Abstract
Today, the decision to treat breast cancer patients with endocrine therapy relies solely on tumor expression of two predictive factors, the estrogen receptor and the progesterone receptor. Expression of these hormone receptors are, however, not a guarantee for a response to treatment and patients who experience response at first may become resistant after prolonged treatment. This paper describes the use of preclinical models to identify mechanisms and new markers for endocrine sensitivity and resistance and the translation of these data to clinical utility.
Expression of a predictive factor is associated with response to a specific therapy. The only well-established predictive factors for endocrine therapy are the estrogen receptor (ER) and the progesterone receptor (PR) Citation[1]. Patients whose tumors are both ER positive (ER + ) and PR positive (PR + ) have more than 70% likelihood of responding to endocrine therapy, while those with both ER negative (ER − ) and PR negative (PR − ) tumors have less than 10% response rate. However, despite of tumor ER and/or PR expression, many patients do not respond to endocrine therapy, either at the beginning of treatment (de novo or intrinsic resistance) or after prolonged use (acquired resistance). All endocrine therapies for postmenopausal women are designed to counteract the growth effects of estrogen, which are mediated via estrogen binding to the ER, resulting in activation of ER-regulated signaling in the cancer cells. The two most common endocrine treatment modalities, aromatase inhibitor therapy and antiestrogen therapy, target estrogen biosynthesis and ER activation, respectively.
Predictive markers for endocrine therapy presently used in the clinic
Many resources are put into understanding the biology of hormone-responsive breast cancer. In spite of this, only ER and PR have been established as predictive markers for endocrine therapy and are recommended for routine analyzis in primary tumors from patients with breast cancer using immunohistochemistry (IHC). Hormone receptor positive (HR + ) tumors are defined as those with ER or PR expression detectable above a pre-set limit. When at least 10% of the invasive tumor cells express either ER and/or PR, the tumor is defined as HR+ and the patient belongs to a group categorized as endocrine responsive. Unless these patients are categorized as low risk patients they are recommended endocrine therapy Citation[1]. This pre-set limit of 10% may not be ideal since it has been shown that patients with as few as 1–10% HR+ tumor cells can benefit from endocrine therapy Citation[2].
PR is an estrogen induced protein and PR expression is associated with a better prognosis, furthermore some studies have found it to be a stronger predictor of the effectiveness of endocrine therapy than ER. In the ATAC trial, the benefit from aromatase inhibitor therapy was substantially greater than the benefit from antiestrogen in the ER + /PR− subgroup. This indicated that expression of PR could be used to select the patients, who would benefit more from AI than from antiestrogen. However, this finding could not be confirmed in a later central assessment of the ATAC study Citation[3] nor in central assessment of the BIG 1–98 study Citation[4].
A third predictive marker that is routinely analyzed in breast tumors is the growth factor receptor HER-2. It has been suggested that HER-2 overexpression, which is a predictive marker for treatment with trastuzumab (Herceptin®), may be a selection criteria for aromatase inhibitor therapy versus treatment with antiestrogen. However, in the BIG 1–98 study aromatase inhibitor treatment was superior to antiestrogen irrespective of HER-2 status Citation[5]. Thus, new markers are required, both to have better endocrine treatment response and to select patients for aromatase inhibitor or antiestrogen therapy.
Identification of mechanisms and markers for endocrine sensitivity and resistance
Preclinical models of endocrine sensitivity
The first preclinical model for estrogen responsive human breast cancer was a cell culture model with the human breast cancer cell line MCF-7, which could be growth stimulated with estradiol and growth inhibited with the antiestrogen tamoxifen Citation[6]. Cell culture studies have shown that presence of ER in the tumor cells and activation of ER by binding of ligand is a prerequisite for estrogen stimulated growth, confirming the clinical observation that the responders to endocrine therapy are almost exclusively patients with ER positive tumors.
