Abstract
Complement activation contributes to a pathological process in a number of autoimmune and inflammatory diseases, including rheumatoid arthritis (RA). In this review we summarize current knowledge of complement contribution to RA, based on clinical observations in patients and in vivo animal models, as well as on experiments in vitro aiming at elucidation of underlying molecular mechanisms. There is strong evidence that both the classical and the alternative pathways of complement are pathologically activated during RA as well as in animal models for RA. The classical pathway can be initiated by several triggers present in the inflamed joint such as deposited autoantibodies, dying cells, and exposed cartilage proteins such as fibromodulin. B cells producing autoantibodies, which in turn form immune complexes, contribute to RA pathogenesis partly via activation of complement. It appears that anaphylatoxin C5a is the main product of complement activation responsible for tissue damage in RA although deposition of membrane attack complex as well as opsonization with fragments of C3b are also important. Success of complement inhibition in the experimental models described so far encourages novel therapeutic approaches to the treatment of human RA.
| Abbreviations | ||
| CII | = | type II collagen |
| C5aR | = | C5a receptor |
| CAIA | = | collagen‐antibody‐induced arthritis |
| CIA | = | collagen‐induced arthritis |
| CR1 (2, 3) | = | complement receptor 1 (2, 3) |
| CVF | = | cobra venom factor |
| FH | = | factor H |
| FM | = | fibromodulin |
| G6PI | = | glucose 6 phosphoisomerase |
| IC | = | immune complex |
| IgG | = | immunoglobulin G |
| IL‐1 | = | interleukin 1 |
| LRR | = | leucine‐rich repeat (protein family) |
| MHC | = | major histocompatibility complex |
| MAC | = | membrane attack complex |
| MBL | = | mannan‐binding lectin |
| RA | = | rheumatoid arthritis |
| RF | = | rheumatoid factor |
| TNF‐α | = | tumor necrosis factor |
The complement system
The human complement system was first described more than a century ago as a bactericidal aid to antibodies, and most of its components have been known for over 30 years. This has allowed thorough investigation of various physiological functions of complement. The complement system is a part of innate immunity that is able to remove pathogens without need for previous exposure. Human complement not only protects against invading pathogens due to its opsonic, inflammatory, and lytic activities but also contributes to the regulation of other biological systems, particularly the adaptive immune response Citation1–3.
The complement cascade can proceed by the classical, the alternative, and the lectin pathways. The classical pathway is initiated by several mechanisms such as by clustered antibodies bound to their targets, the lectin pathway begins due to recognition of certain saccharides by mannan‐binding lectin (MBL) or ficolins, and the alternative pathway is started by autoactivation of unstable complement factor C3 and its subsequent deposition on activating pathogen surfaces. Central to complement activation is the formation of enzymatic complexes such as C3‐ and C5‐convertases following cleavage of proenzymes leading to their activation and concomitant release of chemoattractant anaphylatoxin fragments. Upon activation of C5, the C5a fragment is released and acts as chemoattractant for neutrophils, monocytes, macrophages, and eosinophils, induces production of cytokines and other proinflammatory mediators, and enhances vascular permeability Citation4. Similar but less pronounced effects are mediated by C3a Citation5. The other product of C5 activation, C5b, initiates assembly of membrane attack complex (MAC), which can further contribute to inflammation due to its ability to lyse cells and also to induce drastic changes in cells when present at sublytic concentrations Citation6–8. Complement factors involved in the complement cascade are controlled by a number of inhibitors, which are described in more detail below. A schematic illustration of events in the complement system is presented in .
Figure 1. Scheme of the complement system and its inhibitors. Three pathways by which the human complement system can be activated and their physiological effects: 1) clearance of apoptotic cells, 2) opsonization of pathogens and immune complexes for phagocytosis, 3) release of anaphylatoxins and lysis. Furthermore, sites of action of soluble (italics) and membrane‐bound (regular font) complement inhibitors are indicated.
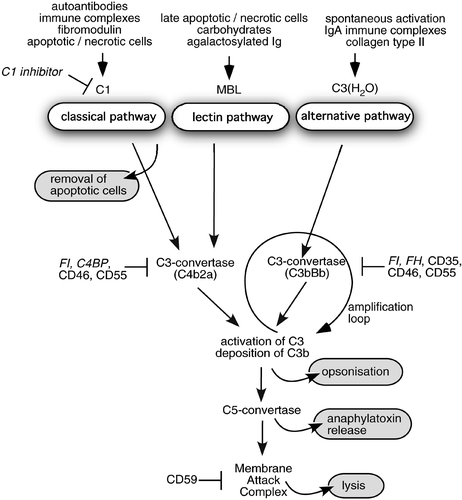
Major progress has been made regarding understanding of various functions of complement from the pathology of conditions with deficiency of various components of the complement cascade. These are often associated with recurrent infections with encapsulated bacteria and immune complex diseases such as systemic lupus erythematosus and glomerulonephritis Citation9. However, deficiencies and defects in complement inhibitors also cause diseases such as atypical hemolytic uremic syndrome Citation10,11, membranoproliferative glomerulonephritis Citation12 and age‐related macular degeneration Citation12,13. In general, the complement system is a double‐edged sword. Misguided or excessive complement activation contributes to pathogenesis of glomerulonephritis Citation14, ischemia/reperfusion injury Citation15, rheumatoid arthritis (RA) Citation16,17, multiple sclerosis Citation18, and many more acute and chronic inflammatory diseases.
Considering the rapidly activated and potentially destructive character of the complement cascade, there is an obvious need for inhibitors for protection of own tissues of the host. Complement inhibitors are either soluble or membrane‐bound and act at various levels of the complement cascades. There are four principal kinds of complement inhibitors—the first one is a proteinase inhibitor, C1 inhibitor, which belongs to the serpin family Citation19. The second type is serum carboxypeptidase N, which proteolytically inactivates anaphylatoxins generated from C5 and C3 Citation20. The third is CD59 (protectin), a glycosylphosphatidylinositol (GPI)‐anchored cell surface inhibitor of MAC Citation21. Finally a number of inhibitors act on C3‐ and C5‐convertases either by increased dissociation of enzyme complexes (acceleration of decay) or by promoting enzymatic degradation of C3b or C4b mediated by the serine proteinase factor I Citation21,22. The latter inhibitors are composed almost exclusively of 4–35 complement control protein (CCP) domains Citation23.
All these CCP‐containing complement inhibitors are encoded by genes on chromosome 1. The gene cluster is referred to as the Regulators of Complement Activation (RCA) cluster Citation24. C4b‐binding protein is the major soluble inhibitor of the classical and lectin pathways, whereas factor H (FH) inhibits the alternative route Citation25. In recent years, a whole family of FH‐like and related molecules has been identified, but it is still unclear if the major role of these molecules is to inhibit complement Citation26,27. Membrane‐bound complement inhibitors belonging to RCA include complement receptors 1 (CR1) and 2 (CR2), membrane cofactor protein (CD46), and decay‐accelerating factor (CD55).
Several complement inhibitors have already gained recognition as potential therapeutics due to their cardioprotective roles Citation28, anti‐inflammatory actions Citation29, and their potential to counteract paroxysmal nocturnal hemoglobinuria Citation30 or hyper‐acute rejection during xenotransplantation Citation31. However, although complement inhibitors have therapeutic potential, a major obstacle is that our knowledge of the intermolecular interactions involving complement regulatory proteins is limited.
Key messages
Clinical observations in patients and in vivo animal models show that activation of the complement system contributes to a pathological process in rheumatoid arthritis.
The arthritic joint contains several compounds like deposited autoantibodies, immune complexes, dying cells or exposed cartilage proteins capable of activating complement and thus mediating local inflammation mainly in a anaphylatoxin‐dependent manner.
Complement inhibition in experimental models of rheumatoid arthritis results in improved clinical score and reverses even established disease.
