

ABSTRACT
Organocatalysis is the use of non-stoichiometric amounts of low-molecular-weight organic molecules made up of C, H, N, O, S, and P to accelerate stereoselective chemical transformations. Despite having a long history, the use of small organic molecules as chiral catalysts in enantioselective synthesis has only recently intrigued scientists interest. However, in synthetic and medicinal chemistry, there has been a resurgence of interest in performing asymmetric synthesis of a variety of important targets by utilizing an organocatalytic approach as a leading reaction. The development of an asymmetric protocol for the synthesis of enantioenriched molecules of therapeutic relevance is crucial in the development of new drug candidates.
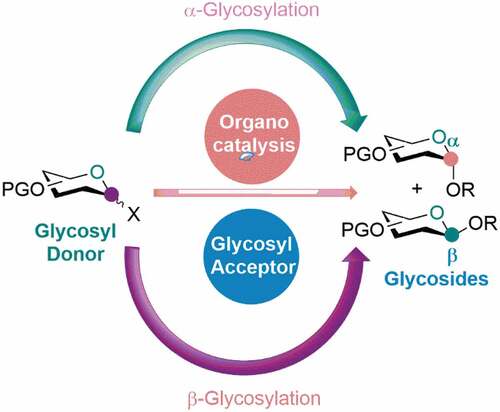
Modern asymmetric catalysis is based on three pillars: (i) biocatalysts (i.e., enzymes), (ii) transition metal catalysis, and (iii) organocatalysis. Among these catalytic systems, organocatalysis has become the most flourishing field within the field of a catalytic regime that offers many significant benefits to synthetic and medicinal chemists. In contrast to many metal-based catalytic models, most organocatalysts have a high tolerance capability for air and moisture, easy availability, easy handling, non-toxic and enantiomeric purity, etc. They are often derived from natural resources of high enantiomeric virtue with the desired stereocentre or easily synthesized in the laboratory in some simple synthetic operations. This perspective aims to draw readers’ attention to the power of asymmetric organocatalysis in assembling glycosyl donors and acceptors in the synthesis of biologically significant stereodefined O-glycosides. Glycosylation is a fundamental chemical transformation and is ubiquitous in both nature and chemical laboratories. In nature, the glycosylation processes are carried out by enzyme catalysis. We were interested in portraying the recent impressive progress achieved by the scientific community in the field of chemical glycosylation, specifically O-glycosylation. Poly-functionalized sugar moieties offer many opportunities to discover new drug candidates in which the modification of aglycone through the glycosylation process is a key reaction.
Graphical Abstract
Acknowledgments
RNY and AKS thanks to VBS purvanchal University and TEQIP Phase III for the necessary facilities in the preparation of the manuscript. BKB is grateful to the Department of Mathematics and Natural Sciences, Prince Mohammad Bin Fahad University (PMU), Saudi Arabia. Financial support from University Grants Commission, India [UGC-BSR/Start-Up-Grant/2019-2020 (No. F. 30-515/2020(BSR), FD Diary No. 9718)] is gratefully acknowledged by MFH. The sincere input of Prof. R. Latif of Department of Mathematics and Natural Sciences, Prince Mohammad Bin Fahad University (PMU), Saudi Arabia and Prof. Rajib Bhaumik, Department of English, Alipurduar University, W.B Kolkata, India is also gratefully acknowledged.
Disclosure statement
No potential conflict of interest was reported by the author(s).