ER acts as a transcription factor and in the classical genomic pathway, ER activation occurs via binding of ligand. The most potent ligand is estradiol, which is produced by aromatization of androgens either in situ in the cancer cells () or in surrounding stromal cells and in peripheral tissues. ER may also be activated via growth factor receptor signaling resulting in phosphorylation of ER and thus activation (). Besides the function as a direct transcription factor, ER may also act indirectly as a transcription factor via association with other transcription factors. Finally, ER may be located in the membrane and upon ligand binding capable of associating with growth factor receptors and thereby stimulate growth factor receptor signaling Citation[7]. These different pathways, by which ER is able to stimulate tumor growth, should be considered in new therapy targeting the ER.
Figure 1. ER signaling pathways. The figure illustrates a simplistic model of estrogen receptor (ER) signaling in a breast tumor cell. The aromatase enzyme converts androgens to estrogens (estrone; E1 and estradiol; E2). E2 binds to the ER and mediates classical genomic gene transcription of estrogen-regulated genes such as progesterone receptor (PR), Bcl-2 and insulin like growth factor receptor 1 (IGF-IR). In addition, the ER can be phosphorylated and activated by growth factor receptors such as human epidermal growth factor receptor 1 and 2 (EGFR and HER-2), and IGF-IR. A further description of the signaling pathways is given in the text.
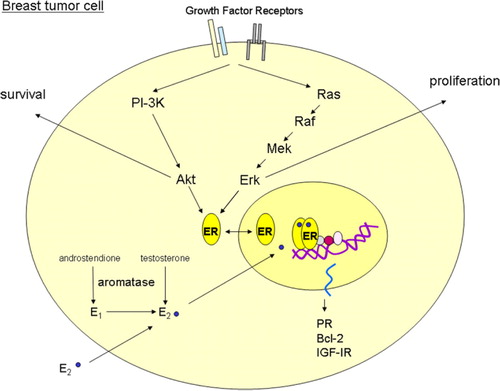
The responders to endocrine therapy are the patients with tumors in which ER is driving tumor growth and identification of markers reflecting ER-driven tumor growth will improve the response rate to endocrine therapy. Thus, addition of the marker PR which is an estrogen stimulated protein adds to the predictive value in combination with ER Citation[8]. Gene expression analysis of estrogen-stimulated human breast cancer cells have revealed that as many as about 3% of all genes may be regulated by estrogen Citation[9] and ER+ human breast tumors have a remarkably distinct gene expression pattern. Currently, clinical studies are in progress to test the predictive value of gene expression profiles. Studies evaluating the predictive value of a combination of a limited number of selected estrogen regulated genes are also underway.
Preclinical models of endocrine resistance
De novo resistance to endocrine therapy is associated with ER negativity, but as mentioned, a considerable fraction of the ER+ tumors do also not respond to endocrine therapy and patients with advanced breast tumors will eventually develop acquired resistance. Human breast cancer cell lines grown in culture or in athymic nude mice are useful to study the underlying molecular mechanisms of resistance and thereby identify new markers for treatment response and targets for treatment of resistant tumor cells. In the following, we have focused on model systems for resistance to the two major endocrine treatment modalities; antiestrogens and aromatase inhibitors.