Rheumatoid arthritis
RA is a chronic inflammatory disease affecting approximately 1% of the adult human population with three times more females than males affected Citation32. RA is associated with persistent inflammatory polyarticular synovitis affecting mainly peripheral joints. In the early stages of disease there is proliferation and edema of synovial lining cells. The synovial lining of the affected joint is infiltrated by large numbers of T and B cells, macrophages, and granulocytes. Normally thin synovium becomes thicker resulting in swollen and tender joints. As the disease progresses, the synovium invades the joint cartilage and bone to form the ‘pannus’ Citation33. This causes irreversible joint destruction through bone resorption by osteoclasts and breakdown of bone, in particular beneath the cartilage. Release of many proteolytic enzymes such as metalloproteinases, aggrecanases, and cathepsins leads to degradation of a number of matrix constituents including proteoglycans in both bone and cartilage Citation34. Furthermore, there is neovascularization of the synovial lining surrounding the joint as well as of the pannus Citation35. As results of these processes, the joint becomes deformed, unstable, inflamed, and painful causing major disability of the patient. Cartilage as well as bone are affected, with destruction of normal architecture and function Citation36.
The pathogenesis of RA is not yet fully understood. RA has a clear genetic predisposition and part of the disease development is contributed by HLA‐DR alleles within the major histocompatibility complex (MHC) Citation37, implicating an important role for T cell autoimmunity. However, RA is a polygenic disease with a complex genetics, and it is likely that many genes in the genome will be found to contribute to the pathogenesis Citation38. In monozygotic twins the RA concordance rate ranges from 12% to 15%. When compared to disease incidence in the general population, this high value predicts that genetic predisposition could be a dominant factor in RA susceptibility. However, for dizygotic twins and non‐twin siblings the concordance rate was 2%–4% implying that there are multiple genes contributing to the genetic predisposition Citation39. Also environmental factors like exposure to tobacco smoke, mineral oil, or silica dust have been shown to increase the risk of developing RA Citation40.
Evidence for complement involvement in RA from patients
RA is considered to be an autoimmune disease since the presence of autoantibodies to a variety of antigens has been reported for patients. The classical serological marker of RA is rheumatoid factor (RF), an autoantibody that reacts with Fc parts of immunoglobulin G (IgG) Citation41. Detection of RF is one of the diagnostic criteria of RA but has relatively low specificity as it can be detected in healthy individuals as well as in chronic infections and other inflammatory conditions. Importantly, the titers of RF have been shown to predict the development of RA Citation42. However, despite intense investigations there is no strong evidence that RF has any pathogenic role. More recently, it has been found that antibodies specific for protein epitopes containing a citrullinated arginine side chain have a remarkably high sensitivity and specificity to RA. In fact, these antibodies can be detected many years before the clinical onset of RA Citation43. It is, however, unclear whether such antibodies are a result of or cause inflammation. There is a number of other autoantibody responses reported with lower degrees of sensitivity Citation44. Antibodies specific for type II collagen (CII) can be detected in 10%–50% of RA patients and has clearly been shown to be pathogenic as such antibodies induce arthritis in mice. Antibodies to glucose 6 phosphoisomerase (G6PI) may occur in patients with very severe arthritis. Importantly, both CII and G6PI are specifically targeted in the joints by antibodies and these antibodies trigger arthritis after binding to the joint cartilage. Taken together, pathogenesis of RA involves a wide range of antibodies reacting with antigens in the joints and with the potential ability to form immune complexes (IC) within cartilage and in the synovial pannus tissue. IC activate complement, become opsonized by early complement components and finally taken up by phagocytes carrying the complement receptors Citation45. Indeed, elevated levels of complement activation products and increased consumption of C3 and C4 can be detected in synovial fluids of patients suffering from RA Citation46–52. Therefore complement activation by IC in the arthritic joint is likely to be a mediator, which attracts effector cells (). Activation of infiltrated macrophages, mast cells, fibroblasts, and granulocytes by proinflammatory cytokines leads to their degranulation and subsequent detrimental release of proteolytic enzymes with ensuing joint erosion Citation34,Citation53,54. Furthermore, in vitro studies show that sublytic concentrations of MAC can stimulate collagenase production in synovial fibroblasts Citation55. Also C3b and its degradation product iC3b can activate complement receptors CR1 and CR3 normally present on synovial fluid neutrophils. These receptors are upregulated in RA as well as in other forms of arthritis Citation56. Complement activation in RA is likely to cause local inflammation due to release of small anaphylatoxins C3a and C5a. A study with 41 participating RA patients showed a 7‐fold higher concentration of C3a in synovial fluid compared to patients with degenerative joint disease or traumatic arthritis Citation51. Another study performed on synovial fluid from 22 patients demonstrated C5a concentration sufficient to induce two of the characteristic features of the acute inflammatory phases of RA: neutrophil accumulation and leakage of plasma proteins from microvessels Citation52. Also the C5a receptor (C5aR) was found on synovial macrophages and on synovial fibroblasts from RA patients. Analysis of the relationship between C5aR expression and clinical data showed significant correlation with the number of swollen joints Citation57.
Figure 2. Schematic diagram of possible contributions of the complement system to development of rheumatoid arthritis(RA) in a joint. Complement can be activated in several ways in the RA joint. Examples are deposited autoantibodies and immune complexes (IC), apoptotic and necrotic cells, certain cartilage molecules such as fibromodulin (FM) exposed upon initial cartilage damage. Activation of complement leads to a release of proinflammatory anaphylatoxin C5a that attracts and activates neutrophils and macrophages. These cells in turn release proteases and cytokines attracting T and B lymphocytes and other inflammatory cells, further fuelling the process of inflammation. As a result of complement activation membrane attack complex (MAC) is formed and has a plethora of effects on cells even at sublytic concentrations.
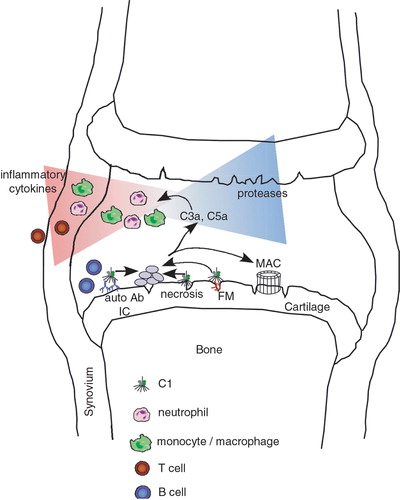
Complement components resulting from early events in the classical pathway activation were shown to reflect disease activity since the plasma level of C1q‐C4 complex was much higher in patients with active RA than in those with clinical remission Citation58. Synovial fluid of RA patients contains high concentrations of microparticles originating from apoptotic leukocytes Citation59, which activate the classical complement pathway, as shown by studies of Ig, C1q, C3, and C4 deposition on the microparticles Citation16. Noticeably, there was a positive correlation between circulating IC and C4/C4b but not C3/C3b Citation60. However, complement activation during RA seems not to be limited to the classical pathway as there is evidence that the concentration of Bb fragments generated during formation of the C3 convertase of the alternative pathway also increases in the synovial fluid of affected patients Citation48. It is still discussed which complement activation pathway is the most relevant in RA. Some data suggest that in juvenile RA the alternative pathway is the main contributor Citation61, and factor Bb concentrations were found to correlate better with the level of circulating IC Citation62. One possible explanation depends on activation of the alternative pathway by IC bearing IgA RF. Indeed, direct activation of the alternative pathway by IgA has been described Citation63, and high‐molecular‐weight IgA RF‐containing IC have been reported in children with polyarticular disease Citation64 as well as adults with RA Citation65. Another explanation relates to the fact that the alternative pathway can be activated directly by collagen type II Citation66, which is specific for cartilage and becomes exposed as a result of proteolysis during the course of disease. Furthermore, one has to remember that the alternative pathway always acts as an efficient amplification loop for the classical one at the stage of generation of C3b. Therefore the alternative pathway is crucial also for processes that were initiated via activation of the classical pathway.
