This article refers to:
Ismail I. Fasfous, Jamal N. Dawoud, Abdulwahab K. Sallabi, Taghreed S. Hassouneh. A density functional theory study of the Cu+·(CO)n (n = 1–3) complexes. J. Coord. Chem. doi: 10.1080/00958972.2015.1021343
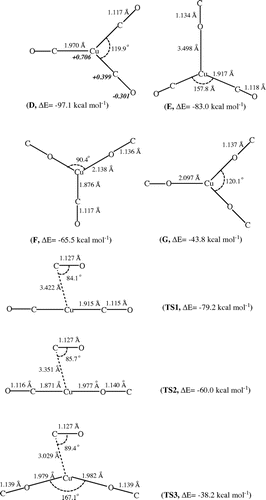
The authors of the above article report that, when first published online, it contained an incomplete version of Figure . This has been corrected in both print and online versions as follows.
Figure 4. Optimized structures for the minima and transition states of the Cu+⋅(CO)3 complex obtained on the basis of the B3P86/LANL2TZ+/6-311+G(df) method. The solid italic numbers represent the NBO analysis of the atomic charges of the global minimum structure D.