Antiestrogen resistance
Several laboratories have developed model systems with human breast cancer cell lines which are resistant to treatment with antiestrogens. The cell lines have been developed from endocrine sensitive breast cancer cells using two different experimental approaches; either by ectopic overexpression of candidate causative genes for endocrine resistance, or by treatment of sensitive breast cancer cell lines with antiestrogen followed by selection of the surviving cells with acquired resistance. Examples of candidate genes that upon overexpression may render breast cancer cells resistant to antiestrogen therapy, are ER co-activators, growth factors, growth factor receptors, signaling molecules and cell cycle regulators. This type of studies have demonstrated decreased sensitivity to antiestrogen treatment in human breast cancer cell lines that overexpress e.g. the ER co-activator Amplified In Breast cancer 1 (AIB1), the epidermal growth factor receptor (EGFR/HER-1) or HER-2, the intracellular kinases protein kinase B (PKB/Akt), protein kinases C (PKCδ and PKCα), or the cell cycle regulator cyclin D1 Citation[10]. Such studies demonstrate the ability of a given protein to render the cells less sensitive to antiestrogen treatment and the protein may be a useful marker for prediction of response to treatment and a potential target for treatment. Yet, changes in a single protein may be to simplistic to explain the process of resistance development in breast cancer. The model systems in which endocrine sensitive cells have been treated for a long time with antiestrogen in order to select cells with acquired resistance allow multiple biological changes to occur. Direct comparison between the sensitive and the resistant cells can be performed in order to identify the molecular changes in the resistant cells. This can be done at the DNA, RNA and protein level. Recent techniques have also allowed a detection of the activated forms of the proteins in the cells, thereby allowing identification of the signaling pathways which may be involved in resistance. Single proteins; the EGFR, HER-2, PKB/Akt, PTEN, PKCδ, PKCα have been described to be causally involved in the ability of resistant cells to grow in presence of antiestrogens Citation[10]. Accordingly, the activated forms of these proteins are potential new markers for prediction of resistance to the antiestrogens tamoxifen and/or fulvestrant and are also potential targets for treatment. Studies are in progress to evaluate the utility of these new markers to predict response to antiestrogen therapy.
Knowledge of the mechanisms involved in antiestrogen resistance has initiated model studies with combined treatments of the endocrine agent and compounds targeting the molecules which appear to be causally involved in antiestrogen resistance. In a model with human breast cancer cells overexpressing the HER-2 receptor and grown in athymic nude mice, it was found that tamoxifen stimulated tumor growth, presumably by acting as an ER agonist as a result from crosstalk between ER and HER-2. Combined treatment targeting ER and HER-2 gave superior result compared to single therapy and similarly patients with ER + /HER-2+ tumors, who respond poorly to tamoxifen, may benefit from combined AI and trastuzumab therapy Citation[7].
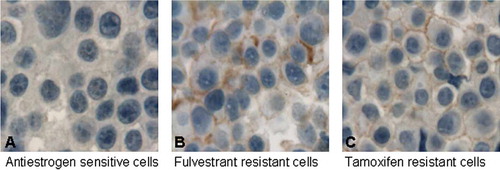
Although much focus has been on overexpression of HER-2 in tamoxifen resistant human breast cancer Citation[7], overexpression of EGFR appears to be more common in the model systems with tamoxifen resistant tumor growth Citation[10]. shows immunocytochemical staining of the active phosphorylated form of EGFR in a model of antiestrogen resistance, clearly showing an elevated level of active EGFR in the resistant cell lines. Thus, markers for active signaling from EGFR may be useful to select patients who may require combined treatment with antiestrogen and inhibitors of EGFR signaling Citation[10]. The EGFR family member HER-3 may also be important in combination with HER-2, whereas the importance of the EGFR family member HER-4 in antiestrogen resistance is not yet understood.
Figure 2. Immunocytochemical (ICC) analysis of pEGFR. Antibody staining of pEGFR in paraffin embedded human breast cancer cells. The antibody used only recognizes EGFR if a specific tyrosine residue required for signal transduction is phosphorylated. A: ICC analysis of pEGFR in antiestrogen sensitive breast cancer cells showing no staining. B: ICC analysis of pEGFR in the antiestrogen fulvestrant resistant breast cancer cells showing brown membrane staining. C: ICC analysis of pEGFR in tamoxifen resistant breast cancer cells demonstrating brown membrane staining.