Relatively little is known about contributions of the lectin pathway to RA. Among other sugar moieties of glycopeptides, MBL is able to bind agalactosylated glycoforms of IgG (IgG‐G0) and certain glycoforms of IgM, which have a high incidence of glycans terminated by GlcNAc. In RA, serum IgG‐G0 concentrations can increase significantly compared to healthy individuals Citation67. When sera of RA patients and healthy donors were examined for MBL‐binding proteins, IC containing both IgG and IgM‐RF were identified in patients' sera whereas only IgGs were found in controls. Once bound to MBL, these IC could consume C4 Citation68. These results suggest that the lectin pathway may contribute to inflammation during RA. However, the clinical data obtained so far have not verified this theory, since MBL insufficiency is positively associated with clinical symptoms and radiographic signs indicating a poor prognosis Citation69. Also, RA patients with MBL deficiency are younger at disease onset Citation70. This can perhaps be explained by the fact that MBL is one of the molecules recognizing late apoptotic and necrotic cells Citation71. In the absence of MBL, clearance of apoptotic cells and necrotic debris in the inflamed joint may be impaired leading to exacerbation of the disease.
It is worth mentioning that the synovial membrane is especially vulnerable not only to damage mediated by attracted immune cells but also by MAC formation and lysis of synovial cells. Major complement‐mediated lysis inhibitor CD59 is underexpressed in synovial‐lining cells compared to nearby stroma or endothelium. This is reflected by histological evidence from RA patients, where C5b‐9 deposition is located mainly in the synovial membrane but not in the vessel wall Citation72.
Evidence from genetic and pharmacologic studies in animal models
The importance of complement for RA in animal models was first implied when it was found that rats temporarily decomplemented with cobra venom factor were resistant to collagen‐induced arthritis (CIA) Citation73. CIA is perhaps the most widely used model, which meets many of the criteria for diagnosis of RA Citation74. A well established strategy to explore the genetic causative factors and molecular mechanism for common disease is to use inbred animal models. Various genetic linkage studies have located more than 30 genomic loci potentially harboring genes predisposing to CIA and other models for human RA in mice and rats. The Cia2 and Cia9 loci were identified as the main genetic factors influencing the arthritis development in an F2 intercross between the arthritis susceptible strain C57Bl/10.Q (B10.Q) and the resistant NOD.Q strain Citation75. The Cia2 locus associated with CIA was also identified in (DBA/1×SWR/J) F2 crosses Citation76 and also associated with arthritis in the K/B×N arthritis model in a cross between B6 and NOD Citation77. The Cia2 locus harbors a strong candidate gene for arthritis susceptibility, the complement component C5 encoding gene Hc. CIA was also used to show that SWR mice expressing the I‐Aq susceptibility gene but naturally lacking Hc gene were resistant to the disease Citation78. Both the NOD and the SWR/J strains carry a 2 bp deletion in an exon near the 5' end of the Hc gene resulting in frame‐shift and termination resulting in C5 protein deficiency Citation79,80.
Gene inactivation technology was also used to ascertain the role of complement in arthritis. Genetic deletion of C5, C3, or factor B in DBA/1 mice in each case resulted in mice being resistant to CIA despite high titers of anti‐collagen type II antibodies Citation81,82. C3−/− mice developed mild arthritis whereas arthritis in FB−/− mice was intermediate with regard to C3−/− and control mice. In other studies no role for the classical pathway was found, and it was postulated that only the alternative pathway operated to mediate arthritis in CIA Citation83. Thus, the relative importance of the classical versus the alternative pathway in CIA is not yet established. C3 and factor B knockout mice were shown to be resistant to induction of disease in the anti‐collagen antibody‐induced arthritis (CAIA) model Citation84. This model bypasses the complexity of CIA, which consists of both the priming and effector phase by focusing on the effector phase only as it is induced with monoclonal antibodies without the need for T cells or B cells Citation85. Regarding C5 component, both deficiency and systemic administration of anti‐C5 antibodies ameliorated CIA Citation86. The antibodies prevent cleavage of C5 and release of C5a. C5a induced chemotaxis as well as a wide array of effects such as increase in vascular permeability and cellular degranulation. Importantly, antibody against C5a prevented not only arthritis but also reversed ongoing disease when injected several days after disease onset Citation86.
Recently another model of RA was used to systematically assess the role of complement. K/B×N mice produce high titers of IgG1 antibodies to G6PI and develop arthritis spontaneously Citation87. Injection of serum from sick into healthy animals provoked arthritis with clinical and histological symptoms similar to human RA Citation88. No difference was induced by crossing the K/B×N mice with C1q and C4 knockout mice showing that the classical pathway is not crucial for disease progression in this model. However, progeny of K/B×N mice and knockout mice deficient in factor B, C3, C5, or C5aR were completely resistant to arthritis. It appears that this model is dependent on the alternative pathway Citation87. An important difference in this model, as compared with CAIA, is that the serum from K/B×N mice contains antibodies to G6PI predominantly of the IgG1 isotype, which do not activate complement, whereas CAIA is induced by monoclonal antibodies of different isotypes including IgG2a/c and IgG2b Citation85. Most importantly, it was observed that C6 deficiency did not influence disease progression in this model, showing that MAC is not a mediator of the tissue damage Citation87. summarizes the evidence for involvement of complement in RA observed in animal models.
Table I. Evidence for involvement of complement in rheumatoid arthritis (RA) observed in animal models.
Cartilage molecules contributing to the regulation of complement activation in RA
A well established clinical observation, pertinent to understanding of mechanisms in joint disease, is that joint replacement surgery ameliorates inflammation. It appears that molecules present in or released from cartilage may have a role in propagating this inflammation by activating complement. One candidate is fibromodulin (FM) Citation89.
Cartilage contains low numbers of cells separated by a predominating extracellular matrix with several distinct supramolecular assemblies. These include extremely negatively charged proteoglycans (aggrecan) entrapped in networks of collagen fibers. The latter contain many additional molecules bound at the fiber surface to provide stability and interactions with surrounding structures. Among these molecules there are members of the leucine‐rich repeat (LRR) protein family, such as decorin, biglycan, osteoadherin, FM, lumican, and chondroadherin Citation90. Some LRR proteins, present in several tissues, including cartilage, were found to interact with complement factors. Thus, C1q binds to decorin Citation91 and biglycan Citation91 as well as to non‐related fibronectin Citation92 and laminin Citation93. However, in none of these examples does the interaction lead to activation of the complement cascade.
FM was first described as a 59‐kDa protein with substituents of highly anionic keratan sulphate glycosaminoglycan chains. The protein interacts with collagen types I and II Citation94. It localizes to collagen fibers in cartilage Citation95. FM is thought to play an important role in collagen fiber formation as shown by the observation that FM knockout mice form abnormal collagen fibrils in e.g. tendons Citation96. During inflammatory joint disease proteinases, particularly of the metalloproteinase family (MMPs) degrade cartilage tissue molecules. In this process an initial event is liberation of the N‐terminal portion of FM by the actions of MMP‐13 Citation97, while at later stages the remainder of the molecule is released.
FM interacts with the globular heads of C1q. This interaction, typical for ligands like IC, leads to the activation of complement. FM mainly activates the classical pathway of complement Citation89. The activation initiated by FM results in pronounced deposition of C3b and C4b but less pronounced deposition of MAC. The failure to induce MAC deposition is most probably due to binding between FM and the complement inhibitor FH. C1q and FH bind to non‐overlapping sites on FM, and none of these interactions appear to be mediated by the highly polyanionic polysaccharide keratan sulfate decorating FM Citation89. Taken together, it appears that FM is one of the factors fuelling joint inflammation when the initial damage to the cartilage has occurred.
