toΔrS0m/2F = −0.3 ± 0.1 mV K− 1.
![Figure 5. Determination of the molar entropy for the HSeO4−/H2SeO3 couple. The HSeO4−/H2SeO3 standard redox potential as a function of temperature is shown as the filled circle with vertical bar showing the standard deviation in each data point. The solid line is a weighted linear regression [Citation24] of data. The slope of this line corresponds toΔrS0m/2F = −0.3 ± 0.1 mV K− 1.](/cms/asset/45112fff-cfdd-45f3-a693-e419364ad7a7/tnst_a_872059_f0005_b.gif)
Table 4. Standard thermodynamic data for the Se(VI)/(IV) couple.
The value of was determined to be 0.29 ± 0.03 kg mol−1 in this study. Available literature values for
are −0.01 ± 0.02 kg mol−1 [Citation2] and 0.11 ± 0.05 kg mol−1 [Citation25]. The difference was found between
and
. The ion interaction coefficient is dependent on the size and charge of the ion [Citation8]. The relationship between the size of monovalent anions and the ion interaction coefficient for Na+ was surveyed. We compare the ionic radius of F−, Cl−, Br− and I−, which increase in that order [Citation26]. This order is the same as found for the magnitude of the ion interaction coefficient for Na+, i.e.
[Citation8]. Among these monovalent anions, the larger the size, the larger the magnitude of the ion interaction coefficient for Na+ is. The ionic radius of Se6+ is longer than that of S6+ [Citation26]. From the fact that the interatomic distances of H2Se, SeO42− and SeO2 are longer than those of H2S, SO42− and SO2, respectively [Citation27], the interatomic distances of HSeO4− are considered to be longer than those of HSO4−. Therefore, the size of HSeO4− is likely to be larger than that of HSO4−, which is consistent with the relationship of
.
A plot of the ϵT(HSeO4−, Na+) values vs. temperature is shown in . The value of ∂ϵ/∂T was obtained by using the extrapolation of weighted linear regression [Citation24] through ϵT(HSeO4−, Na+) obtained at 288, 298, 308 and 323 K, which corresponds to the line in . The slope of this line corresponds to ∂ϵ/∂T given by
(30)
Figure 6. Dependence of the ion interaction coefficient ϵT(HSeO4−, Na+) on temperature. The ion interaction coefficient ϵT(HSeO4−, Na+) as a function of temperature is shown as the filled circle with vertical bar showing the standard deviation in each data point. The solid line is a weighted linear regression [Citation24] of data. The slope of this line corresponds to ∂ϵ/∂T = −0.002 ± 0.002 kg mol−1 K−1.
![Figure 6. Dependence of the ion interaction coefficient ϵT(HSeO4−, Na+) on temperature. The ion interaction coefficient ϵT(HSeO4−, Na+) as a function of temperature is shown as the filled circle with vertical bar showing the standard deviation in each data point. The solid line is a weighted linear regression [Citation24] of data. The slope of this line corresponds to ∂ϵ/∂T = −0.002 ± 0.002 kg mol−1 K−1.](/cms/asset/d8f91ca6-0689-4f84-9b7e-206fde082fdc/tnst_a_872059_f0006_b.gif)
5. Conclusion
An electrochemical investigation of selenium species, using cyclic voltammetry, has been carried out for the purpose of determining the molar entropy of the Se(VI)/(IV) couple. To obtain the value for ϵT(HSeO4−, Na+), the following reaction involving HSeO4− as the oxidant and the H2SeO3 uncharged species as the reductant was used.