Analyses in cell culture models have revealed that breast cancer cell lines, which acquire resistance to tamoxifen therapy, often remain ER+ and estrogen-responsive. Presence of ER and estrogen-regulated genes indicate that tamoxifen resistant cells may be growth inhibited by a second line treatment with another potent antiestrogen. Several laboratories have confirmed this hypothesis as tamoxifen resistant human breast cancer cell lines respond to treatment with the pure steroidal antiestrogen fulvestrant Citation[11]. Clinical studies also demonstrate response to fulvestrant in patients who have relapsed on tamoxifen therapy Citation[12]. Yet, a reduced level of ER is often seen in antiestrogen resistant cell culture models and also in breast tumors resistant to tamoxifen therapy Citation[13]. This results in decreased signaling from the ER, and a reduced level of several estrogen-regulated proteins like PR, Bcl-2 and IGF-IR Citation[14]. A reduced level of the anti-apoptotic protein Bcl-2 appears to be an advantage in relation to treatment of resistant tumor cells. Thus, antiestrogen resistant cells were more sensitive to treatment with cisplatin and this was ascribed to the reduced Bcl-2 level Citation[15]. Accordingly, low Bcl-2 expression in the metastatic lesion may be a predictive factor for response to cisplatin treatment.
Aromatase inhibitor resistance
When turning to aromatase inhibitors (AIs), only little is known of the mechanisms underlying development of resistance. This is partly due to the relatively short period that AIs have been used as first or second line endocrine treatment, but also because of methodological problems in developing appropriate model systems. In contrast to antiestrogens, AIs may not only act directly on the cancer cells, but also on tumor-associated stromal cells that have been shown to express aromatase protein Citation[16]. In postmenopausal women, estrogens are synthesized in peripheral tissues from androgens via the aromatase enzyme and in situ estrogen production in breast tumors is likely to be important for growth stimulation of the cancer cells (). A theory that has gained major support is that estrogens are primarily synthesized in the stromal compartment of a breast tumor, in turn supplying the cancer cells with estrogen. However, breast cancer cell lines have been shown to convert enough androgen to estrogen in vitro to cause estrogen-mediated protein expression and cell proliferation Citation[17]. Furthermore, aromatase is expressed in epithelial breast cancer cells in vivo, and the aromatase activity measured in tumor tissue was correlated to the aromatase expression in the carcinoma cells Citation[16]. Yet, an interaction between the epithelial cancer cells and the tumor stromal cells may be important for aromatase activity, inhibition with AI and development of treatment resistance. This makes it complicated to create an appropriate in vitro model for studies of AI resistance. A simplistic model could be to develop AI resistant cells from aromatase expressing breast cancer cell lines, however to date, this has not been successful. Instead, two approaches have been applied for studying AI resistance, one using ectopic overexpression of aromatase in MCF-7 breast cancer cells (MCF-7-Ca), the other using breast cancer cells long term deprived of estrogen (LTED), in theory mimicking the effect of AI blockade of estrogen synthesis.
The LTED model is compelling to use, as adaptation to estrogen-withdrawal is apparent to be involved in AI resistance. Studies have shown that adaptation to LTED causes activation of growth factor pathways, resulting in increased MAPK and PI3-K/Akt signaling and ER activation by phosphorylation. The estrogen-independent growth of AI resistant MCF-7-Ca cells was likely due to increased MAPK signaling, mediating ligand-independent activation of the ER Citation[18]. Proliferation of resistant cells and LTED cells could be inhibited with the EGFR inhibitor gefitinib (Iressa®) Citation[18], Citation[19] demonstrating that AI resistance results in a switch from ER to growth factor receptor signaling. Again, increased expression of growth factors and their receptors e.g. in the ErbB system is potential markers for resistance to AI therapy. It should be considered that different classes of AIs may have unique resistance mechanisms as partial non-cross resistance has been observed in patients treated sequentially with non-steroidal and steroidal AIs.
The knowledge that AI resistance is often associated with a switch from ER signaling to growth factor receptor mediated signaling has initiated both preclinical and clinical studies in which sequential and combination therapy against ER and ErbB receptors has been compared, and the combination was most effective Citation[20].
The mentioned studies of antiestrogen and AI resistance illustrate the utility of model systems to suggest new predictive markers and new targets for treatment. Model systems may also be useful to study whether combined or sequential single agent therapy will give the longest lasting response for the patients.