Interestingly, decorin Citation91,Citation98 and biglycan Citation91 bind C1q, but instead of triggering complement activation they inhibit the ability of C1 to activate the classical pathway. These interactions take place at physiological ionic strength and may be relevant for regulation of complement activation and C1‐mediated effects on cells in the inflamed joint Citation91. One putative mechanism involves the large and extended anionic glycosaminoglycan chain, which may modulate other interactions, also at a distance from its binding site, with C1q. Immunohistochemical studies revealed that decorin concentrates in the same region of articular cartilage where IC and C3 have been found Citation98,99. Moreover, biglycan was shown to inhibit C1q‐induced production of proinflammatory cytokines in endothelial cells Citation91. Summing up, there are several molecules in cartilage able to bind C1q. While one of them (FM) activates complement the others may act as local complement inhibitors, and the final outcome will depend on many local parameters, such as how fragmentation of proteoglycans during the tissue destruction will affect the binding site and possibly separate a binding site from e.g. an anionic domain modulating interactions and effects.
RA, B cells, complement, and cell death
Substantial numbers of B cells can be found within the infiltrate attracted into RA joints. B cell infiltrates may either be diffuse and lack any structural organization such as form small clusters and large follicle‐like structures Citation100. Most of these B cells can take part in antigen‐driven immune response and differentiate into plasma cells Citation101. Since the RF present in arthritic joints form IC opsonized with complement, such complexes are internalized by B cells, which in turn generate a broad spectrum of peptides enhancing the antigen‐presenting function Citation102. In this way a positive feedback increasing the production of high‐affinity antibodies with RF specificity could develop, thus sustaining the chronic inflammatory process in RA Citation100. The role of B cells in the context of autoimmune disease seems to be important not only due to Ig production but also because of their antigen‐presenting function. This was confirmed in a mouse lupus model, in which MRL/lpr mice that transgenically expressed surface immunoglobulins, but did not permit the secretion of circulating Ig, did develop nephritis Citation103. Moreover, another study showed that fibroblast‐like synoviocytes are able to bind B cells and prevent them from entering into apoptosis. This phenomenon was observed only for synoviocytes from RA patients but not patients with other types of arthritis Citation104.
Since a role of B cells in autoimmune disease was suggested, B cell depletion therapy was proposed for patients with RA Citation105. One of the targets of such therapy is the CD20 molecule present on more than 95% of normal or neoplastic B cells Citation106 but not stem cells and B cells differentiating into plasma cells Citation107. Very promising results of B cell depletion therapy in RA patients were achieved with a genetically engineered, chimeric anti‐CD20 monoclonal antibody—rituximab Citation107. A study performed on more than 100 patients subjected to a single short course of treatment with rituximab, either alone or in combination with cyclophosphamide or continuing methotrexate, provided significant improvements in disease symptoms. The mechanism suppressing arthritis is, however, not yet clarified and may involve decrease of pathogenic antibodies or may be due to other B cell functions than the production of antibodies. The mechanisms whereby the injected antibodies delete the B cells seem to be better clarified and complement appears to be involved in B cell elimination. Several observations testify to this theory: 1) After ligation of CD20 with mAb cell death is independent of caspases Citation108; 2) treatment with rituximab did not cure C1q‐deficient mice bearing human CD20‐positive lymphomas Citation109; 3) elevated expression of CD55 and CD59 was found on cells from patients not responding to rituximab Citation110; and 4) substantial complement consumption was observed during rituximab treatment Citation111.
Taken together, B cell activity in RA appears to sustain the process of acute inflammation already initiated in the joint tissue. However, depletion of reactive B cells proves to be promising in treatment, and is at least able to diminish activity at clinical onset or delay it. When mAb directed against B cells are introduced, their therapeutic potential is likely to be dependent on the decrease of production of pathogenic antibodies and the downstream dependence on complement.
Developing therapeutic strategies based on complement contribution to RA
Advances in understanding the roles of cytokines are directed to new developments in the treatments available to RA patients. Nowadays there are several approved compounds based on monoclonal antibodies or binding proteins, which target inflammatory cytokines. The anti‐tumor necrosis factor (TNF‐α) clinically approved drugs are infliximab (Remicade), etanercept (Enbrel), and adalimumab (Humira), and the interleukin 1 (IL‐1) inhibitor is anakinra (Kineret) Citation112. Anticytokine therapy in RA was found to reduce the symptoms of inflammation as well as slow or stop structural joint damage Citation113. However, although targeting of these key cytokines is very efficient in the two‐thirds of patients that react, they have inherent complications as these factors are important in immune defense and their inhibition compromises immune regulation with ensuing rare infections Citation114, lupus‐like syndrome, or re‐activation of chronic hepatitis B Citation115. TNF‐α antagonists should be avoided in patients with a preexisting demyelinating disease Citation115. Considering these numbers of restrictions, new drugs targeting upstream mediators of joint inflammation would provide new openings for more specific therapy.
As the complement system is one of the significant cascades generating inflammation in the effector phase of RA, it is feasible that complement inhibitors will be able to ameliorate the disease. Up to date, several types of compounds interfering with the complement cascade were extensively studied and entered into clinical trials (for review see Citation116). One example is the soluble CR1 (sCR1) receptor derivatives. Membrane‐bound CR1 acts as cofactor for factor I‐mediated cleavage of C3b or C4b and decay‐accelerating factor for classical and alternative C3 and C5 convertases Citation117. The protein is expressed mainly on erythrocytes and other blood cells. Negligible quantities (up to 30 ng/mL) of soluble CR1 are found in blood Citation118. Since sCR1 retains its powerful complement inhibitory activity, it appeared as potential candidate for therapeutic complement regulator. Two strategies were employed to improve and target the CR1 potential to cells under complement attack. First was the decoration of sCR1 with sialyl‐Lewisx tetrasaccharides. This modification introduced the affinity to selectins, which are overexpressed by cells at the sites of inflammation Citation29. Another modification concerns truncation of sCR1 to three N‐terminal CCP domains and coupling to a membrane‐targeting peptide Citation119. This compound, named APT070 was successfully tested in a rat model of antigen‐induced arthritis Citation119. TP10, a sCR1‐derived compound, was tested in patients with myocardial infarction, acute respiratory distress syndrome, and postcardiopulmonary bypass syndrome Citation120,121. A second‐generation of this drug named TP20 was tested successfully in animals Citation122. Another anticomplement drug, monoclonal antibody against C5 (eculizumab, 5G1.1), targets the C5 molecule in a way which disables its splitting into C5a and C5b, thus preventing both enhancement of inflammation by anaphylatoxin release and formation of MAC Citation123. Previous studies showed that mouse anti‐C5 Ab (BB5.1), which is functionally related to eculizumab, gave promising results in CIA Citation86. It inhibited both the onset of disease after immunization and ameliorated already established disease. Up to date, the most beneficial application of eculizumab was reported in patients suffering from paroxysmal nocturnal hemoglobinuria Citation30. Furthermore, the results of eculizumab's clinical phase II studies in RA patients are available Citation124. They revealed significant improvement in RA clinical score when the drug was given intravenously at certain intervals for 3 months. The best results were obtained for patients who exhibited high basal level of C5b‐9 in serum Citation125.
Additionally to those anticomplement drugs already tested clinically, there are new compounds targeting the complement system, and some of them have passed trials in animal models of arthritis. They can be grouped into several types, e.g. serine protease inhibitors (FUT‐175, BCX‐1470), C3 inhibitors (compstatin), C5 inhibitors (TS‐A12/22, K76COONa), anaphylatoxin receptor antagonists (SB290157, AcF‐[OPdChaWR], PMX‐53); for review see Citation126. The most promising therapeutic approaches targeting complement for RA treatment which are presently under development are listed in .
Table II. Therapeutic approaches to complement inhibition in rheumatoid arthritis.