The Se(VI)/(IV) half-wave potentials were measured in acidic sodium nitrate solutions as functions of mNa+ ranging from 0.500 to 2.00 mol kg−1 and temperature ranging from 288 to 323 K. SIT was used to calculate
and ϵT(HSeO4−, Na+). The following molar entropy was derived from the temperature dependence of
:
The value of ϵT(HSeO4−, Na+) at 298 K was determined to be 0.29 ± 0.03 kg mol−1. The following ∂ϵ/∂T was derived from the temperature dependence of ϵT(HSeO4−, Na+):
Nomenclature
| ai | = | activity of ion i |
| γi | = | activity coefficient of ion i |
| zi | = | charge of ion i |
| mi | = | molality of ion i (amount of ion i divided by the mass of the solvent, mol kg−1) |
| [i] | = | molarity or concentration of ion i (amount of ion i divided by the volume of the solution, mol dm−3) |
| T | = | absolute temperature (K) |
| T0 | = | reference temperature (= 298.15 K) |
| Im | = | ionic strength of the working solution |
| IRef | = | ionic strength of a solution contained in the reference electrode (mol kg−1) |
| D(T, Im) | = | Debye–Hückel term |
| A(T) | = | Debye–Hückel constant (values taken from TDB-2 [Citation8]) |
| ϵT(i, j) | = | specific interaction coefficient between ion i and ion j of opposite charge (kg mol−1) |
| EOx/Red(T, 0) | = | standard redox potential of the Ox/Red couple |
| ERef(T, IRef) | = | potential of the silver–silver chloride (Ag/AgCl) reference electrode with a saturated solution of potassium chloride (KCl) |
| Ep(Ox) | = | anodic peak potential against the Ag/AgCl reference electrode |
| Ep(Red) | = | cathodic peak potential against the Ag/AgCl reference electrode |
| E1/2(T, Im) | = | half-wave potential {E1/2(T, Im) = [Ep(Ox) + Ep(Red)]/2} against the Ag/AgCl reference electrode |
| J | = | RT/2Flog10e (= 0.02958 V at 298.2 K) |
| E (V vs. Ag/AgCl) | = | potential against the Ag/ACl reference electrode |
| E (V vs. SHE) | = | potential against the standard hydrogen electrode |
| v | = | scan rate (V s−1) |
| ΔrG0m | = | the molar Gibbs energy of reaction (kJ mol−1) |
| ΔrH0m | = | the molar enthalpy of reaction (kJ mol−1) |
| ΔrS0m | = | the molar entropy of reaction (J K−1 mol−1) |
| ΔrC0p, m | = | the molar heat capacity of reaction (J K−1 mol−1) |
| K | = | the equilibrium constant of the reaction |
| σ | = | standard deviation |
Acknowledgements
The author would like to thank Prof. Yokoyama (Kanazawa University), Dr Kamei (Japan Atomic Energy Agency) and Dr Rai (Rai Enviro-Chem, LLC) for the valuable discussions and suggestions.
References
- Jorg G, Buhnemann R, Hollas S, Kivel N, Kossert K, Van Winckel S, Gostomski CL. Preparation of radiochemically pure 79Se and highly precise determination of its half-life. Appl Radiat Isotopes. 2010;68:2339–2351.
- Olin Å, Noläng B, Öhman LO, Osadchii EG, Rosén E. Chemical thermodynamics of selenium. Amsterdam: Elsevier; 2005.
- Iida Y, Yamaguchi T, Tanaka T, Nakayama S. Solubility of selenium at high ionic strength under anoxic conditions. J Nucl Sci Technol. 2010;47:431–438.
- Japan Nuclear Cycle Development Institute (JNC). H12: project to establish the scientific and technical basis for HLW disposal in Japan – second progress report on research and development for the geological disposal of HLW in Japan. Tokai-mura (Japan): JNC; 2000.
- Chunming S, Donald LS. Selenate and selenite sorption on iron oxides an infrared and electrophoretic study. Soil Sci Soc Am J. 2000;64:101–111.
- Cui D, Puranen A, Devoy J, Scheidegger A, Leupin OX, Wersin P, Gens R, Spahiu K. Reductive immobilization of 79Se by iron canister under simulated repository environment. J Radioanal Nucl Chem. 2009;282:349–354.
- Doi R. Determination of the selenium (VI)/(IV) standard redox potential by cyclic voltammetry. J Nucl Sci Technol. 2013;51:56–63.
- Grenthe I, Mompean F, Spahiu K, Wanner H. TDB-2 guidelines for the extrapolation to zero ionic strength. Issy-les-Moulineaux (France): OECD Nuclear Energy Agency; 2013. Available from: http://www.oecd-nea.org/dbtdb/guidelines/tdb2.pdf
- Kitamura A, Fujiwara K, Doi R, Yoshida Y, Mihara M, Terashima M, Yui M. JAEA thermodynamic database for performance assessment of geological disposal of high-level radioactive and TRU wastes. Tokai-mura (Japan): Japan Atomic Energy Agency; 2009.
- Philippini V, Aupiais J, Vercounter T, Moulin C. Formation of CaSO4(aq) and CaSeO4(aq) studied as a function of ionic strength and temperature by CE. Electrophoresis. 2009;20:3582–3590.
- Riglet CH, Robouch P, Vitorge P. Standard potentials of the (MO2 +2/MO2+) and (M4+/M3+) redox systems for neptunium and plutonium. Radiochimica Acta. 1989; 46:85–94.