Clinical studies of endocrine sensitivity and resistance
The exploitation of basic research, preclinical and clinical studies for patient benefit is referred to as translational research. For studies of endocrine sensitivity and resistance, knowledge of molecular resistance mechanisms gained from preclinical models can suggest new predictive markers to be tested in clinical studies. The utility of new markers in patient management requires robust and quality controlled assays as well as standardized interpretation of the data. Several potential markers for response to endocrine therapy have been tested in retrospective studies. A DBCG study has tested the value of EGFR, HER-2 and p53 in predicting the efficacy of tamoxifen in high-risk postmenopausal breast cancer patients. Neither the single markers nor combination of EGFR and HER-2 disclosed statistical significant difference in response to tamoxifen Citation[21]. Markers like the vascular endothelial growth factor (VEGF), the urokinase-type plasminogen activator (uPA), its receptor (uPAR), its main inhibitor (PAI-1), Bcl-2, pS2, as well as growth factor receptors have been tested in other retrospective studies and shown potential as new predictive markers for response to endocrine Citation[22]. However, none of these single markers have yet reached level 1 evidence.
Preclinical studies have indicated that response and resistance to endocrine therapy depend on interplay between multiple proteins and consequently multiple markers may be required for prediction of therapy. Clinical studies are in progress to establish a custom-made cDNA microarray reflecting estrogen responsiveness of breast tumors. One Danish study is in progress with establishment of a predictive gene expression profile based on measurement of a selected combination of important estrogen-regulated genes e.g. PR, IGF-IR and Bcl-2. By the use of quantitative RT-PCR an initial profile of 18 genes has been selected among 59 candidate genes Citation[23]. In another Danish study a profile based on IHC detection of selected estrogen stimulated proteins on tissue micro arrays is also in progress Citation[24]. The profiles obtained from these retrospective studies will be evaluated in larger clinical studies.
It is now well established that AI therapy is superior to tamoxifen therapy for HR+ patients with advanced and early breast cancer. Retrospective studies have shown that neither PR nor HER-2 were able to be a marker for selection of patients for either AI or tamoxifen therapy Citation[4], Citation[5]. Regarding predictive markers for resistance to endocrine therapy, preclinical studies clearly showed that both tamoxifen resistance and resistance to AI was associated with a shift from ER driven growth to growth factor receptor driven growth and IHC analyses for activated ErbB receptors are in progress to establish marker combinations which will reflect resistance.
Large clinical trials comparing treatment modalities are ideal for retrospective identification of possible predictive markers and inclusion of such investigations in the planning of clinical studies would hopefully speed up the process of selecting markers for endocrine treatment. Another valuable clinical set-up in relation to the discovery of predictive markers is neoadjuvant studies in which a pre-treatment biopsy and a biopsy taken at the time of operation can be used to study marker profiles – and most importantly find changes in marker expression which reflect response to treatment. Several neoadjuvant trials have suggested that suppression of the proliferation marker Ki67 predicts response to neoadjuvant endocrine treatment Citation[25] and Ki67 expression has even been suggested as a more relevant end-point in such trials than clinical response. Further, Ki67 may be predictive of long term outcome in the adjuvant setting. EGFR and HER2 expression has also been examined in the neoadjuvant setting, but a clear correlation to treatment response has not been established. In addition to single marker investigations new planned studies will also include gene expression profiling conducted before and after onset of neoadjuvant endocrine therapy. An example of a DBCG study that includes endocrine neoadjuvant therapy is the REAL study. This randomized trial is expected to enroll 1800 HR+ postmenopausal patients with primary tumors larger than 2 cm. Patients in one arm of the trial will be offered primary surgery followed by adjuvant treatment with an AI whereas patients in the neoadjuvant treatment arm will be treated for 4 months with an AI before removal of the primary tumor. In this way, patients with tumors that are responsive or non-responsive to endocrine therapy may possibly be identified before selecting adjuvant treatment, and it offers a great chance to find new predictive markers for endocrine therapy and resistance.
Future perspectives
The need for new predictive markers in breast cancer has enhanced the search for novel techniques in addition to IHC and FISH. Translational research has made its entry into clinical investigations and besides of tumor samples that are routinely collected from each patient, blood samples and/or frozen tissue are collected as well. This has opened up for the use of profiling assays including gene expression profiles and proteomics.