While there is compelling evidence for great benefits of therapeutic complement inhibition, recent advances indicate that complement components are multifunctional. Many of these components are critical for the proper function of mammalian cells and tissues. Thus, prior to clinical translation an understanding of the proper targeting, timing, duration, and potency of anticomplement agents is needed in order to avoid interfering with critical processes related to tissue repair, healing, and function. A putative, ideal therapy based on complement inhibition would act only locally in affected tissue or act systemically but keep the complement activity at the level sustaining its other functions. Systemic complement depletion can be dangerous since it is clear that the complement cascade is made up of intricate systems with widely distinct effects. Any proposed therapy must take this variety into account, particularly in the case of diseases like RA that often requires lifelong treatment.
References
- Walport M. J. Complement. Second of two parts. N Engl J Med 2001; 344: 1140–4
- Seelen M. A., Roos A., Daha M. R. Role of complement in innate and autoimmunity. J Nephrol 2005; 18: 642–53
- Kohl J. Drug evaluation: the C5a receptor antagonist PMX‐53. Curr Opin Mol Ther 2006; 8: 529–38
- Riedemann N. C., Guo R. F., Ward P. A. A key role of C5a/C5aR activation for the development of sepsis. J Leukoc Biol 2003; 74: 966–70
- Law S., Reid K. Complement. IRL Press, OxfordUK 1988
- Kilgore K. S., Flory C. M., Miller B. F., Evans V. M., Warren J. S. The membrane attack complex of complement induces interleukin‐8 and monocyte chemoattractant protein‐1 secretion from human umbilical vein endothelial cells. Am J Pathol 1996; 149: 953–61
- Reiter Y., Ciobotariu A., Fishelson Z. Sublytic complement attack protects tumor cells from lytic doses of antibody and complement. Eur J Immunol 1992; 22: 1207–13
- Fosbrink M., Niculescu F., Rus H. The role of c5b‐9 terminal complement complex in activation of the cell cycle and transcription. Immunol Res 2005; 31: 37–46
- S Reis E., Falcao D. A., Isaac L. Clinical aspects and molecular basis of primary deficiencies of complement component C3 and its regulatory proteins factor I and factor H. Scand J Immunol 2006; 63: 155–68
- Richards A., Kathryn Liszewski M., Kavanagh D., Fang C. J., Moulton E., Fremeaux‐Bacchi V., et al. Implications of the initial mutations in membrane cofactor protein (MCP; CD46) leading to atypical hemolytic uremic syndrome. Mol Immunol 2007; 44: 111–22
- Zipfel P. F., Misselwitz J., Licht C., Skerka C. The role of defective complement control in hemolytic uremic syndrome. Semin Thromb Hemost 2006; 32: 146–54
- Zipfel P. F., Heinen S., Jozsi M., Skerka C. Complement and diseases: defective alternative pathway control results in kidney and eye diseases. Mol Immunol 2006; 43: 97–106
- Schaumberg D. A., Hankinson S. E., Guo Q., Rimm E., Hunter D. J. A prospective study of 2 major age‐related macular degeneration susceptibility alleles and interactions with modifiable risk factors. Arch Ophthalmol 2007; 125: 55–62
- Seelen M. A., Daha M. R. The role of complement in autoimmune renal disease. Autoimmunity 2006; 39: 411–5
- Chan R. K., Ibrahim S. I., Takahashi K., Kwon E., McCormack M., Ezekowitz A., et al. The differing roles of the classical and mannose‐binding lectin complement pathways in the events following skeletal muscle ischemia‐reperfusion. J Immunol 2006; 177: 8080–5
- Biro E., Nieuwland R., Tak P. P., Pronk L. M., Schaap M. C., Sturk A., et al. Activated complement components and complement activator molecules on the surface of cell‐derived microparticles in patients with rheumatoid arthritis and healthy individuals. Ann Rheum Dis 2007 Jan 29, (Epub ahead of print)
- Allegretti M., Moriconi A., Beccari A. R., Di Bitondo R., Bizzarri C., Bertini R., et al. Targeting C5a: recent advances in drug discovery. Curr Med Chem 2005; 12: 217–36
- Rus H., Cudrici C., Niculescu F. C5b‐9 complement complex in autoimmune demyelination and multiple sclerosis: dual role in neuroinflammation and neuroprotection. Ann Med 2005; 37: 97–104
- Harpel P. C., Cooper N. R. Studies on human plasma C1 inactivator‐enzyme interactions. I. Mechanisms of interaction with C1s, plasmin, and trypsin. J Clin Invest 1975; 55: 593–604
- Skidgel R. A., Kawahara M. S., Hugli T. E. Functional significance of the subunits of carboxypeptidase N (kininase I). Adv Exp Med Biol 1986; 198 Pt A: 375–80
- Kim D. D., Song W. C. Membrane complement regulatory proteins. Clin Immunol 2006; 118: 127–36
- Tsiftsoglou S. A., Willis A. C., Li P., Chen X., Mitchell D. A., Rao Z., et al. The catalytically active serine protease domain of human complement factor I. Biochemistry 2005; 44: 6239–49
- Krushkal J., Bat O., Gigli I. Evolutionary relationships among proteins encoded by the regulator of complement activation gene cluster. Mol Biol Evol 2000; 17: 1718–30
- Carroll M. C., Alicot E. M., Katzman P. J., Klickstein L. B., Smith J. A., Fearon D. T. Organization of the genes encoding complement receptors type 1 and 2, decay‐accelerating factor, and C4‐binding protein in the RCA locus on human chromosome 1. J Exp Med 1988; 167: 1271–80
- Blom A. M., Kask L., Ramesh B., Hillarp A. Effects of zinc on factor I cofactor activity of C4b‐binding protein and factor H. Arch Biochem Biophys 2003; 418: 108–18
- Schwaeble W., Zwirner J., Schulz T. F., Linke R. P., Dierich M. P., Weiss E. H. Human complement factor H: expression of an additional truncated gene product of 43 kDa in human liver. Eur J Immunol 1987; 17: 1485–9
- McRae J. L., Murphy B. E., Eyre H. J., Sutherland G. R., Crawford J., Cowan P. J. Location and structure of the human FHR‐5 gene. Genetica 2002; 114: 157–61
- Fu J., Lin G., Zeng B., Wu Z., Wu Y., Chu H., et al. Anti‐ischemia/reperfusion of C1 inhibitor in myocardial cell injury via regulation of local myocardial C3 activity. Biochem Biophys Res Commun 2006; 350: 162–8
- Mulligan M. S., Warner R. L., Rittershaus C. W., Thomas L. J., Ryan U. S., Foreman K. E., et al. Endothelial targeting and enhanced antiinflammatory effects of complement inhibitors possessing sialyl Lewisx moieties. J Immunol 1999; 162: 4952–9
- Hillmen P., Young N. S., Schubert J., Brodsky R. A., Socie G., Muus P., et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med 2006; 355: 1233–43
- Kirschfink M. C1‐inhibitor and transplantation. Immunobiology 2002; 205: 534–41
- Smith C. A., Arnett F. C. Epidemiologic aspects of rheumatoid arthritis. Current immunogenetic approach. Clin Orthop Relat Res 1991; 1991: 23–35
- Feldmann M., Brennan F. M., Maini R. N. Rheumatoid arthritis. Cell 1996; 85: 307–10
- Lark M. W., Bayne E. K., Flanagan J., Harper C. F., Hoerrner L. A., Hutchinson N. I., et al. Aggrecan degradation in human cartilage. Evidence for both matrix metalloproteinase and aggrecanase activity in normal, osteoarthritic, and rheumatoid joints. J Clin Invest 1997; 100: 93–106