- Capdevila H, Vitorge P. Temperature and ionic strength influence on U(VI/V) and U(IV/III) redox potentials in aqueous acidic and carbonate solutions. J Radioanal Nucl Chem. 1990;143:403–414.
- Tomas V, Pierre V, Badia A, Eric G, Solange H, Christophe M. Stabilities of the aqueous complexes Cm(CO3)33− and Am(CO3)33− in the temperature range 10–70 °C. Inorg Chem. 2005;44:5833–5843.
- Pitzer KS, Kim JJ. Thermodynamics of electrolytes. IV. Activity and osmotic coefficients for mixed electrolytes. J Am Chem Soc. 1974;96:5701–5707.
- Pitzer KS, Mayorga G. Thermodynamics of electrolytes. II. Activity and osmotic coefficients for strong electrolytes with one or both ions univalent. J Phys Chem. 1976;77:2300–2308.
- Bard J, Parsons R, Jordan J. Standard potentials in aqueous solution. New York (NY): Marcel Dekker, Inc.; 1986.
- Wagman DD, Evans WH, Parker VB, Schumm RH, Halow I, Bailey SM, Churney KL, Nuttall RL. The NBS tables of chemical thermodynamic properties: selected values for inorganic and C1 and C2 organic substances in SI units. New York (NY): American Chemical Society and American Institute of Physics; 1982.
- Puigdomènech I, Rard JA, Plyasunov AV, Grenthe I. TDB-4 temperature corrections to thermodynamic data and enthalpy calculations. Issy-les-Moulineaux (France): OECD Nuclear Energy Agency; 1999. Available from: http://www.oecd-nea.org/ dbtdb/guidelines/tdb4new.pdf
- Doi R, Yui M. Experimental study by cyclic voltammetry of standard potential of Se(IV)/Se(VI) redox system. J Nucl Fuel Cycle Environ. 2009;16:35–42. Japanese.
- Kim SY, Asakura T, Morita Y. Electrochemical and spectroscopic studies of Pu(IV) and Pu(III) in nitric acid solutions. J Radioanal Nucl Chem. 2013;295:937–942.
- Fujishima A, Aizawa M, Inoue T. Electrochemical measurements. Tokyo: Gihodo Syuppan Co.; 1984.
- Murat A, Umit D, Curtis S. Electrochemical formation of Se atomic layers on Au(111) surface: the role of adsorbed selenate and selenite. J Electroanal Chem. 2004;561:21–27.
- Andrews RW, Johnson DC. Voltammetric deposition and stripping of selenium(IV) at a rotating gold-disk electrode in 0.1M perchloric acid. Anal Chem. 1975;47:294–299.
- Wanner H, Östhols E. TDB-3 guidelines for the assignment of uncertainties. Issy-les-Moulineaux (France): OECD Nuclear Energy Agency; 2000. Available from: http://www.oecd-nea.org/dbtdb/guidelines/ tdb3new.pdf
- Tomas V, Badia A, Christophe M, Eric G, Pierre V. Sulfate complexation of trivalent lanthanides probed by nanoelectrospray mass spectrometry and time-resolved laser induced luminescence.Inorg Chem. 2005;44:7570–7581.
- Shannon RD. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr Section. 1976;A32:751–767.
- The Chemical Society of Japan. Chemical handbook. Tokyo: Maruzen Publishing; 1993.
- Lewis GN, Randall M. Thermodynamics. 2nd ed. Revised by Pitzer KS and Brewer L. New York (NY): McGraw-Hill; 1961.
- Millero FJ. Effects of pressure and temperature on activity coefficients. In: Pytkowicz RM, editor. Activity coefficients in electrolyte solutions. Boca Raton (FL): CRC Press; 1979.
- Helgeson HC, Kirkham DH, Flowers GC. Theoretical prediction of the thermodynamic behavior of aqueous electrolytes at high pressures and temperatures: IV. Calculation of activity coefficients, osmotic coefficients, and apparent molal and standard and relative partial molal properties to 600°C and 5 kb. Am J Sci. 1981;281:1249–1516.
- Oelkers EH, Helgeson HC. Triple-ion anions and polynuclear complexing in supercritical electrolyte solutions. Geochimica Cosmochimica Acta. 1990;54:727–738.
- Grenthe I, Plyasunov A. On the use of semiempirical electrolyte theories for the modeling of solution chemical data. Pure Appl Chem. 1997;69:951–958.
- Xiangliang, Siagian S, Basar K, Sakuma T, Takahashi H, Igawa N, Ishii Y. Inter-atomic distance and temperature dependence of correlation effects among thermal displacements. Solid State Ionics. 2009;180:480–482.