At present, gene expression profiles are not routinely used in the clinic, however, the US Food and Drug administration (FDA) recently approved the use of a 70-gene expression profile, the “MammaPrint”, to determine the outcome of breast cancer recurrence within 5–10 years after prognosis. The MammaPrint was identified using microarray analysis and although the test is a prognostic test and does not have any predictive value, it certainly gives rise to optimism regarding implementing the microarray technology into routine practice, also in the context of predictive markers.
It can be speculated that large expression profiles may be substituted by measurement of a more limited number of proteins especially their active forms, if the proteins are selected based on their biological significance in a specific form of cancer. For endocrine treatment, measurement of a few proteins reflecting ER activity, together with proteins that are often changed in endocrine resistant breast cancer cells, e.g. growth factor receptors and ER co-regulators could possibly be sufficient to predict treatment response. Such an approach is more likely to be implemented in the clinic in the near future, in comparison to large scale gene and protein expression profiles.
Conclusion
Despite significant research in the past 20 years, only ER and PR are internationally accepted as being predictive for endocrine treatment of breast cancer. Yet, preclinical models have improved the understanding of mechanisms involved in endocrine sensitivity and resistance of breast cancer. Together with knowledge gained from neoadjuvant and retrospective clinical studies, this has changed the focus from a “one size fits all” treatment strategy, to the idea of tailored breast cancer therapies for which new predictive markers are currently being explored.
Acknowledgements
We kindly acknowledge the Danish Cancer Society's Scientific Committee (DLNU), “Fonden til fremme af klinisk eksperimentiel cancerforskning specielt vedrørende cancer mammae” and “Lykfeldt legatet” for financial support.
References
- Goldhirsch A, Glick JH, Gelber RD, Coates AS, Thurlimann B, Senn HJ. Meeting highlights: International expert consensus on the primary therapy of early breast cancer 2005. Ann Oncol 2005; 16: 1569–83
- Harvey JM, Clark GM, Osborne CK, Allred DC. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J Clin Oncol 1999; 17: 1474–81
- Dowsett M, Allred C, Knox J, Quinn E, Salter J, Wale C, et al. Relationship between quantitative estrogen and progesterone receptor expression and Human Epidermal Growth Factor Receptor 2 (HER-2) status with recurrence in the arimidex, tamoxifen, alone or in combination trial. J Clin Oncol 2008; Epub ahead of print.
- Viale G, Regan MM, Maiorano E, Mastropasqua MG, Dell'Orto P, Rasmussen BB, et al. Prognostic and predictive value of centrally reviewed expression of estrogen and progesterone receptors in a randomized trial comparing letrozole and tamoxifen adjuvant therapy for postmenopausal early breast cancer: BIG 1–98. J Clin Oncol 2007; 25: 3846–52
- Rasmussen BB, Regan MM, Lykkesfeldt AE, Dell'Orto P, Del CB, Henriksen KL, et al. Adjuvant letrozole versus tamoxifen according to centrally-assessed ERBB2 status for postmenopausal women with endocrine-responsive early breast cancer: Supplementary results from the BIG 1–98 randomised trial. Lancet Oncol 2008; 9: 23–8
- Lippman M, Bolan G, Huff K. The effects of estrogens and antiestrogens on hormone-responsive human breast cancer in long-term tissue culture. Cancer Res 1976; 36: 4595–601
- Schiff R, Massarweh SA, Shou J, Bharwani L, Mohsin SK, Osborne CK. Cross-talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin Cancer Res 2004; 10: 331S–6S