- Szekanecz Z., Gaspar L., Koch A. E. Angiogenesis in rheumatoid arthritis. Front Biosci 2005; 10: 1739–53
- Schett G. Rheumatoid arthritis: inflammation and bone loss. Wien Med Wochenschr 2006; 156: 34–41
- Gregersen P. K., Silver J., Winchester R. J. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum 1987; 30: 1205–13
- Plenge R. M., Padyukov L., Remmers E. F., Purcell S., Lee A. T., Karlson E. W., et al. Replication of putative candidate‐gene associations with rheumatoid arthritis in >4,000 samples from North America and Sweden: association of susceptibility with PTPN22, CTLA4, and PADI4. Am J Hum Genet 2005; 77: 1044–60
- Wandstrat A., Wakeland E. The genetics of complex autoimmune diseases: non‐MHC susceptibility genes. Nat Immunol 2001; 2: 802–9
- Klareskog L., Padyukov L., Ronnelid J., Alfredsson L. Genes, environment and immunity in the development of rheumatoid arthritis. Curr Opin Immunol 2006; 18: 650–5
- Weissmann G. Pathogenesis of rheumatoid arthritis. J Clin Rheumatol 2004; 10: S26–31
- Aho K., Palosuo T., Raunio V., Puska P., Aromaa A., Salonen J. T. When does rheumatoid disease start?. Arthritis Rheum 1985; 28: 485–9
- Rantapaa‐Dahlqvist S., de Jong B. A., Berglin E., Hallmans G., Wadell G., Stenlund H., et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum 2003; 48: 2741–9
- Mewar D., Wilson A. G. Autoantibodies in rheumatoid arthritis: a review. Biomed Pharmacother 2006; 60: 648–55
- Hepburn A. L., Mason J. C., Wang S., Shepherd C. J., Florey O., Haskard D. O., et al. Both Fcgamma and complement receptors mediate transfer of immune complexes from erythrocytes to human macrophages under physiological flow conditions in vitro. Clin Exp Immunol 2006; 146: 133–45
- Morgan B. P., Daniels R. H., Williams B. D. Measurement of terminal complement complexes in rheumatoid arthritis. Clin Exp Immunol 1988; 73: 473–8
- Swaak A. J., Van Rooyen A., Planten O., Han H., Hattink O., Hack E. An analysis of the levels of complement components in the synovial fluid in rheumatic diseases. Clin Rheumatol 1987; 6: 350–7
- Brodeur J. P., Ruddy S., Schwartz L. B., Moxley G. Synovial fluid levels of complement SC5b‐9 and fragment Bb are elevated in patients with rheumatoid arthritis. Arthritis Rheum 1991; 34: 1531–7
- Corvetta A., Pomponio G., Rinaldi N., Luchetti M. M., Di Loreto C., Stramazzotti D. Terminal complement complex in synovial tissue from patients affected by rheumatoid arthritis, osteoarthritis and acute joint trauma. Clin Exp Rheumatol 1992; 10: 433–8
- Hogasen K., Mollnes T. E., Harboe M., Gotze O., Hammer H. B., Oppermann M. Terminal complement pathway activation and low lysis inhibitors in rheumatoid arthritis synovial fluid. J Rheumatol 1995; 22: 24–8
- Moxley G., Ruddy S. Elevated C3 anaphylatoxin levels in synovial fluids from patients with rheumatoid arthritis. Arthritis Rheum 1985; 28: 1089–95
- Jose P. J., Moss I. K., Maini R. N., Williams T. J. Measurement of the chemotactic complement fragment C5a in rheumatoid synovial fluids by radioimmunoassay: role of C5a in the acute inflammatory phase. Ann Rheum Dis 1990; 49: 747–52
- Woolley D. E., Tetlow L. C. Observations on the microenvironmental nature of cartilage degradation in rheumatoid arthritis. Ann Rheum Dis 1997; 56: 151–61
- Tetlow L. C., Harper N., Dunningham T., Morris M. A., Bertfield H., Woolley D. E. Effects of induced mast cell activation on prostaglandin E and metalloproteinase production by rheumatoid synovial tissue in vitro. Ann Rheum Dis 1998; 57: 25–32
- Jahn B., Von Kempis J., Kramer K. L., Filsinger S., Hansch G. M. Interaction of the terminal complement components C5b‐9 with synovial fibroblasts: binding to the membrane surface leads to increased levels in collagenase‐specific mRNA. Immunology 1993; 78: 329–34
- Crockard A. D., Thompson J. M., McBride S. J., Edgar J. D., McNeill T. A., Bell A. L. Markers of inflammatory activation: upregulation of complement receptors CR1 and CR3 on synovial fluid neutrophils from patients with inflammatory joint disease. Clin Immunol Immunopathol 1992; 65: 135–42
- Yuan G., Wei J., Zhou J., Hu H., Tang Z., Zhang G. Expression of C5aR (CD88) of synoviocytes isolated from patients with rheumatoid arthritis and osteoarthritis. Chin Med J (Engl) 2003; 116: 1408–12
- Wouters D., Voskuyl A. E., Molenaar E. T., Dijkmans B. A., Hack C. E. Evaluation of classical complement pathway activation in rheumatoid arthritis: measurement of C1q‐C4 complexes as novel activation products. Arthritis Rheum 2006; 54: 1143–50
- Berckmans R. J., Nieuwland R., Tak P. P., Boing A. N., Romijn F. P., Kraan M. C., et al. Cell‐derived microparticles in synovial fluid from inflamed arthritic joints support coagulation exclusively via a factor VII‐dependent mechanism. Arthritis Rheum 2002; 46: 2857–66
- Makinde V. A., Senaldi G., Jawad A. S., Berry H., Vergani D. Reflection of disease activity in rheumatoid arthritis by indices of activation of the classical complement pathway. Ann Rheum Dis 1989; 48: 302–6
- Aggarwal A., Bhardwaj A., Alam S., Misra R. Evidence for activation of the alternate complement pathway in patients with juvenile rheumatoid arthritis. Rheumatology (Oxford) 2000; 39: 189–92
- Jarvis J. N., Taylor H., Iobidze M., Krenz M. Complement activation and immune complexes in children with polyarticular juvenile rheumatoid arthritis: a longitudinal study. J Rheumatol 1994; 21: 1124–7
- Schaapherder A. F., Gooszen H. G., te Bulte M. T., Daha M. R. Human complement activation via the alternative pathway on porcine endothelium initiated by IgA antibodies. Transplantation 1995; 60: 287–91
- Jarvis J. N., Iobidze M., Taylor H., DeJonge J., Chang S. A comparison of immunoglobulin G‐containing high‐molecular‐weight complexes isolated from children with juvenile rheumatoid arthritis and congenital human immunodeficiency virus infection. Pediatr Res 1993; 34: 781–4
- van Zeben D., Hazes J. M., Zwinderman A. H., Cats A., van der Voort E. A., Breedveld F. C. Clinical significance of rheumatoid factors in early rheumatoid arthritis: results of a follow up study. Ann Rheum Dis 1992; 51: 1029–35
- Hanauske‐Abel H. M., Pontz B. F., Schorlemmer H. U. Cartilage specific collagen activates macrophages and the alternative pathway of complement: evidence for an immunopathogenic concept of rheumatoid arthritis. Ann Rheum Dis 1982; 41: 168–76
- Arnold J. N., Dwek R. A., Rudd P. M., Sim R. B. Mannan binding lectin and its interaction with immunoglobulins in health and in disease. Immunol Lett 2006; 106: 103–10
- Sato R., Matsushita M., Miyata M., Sato Y., Kasukawa R., Fujita T. Substances reactive with mannose‐binding protein (MBP) in sera of patients with rheumatoid arthritis. Fukushima J Med Sci 1997; 43: 99–111
- Kilpatrick D. C. Mannan‐binding lectin and its role in innate immunity. Transfus Med 2002; 12: 335–52
- Arend W. P. The innate immune system in rheumatoid arthritis. Arthritis Rheum 2001; 44: 2224–34
- Nauta A. J., Raaschou‐Jensen N., Roos A., Daha M. R., Madsen H. O., Borrias‐Essers M. C., et al. Mannose‐binding lectin engagement with late apoptotic and necrotic cells. Eur J Immunol 2003; 33: 2853–63
- Kemp P. A., Spragg J. H., Brown J. C., Morgan B. P., Gunn C. A., Taylor P. W. Immunohistochemical determination of complement activation in joint tissues of patients with rheumatoid arthritis and osteoarthritis using neoantigen‐specific monoclonal antibodies. J Clin Lab Immunol 1992; 37: 147–62
- Morgan K., Clague R. B., Shaw M. J., Firth S. A., Twose T. M., Holt P. J. Native type II collagen‐induced arthritis in the rat: the effect of complement depletion by cobra venom factor. Arthritis Rheum 1981; 24: 1356–62
- Joe B., Wilder R. L. Animal models of rheumatoid arthritis. Mol Med Today 1999; 5: 367–9
- Lindqvist A. K., Johannesson M., Johansson A. C., Nandakumar K. S., Blom A. M., Holmdahl R. Backcross and partial advanced intercross analysis of nonobese diabetic gene‐mediated effects on collagen‐induced arthritis reveals an interactive effect by two major loci. J Immunol 2006; 177: 3952–9
- Spinella D. G., Jeffers J. R., Reife R. A., Stuart J. M. The role of C5 and T‐cell receptor Vb genes in susceptibility to collagen‐induced arthritis. Immunogenetics 1991; 34: 23–7
- Ji H., Gauguier D., Ohmura K., Gonzalez A., Duchatelle V., Danoy P., et al. Genetic influences on the end‐stage effector phase of arthritis. J Exp Med 2001; 194: 321–30
- McIndoe R. A., Bohlman B., Chi E., Schuster E., Lindhardt M., Hood L. Localization of non‐Mhc collagen‐induced arthritis susceptibility loci in DBA/1j mice. Proc Natl Acad Sci U S A 1999; 96: 2210–4
- Baxter A. G., Cooke A. Complement lytic activity has no role in the pathogenesis of autoimmune diabetes in NOD mice. Diabetes 1993; 42: 1574–8
- Wetsel R. A., Fleischer D. T., Haviland D. L. Deficiency of the murine fifth complement component (C5). A 2‐base pair gene deletion in a 5'‐exon. J Biol Chem 1990; 265: 2435–40
- Wang Y., Kristan J., Hao L., Lenkoski C. S., Shen Y., Matis L. A. A role for complement in antibody‐mediated inflammation: C5‐deficient DBA/1 mice are resistant to collagen‐induced arthritis. J Immunol 2000; 164: 4340–7
- Hietala M. A., Jonsson I. M., Tarkowski A., Kleinau S., Pekna M. Complement deficiency ameliorates collagen‐induced arthritis in mice. J Immunol 2002; 169: 454–9
- Banda N. K., Thurman J. M., Kraus D., Wood A., Carroll M. C., Arend W. P., et al. Alternative complement pathway activation is essential for inflammation and joint destruction in the passive transfer model of collagen‐induced arthritis. J Immunol 2006; 177: 1904–12
- Hietala M. A., Nandakumar K. S., Persson L., Fahlen S., Holmdahl R., Pekna M. Complement activation by both classical and alternative pathways is critical for the effector phase of arthritis. Eur J Immunol 2004; 34: 1208–16
- Nandakumar K. S., Holmdahl R. Antibody‐induced arthritis: disease mechanisms and genes involved at the effector phase of arthritis. Arthritis Res Ther 2007; 8: 223
- Wang Y., Rollins S. A., Madri J. A., Matis L. A. Anti‐C5 monoclonal antibody therapy prevents collagen‐induced arthritis and ameliorates established disease. Proc Natl Acad Sci U S A 1995; 92: 8955–9
- Ji H., Ohmura K., Mahmood U., Lee D. M., Hofhuis F. M., Boackle S. A., et al. Arthritis critically dependent on innate immune system players. Immunity 2002; 16: 157–68
- Matsumoto I., Staub A., Benoist C., Mathis D. Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science 1999; 286: 1732–5
- Sjoberg A., Onnerfjord P., Morgelin M., Heinegard D., Blom A. M. The extracellular matrix and inflammation: fibromodulin activates the classical pathway of complement by directly binding C1q. J Biol Chem 2005; 280: 32301–8
- Bengtsson E., Neame P. J., Heinegard D., Sommarin Y. The primary structure of a basic leucine‐rich repeat protein, PRELP, found in connective tissues. J Biol Chem 1995; 270: 25639–44
- Groeneveld T. W., Oroszlan M., Owens R. T., Faber‐Krol M. C., Bakker A. C., Arlaud G. J., et al. Interactions of the extracellular matrix proteoglycans decorin and biglycan with C1q and collectins. J Immunol 2005; 175: 4715–23
- Barilla M. L., Carsons S. E. Fibronectin fragments and their role in inflammatory arthritis. Semin Arthritis Rheum 2000; 29: 252–65
- Bohnsack J. F., Tenner A. J., Laurie G. W., Kleinman H. K., Martin G. R., Brown E. J. The C1q subunit of the first component of complement binds to laminin: a mechanism for the deposition and retention of immune complexes in basement membrane. Proc Natl Acad Sci U S A 1985; 82: 3824–8
- Oldberg A., Antonsson P., Lindblom K., Heinegard D. A collagen‐binding 59‐kd protein (fibromodulin) is structurally related to the small interstitial proteoglycans PG‐S1 and PG‐S2 (decorin). EMBO J 1989; 8: 2601–4
- Benjamin M., Ralphs J. R. Biology of fibrocartilage cells. Int Rev Cytol 2004; 233: 1–45
- Svensson L., Aszodi A., Reinholt F. P., Fassler R., Heinegard D., Oldberg A. Fibromodulin‐null mice have abnormal collagen fibrils, tissue organization, and altered lumican deposition in tendon. J Biol Chem 1999; 274: 9636–47
- Heathfield T. F., Onnerfjord P., Dahlberg L., Heinegard D. Cleavage of fibromodulin in cartilage explants involves removal of the N‐terminal tyrosine sulfate‐rich region by proteolysis at a site that is sensitive to matrix metalloproteinase‐13. J Biol Chem 2004; 279: 6286–95
- Krumdieck R., Hook M., Rosenberg L. C., Volanakis J. E. The proteoglycan decorin binds C1q and inhibits the activity of the C1 complex. J Immunol 1992; 149: 3695–701
- Poole A. R., Webber C., Pidoux I., Choi H., Rosenberg L. C. Localization of a dermatan sulfate proteoglycan (DS‐PGII) in cartilage and the presence of an immunologically related species in other tissues. J Histochem Cytochem 1986; 34: 619–25
- Kim H. J., Berek C. B cells in rheumatoid arthritis. Arthritis Res 2000; 2: 126–31
- Schroder A. E., Greiner A., Seyfert C., Berek C. Differentiation of B cells in the nonlymphoid tissue of the synovial membrane of patients with rheumatoid arthritis. Proc Natl Acad Sci U S A 1996; 93: 221–5
- Roosnek E., Lanzavecchia A. Efficient and selective presentation of antigen‐antibody complexes by rheumatoid factor B cells. J Exp Med 1991; 173: 487–9
- Chan O. T., Hannum L. G., Haberman A. M., Madaio M. P., Shlomchik M. J. A novel mouse with B cells but lacking serum antibody reveals an antibody‐independent role for B cells in murine lupus. J Exp Med 1999; 189: 1639–48
- Lindhout E., van Eijk M., van Pel M., Lindeman J., Dinant H. J., de Groot C. Fibroblast‐like synoviocytes from rheumatoid arthritis patients have intrinsic properties of follicular dendritic cells. J Immunol 1999; 162: 5949–56
- Clark E. A., Ledbetter J. A. How does B cell depletion therapy work, and how can it be improved?. Ann Rheum Dis 2005; 64(Suppl 4)77–80