- Bardou VJ, Arpino G, Elledge RM, Osborne CK, Clark GM. Progesterone receptor status significantly improves outcome prediction over estrogen receptor status alone for adjuvant endocrine therapy in two large breast cancer databases. J Clin Oncol 2003; 21: 1973–9
- Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology 2003; 144: 4562–74
- Nicholson RI, Hutcheson IR, Jones HE, Hiscox SE, Giles M, Taylor KM, et al. Growth factor signalling in endocrine and anti-growth factor resistant breast cancer. Rev Endocr Metab Disord 2007; 8: 241–53
- Lykkesfeldt AE, Madsen MW, Briand P. Altered expression of estrogen-regulated genes in a tamoxifen-resistant and ICI 164,384 and ICI 182,780 sensitive human breast cancer cell line, MCF-7/TAMR-1. Cancer Res 1994; 54: 1587–95
- Howell A, DeFriend D, Robertson J, Blamey R, Walton P. Response to a specific antioestrogen (ICI 182780) in tamoxifen-resistant breast cancer. Lancet 1995; 345: 29–30
- Johnston SR, Saccani-Jotti G, Smith IE, Salter J, Newby J, Coppen M, et al. Changes in estrogen receptor, progesterone receptor, and pS2 expression in tamoxifen-resistant human breast cancer. Cancer Res 1995; 55: 3331–8
- Frogne T, Jepsen JS, Larsen SS, Fog CK, Brockdorff BL, Lykkesfeldt AE. Antiestrogen-resistant human breast cancer cells require activated protein kinase B/Akt for growth. Endocr Relat Cancer 2005; 12: 599–614
- Yde CW, Gyrd-Hansen M, Lykkesfeldt AE, Issinger OG, Stenvang J. Breast cancer cells with acquired antiestrogen resistance are sensitized to cisplatin-induced cell death. Mol Cancer Ther 2007; 6: 1869–76
- Sasano H, Anderson TJ, Silverberg SG, Santen RJ, Conway M, Edwards DP, et al. The validation of new aromatase monoclonal antibodies for immunohistochemistry–a correlation with biochemical activities in 46 cases of breast cancer. J Steroid Biochem Mol Biol 2005; 95: 35–9
- Sonne-Hansen K, Lykkesfeldt AE. Endogenous aromatization of testosterone results in growth stimulation of the human MCF-7 breast cancer cell line. J Steroid Biochem Mol Biol 2005; 93: 25–34
- Brodie A, Jelovac D, Sabnis G, Long B, Macedo L, Goloubeva O. Model systems: Mechanisms involved in the loss of sensitivity to letrozole. J Steroid Biochem Mol Biol 2005; 95: 41–8
- Martin LA, Farmer I, Johnston SR, Ali S, Marshall C, Dowsett M. Enhanced estrogen receptor (ER) alpha, ERBB2, and MAPK signal transduction pathways operate during the adaptation of MCF-7 cells to long term estrogen deprivation. J Biol Chem 2003; 278: 30458–68
- Johnston SR, Martin LA, Head J, Smith I, Dowsett M. Aromatase inhibitors: Combinations with fulvestrant or signal transduction inhibitors as a strategy to overcome endocrine resistance. J Steroid Biochem Mol Biol 2005; 95: 173–81
- Knoop AS, Bentzen S, Nielsen MM, Rasmussen BB, Rose C. Value of Epidermal Growth Factor Receptor, HER2, p53 and steroid receptors in predicting the efficacy of tamoxifen in high-risk postmenopausal breast cancer patients. J Clin Oncol 2001; 19: 3376–84
- Duffy MJ. Predictive markers in breast and other cancers: A review. Clin Chem 2005; 51: 494–503
- Lyng MB, Laenkholm AV, Vach W, Pallisgaard N, Knoop A, Ditzel HJ. Predictive gene expression profile of breast cancer patients treated with tamoxifen. Breast Cancer Res Treat 2007; 106: S167
- Wurtz SO, Henriksen KL, Lykkesfeldt A, Celis J, Brunner NA. Integration af nye og fremtidige biologiske markører i behandling af cancer mammae. Ugeskr Laeger 2007; 169: 2999–3003
- Dowsett M, Smith IE, Ebbs SR, Dixon JM, Skene A, A'Hern R, et al. Prognostic value of Ki67 expression after short-term presurgical endocrine therapy for primary breast cancer. J Natl Cancer Inst 2007; 99: 167–70