- Shan D., Ledbetter J. A., Press O. W. Signaling events involved in anti‐CD20‐induced apoptosis of malignant human B cells. Cancer Immunol Immunother 2000; 48: 673–83
- Edwards J. C., Szczepanski L., Szechinski J., Filipowicz‐Sosnowska A., Emery P., Close D. R., et al. Efficacy of B‐cell‐targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med 2004; 350: 2572–81
- Chan H. T., Hughes D., French R. R., Tutt A. L., Walshe C. A., Teeling J. L., et al. CD20‐induced lymphoma cell death is independent of both caspases and its redistribution into triton X‐100 insoluble membrane rafts. Cancer Res 2003; 63: 5480–9
- Di Gaetano N., Cittera E., Nota R., Vecchi A., Grieco V., Scanziani E., et al. Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol 2003; 171: 1581–7
- Bannerji R., Kitada S., Flinn I. W., Pearson M., Young D., Reed J. C., et al. Apoptotic‐regulatory and complement‐protecting protein expression in chronic lymphocytic leukemia: relationship to in vivo rituximab resistance. J Clin Oncol 2003; 21: 1466–71
- van der Kolk L. E., Grillo‐Lopez A. J., Baars J. W., Hack C. E., van Oers M. H. Complement activation plays a key role in the side‐effects of rituximab treatment. Br J Haematol 2001; 115: 807–11
- Giles J. T., Bathon J. M. Serious infections associated with anticytokine therapies in the rheumatic diseases. J Intensive Care Med 2004; 19: 320–34
- Breedveld F. New tumor necrosis factor‐alpha biologic therapies for rheumatoid arthritis. Eur Cytokine Netw 1998; 9: 233–8
- Strangfeld A., Listing J. Infection and musculoskeletal conditions: Bacterial and opportunistic infections during anti‐TNF therapy. Best Pract Res Clin Rheumatol 2006; 20: 1181–95
- Desai S. B., Furst D. E. Problems encountered during anti‐tumour necrosis factor therapy. Best Pract Res Clin Rheumatol 2006; 20: 757–90
- Quigg R. J. Use of complement inhibitors in tissue injury. Trends Mol Med 2002; 8: 430–6
- Krych‐Goldberg M., Atkinson J. P. Structure‐function relationships of complement receptor type 1. Immunol Rev 2001; 180: 112–22
- Yoon S. H., Fearon D. T. Characterization of a soluble form of the C3b/C4b receptor (CR1) in human plasma. J Immunol 1985; 134: 3332–8
- Linton S. M., Williams A. S., Dodd I., Smith R., Williams B. D., Morgan B. P. Therapeutic efficacy of a novel membrane‐targeted complement regulator in antigen‐induced arthritis in the rat. Arthritis Rheum 2000; 43: 2590–7
- Rioux P. TP‐10 (AVANT Immunotherapeutics). Curr Opin Investig Drugs 2001; 2: 364–71
- Zimmerman J. L., Dellinger R. P., Straube R. C., Levin J. L. Phase I trial of the recombinant soluble complement receptor 1 in acute lung injury and acute respiratory distress syndrome. Crit Care Med 2000; 28: 3149–54
- Schmid R. A., Hillinger S., Hamacher J., Stammberger U. TP20 is superior to TP10 in reducing ischemia/reperfusion injury in rat lung grafts. Transplant Proc 2001; 33: 948–9
- Thomas T. C., Rollins S. A., Rother R. P., Giannoni M. A., Hartman S. L., Elliott E. A., et al. Inhibition of complement activity by humanized anti‐C5 antibody and single‐chain Fv. Mol Immunol 1996; 33: 1389–401
- Tesser J., Kivitz A., Fleischmann R., Mojcik C. F., Bombara M., Phoneix F. B. Safety and efficacy of the humanized anti‐C5 antibody h5G1.1 in patients with rheumatoid arthritis. Arthritis Rheum 2001; S214
- Burch F., Tesser J., Bell L., Kivitz A. Baseline C5b‐9 level correlates with CRP and ACR 20 response to the humanized anti‐C5 antibody hG51.1 in patients with rheumatoid arthritis. Arthritis Rheum 2001; S274
- Mizuno M. A review of current knowledge of the complement system and the therapeutic opportunities in inflammatory arthritis. Curr Med Chem 2006; 13: 1707–17
- Johansson A. C., Sundler M., Kjellen P., Johannesson M., Cook A., Lindqvist A. K., et al. Genetic control of collagen‐induced arthritis in a cross with NOD and C57BL/10 mice is dependent on gene regions encoding complement factor 5 and FcgammaRIIb and is not associated with loci controlling diabetes. Eur J Immunol 2001; 31: 1847–56
- Solomon S., Kolb C., Mohanty S., Jeisy‐Walder E., Preyer R., Schollhorn V., et al. Transmission of antibody‐induced arthritis is independent of complement component 4 (C4) and the complement receptors 1 and 2 (CD21/35). Eur J Immunol 2002; 32: 644–51
- Dreja H., Annenkov A., Chernajovsky Y. Soluble complement receptor 1 (CD35) delivered by retrovirally infected syngeneic cells or by naked DNA injection prevents the progression of collagen‐induced arthritis. Arthritis Rheum 2000; 43: 1698–709
- Koga T., Kakimoto K., Hirofuji T., Kotani S., Ohkuni H., Watanabe K., et al. Acute joint inflammation in mice after systemic injection of the cell wall, its peptidoglycan, and chemically defined peptidoglycan subunits from various bacteria. Infect Immun 1985; 50: 27–34
- van Lent P. L., van den Bersselaar L. A., van den Hoek A. E., van de Loo A. A., van den Berg W. B. Cationic immune complex arthritis in mice—a new model. Synergistic effect of complement and interleukin‐1. Am J Pathol 1992; 140: 1451–61
- Mizuno M., Nishikawa K., Morgan B. P., Matsuo S. Comparison of the suppressive effects of soluble CR1 and C5a receptor antagonist in acute arthritis induced in rats by blocking of CD59. Clin Exp Immunol 2000; 119: 368–75
- Goodfellow R. M., Williams A. S., Levin J. L., Williams B. D., Morgan B. P. Soluble complement receptor one (sCR1) inhibits the development and progression of rat collagen‐induced arthritis. Clin Exp Immunol 2000; 119: 210–6
- Goodfellow R. M., Williams A. S., Levin J. L., Williams B. D., Morgan B. P. Local therapy with soluble complement receptor 1 (sCR1) suppresses inflammation in rat mono‐articular arthritis. Clin Exp Immunol 1997; 110: 45–52
- Ino Y., Sato T., Koshiyama Y., Suzuki K., Oda M., Iwaki M. Effects of FUT‐175, a novel synthetic protease inhibitor, on the development of adjuvant arthritis in rats and some biological reactions dependent on complement activation. Gen Pharmacol 1987; 18: 513–6
- Ames R. S., Lee D., Foley J. J., Jurewicz A. J., Tornetta M. A., Bautsch W., et al. Identification of a selective nonpeptide antagonist of the anaphylatoxin C3a receptor that demonstrates antiinflammatory activity in animal models. J Immunol 2001; 166: 6341–8
- Woodruff T. M., Strachan A. J., Dryburgh N., Shiels I. A., Reid R. C., Fairlie D. P., et al. Antiarthritic activity of an orally active C5a receptor antagonist against antigen‐induced monarticular arthritis in the rat. Arthritis Rheum 2002; 46: 2476–85
- Harris C. L., Williams A. S., Linton S. M., Morgan B. P. Coupling complement regulators to immunoglobulin domains generates effective anti‐complement reagents with extended half‐life in vivo. Clin Exp Immunol 2002; 129: 198–207
- Fraser D. A., Harris C. L., Williams A. S., Mizuno M., Gallagher S., Smith R. A., et al. Generation of a recombinant, membrane‐targeted form of the complement regulator CD59: activity in vitro and in vivo. J Biol Chem 2003; 278: 48921–7