
Abstract
We present molecular dynamics simulations of monolayer graphene under uniaxial tensile loading. The Morse, bending angle, torsion and Lennard-Jones potential functions are adopted within the mdFOAM library in the OpenFOAM software, to describe the molecular interactions in graphene. A well-validated graphene model using these set of potentials is not yet available. In this work, we investigate the accuracy of the mechanical properties of graphene when derived using these simpler potentials, compared to the more commonly used complex potentials such as the Tersoff-Brenner and AIREBO potentials. The computational speed up of our approach, which scales O(1.5N), where N is the number of carbon atoms, enabled us to vary a larger number of system parameters, including graphene sheet orientation, size, temperature and concentration of nanopores. The resultant effect on the elastic modulus, fracture stress and fracture strain is investigated. Our simulations show that graphene is anisotropic, and its mechanical properties are dependant on the sheet size. An increase in system temperature results in a significant reduction in the fracture stress and strain. Simulations of nanoporous graphene were created by distributing vacancy defects, both randomly and uniformly, across the lattice. We find that the fracture stress decreases substantially with increasing defect density. The elastic modulus was found to be constant up to around 5% vacancy defects, and decreases for higher defect densities.
1. Introduction
Graphene is a monolayer of sp2 hybridised carbon atoms arranged in a hexagonal crystal lattice and it is known to possess excellent mechanical,[Citation1] electrical[Citation2] and chemical properties.[Citation3] Graphene has been the focus of substantial research in the past decade with promise that the material can be used for a broad range of emerging technologies and future applications, including, for example, nanoporous graphene membranes for efficient reverse osmosis desalination [Citation4] and atmospheric filtering [Citation5] for the removal of pollutants or the harvesting of atmospheric gases, graphene-based nanocomposites for bulletproof vests,[Citation6] and graphene-based electrodes for high performance lithium-ion batteries.[Citation7] To be able to introduce graphene to novel applications, its properties and behaviour under various conditions need to be understood and accurately predicted. However, fabricated graphene sheets typically tend to contain a range of defects including nanopores.[Citation8,Citation9] The effect that such defects have on the mechanical properties of graphene need a more thorough investigation.
The mechanical properties of graphene have been investigated using both experimental and computational methods, the latter being by far the most common. Through experimentation, Blakslee et al. [Citation10] report a Young’s modulus of for bulk graphite. More recently, Lee et al. [Citation1] used atomic force microscopy to measure the elastic modulus of graphene and the stress and strain at which pristine graphene fractures. The authors concluded that graphene has an elastic modulus of
TPa and a breaking stress in the zigzag (ZZ) direction of
GPa with a maximum strain of 0.25. A similar result with an elastic modulus of roughly
TPa was obtained by Bunch et al. [Citation11].
Computational and theoretical methods have also been used to calculate and predict the mechanical properties of graphene sheets. Among the most common techniques used is molecular dynamics (MD); the AIREBO and Tersoff-Brenner potentials being reportedly the most accurate and hence widely adopted. With the AIREBO potential, Zhao et al. [Citation12] measured the elastic modulus of graphene to be TPa, the fracture stress 90 and 107 GPa, and the fracture strain 0.13 and 0.20 in the AC and ZZ direction, respectively. The relationships between the elastic and fracture properties with sheet size and temperature were also investigated in some literature sources [Citation12–Citation14] using these potentials, but some disagreement is found as to how the mechanical properties change with the length or aspect ratio of a graphene nanoribbon. For example, Wong et al. [Citation13] report a decrease in fracture stress and an increase in fracture strain with an increase in aspect ratio. Zhao et al. [Citation12] claim that the Young’s modulus increases with an increase in nanoribbon length, until it reaches the value for bulk graphene at approximately 10 nm length. On the contrary, Ni et al. [Citation14] find that the elastic modulus is not affected by the sheet size. More agreement is found on the effect high temperatures have on the fracture properties of graphene. It is generally accepted that the elastic modulus, fracture stress and fracture strain decrease, sometimes linearly, with an increase in system temperature.[Citation13,Citation15–Citation17] The introduction of Stone-Wales defects and single, double and larger vacancies was also studied resulting in a general reduction in fracture stress.[Citation18–Citation20] The fracture behaviour of pristine anddefected graphene was also analysed in [Citation14,Citation15,Citation19,Citation22] with the use of different computational and theoretical techniques. The fracture mode of graphene was generally found to be brittle, especially for pristine graphene. Moreover, the fracture path tends to be anisotropic and hence depends on the loading direction.
Although the mechanical properties of graphene have received a lot of attention by the research community, few have investigated the fracture behaviour and mechanical properties of graphene under various conditions while validating simpler, computationally cheaper potentials using MD. We use the Morse, bending angle, torsion and Lennard-Jones potential functions as described by Walther et al. [Citation23] in their work for hydrated fullerenes. In this paper, we implement these potentials for the first time in the OpenFOAM software within the mdFOAM library and validate this simple force field for pristine graphene under uniaxial loading, while studying the dependence of mechanical properties on sheet size, temperature and orientation. We compare our results with experiments and literature MD simulation results that use different potential functions whenever possible. Furthermore, MD simulations are used to study the fracture behaviour and mechanical properties of nanoporous graphene with increasing monovacancy defect concentration. The latter attempts to mimic graphene in the as-fabricated condition, and also porosity which may be intentionally introduced for applications which require non-pristine sheets such as filtration membranes.
The paper is structured as follows; Section 2 gives a detailed description of the molecular dynamics simulations and case setup, Section 3 describes the validation simulations for pristine graphene and presents results for nanoporous graphene, while Section 4 concludes the presented work.
2. Methodology
2.1. Molecular dynamics
The mechanical properties of graphene are studied using standard MD simulation, which solves Newton’s equations of motion for all atoms in the system:(1)
(2)
where ,
,
and
represent the mass, position, velocity and net force on the
atom.
The Velocity-Verlet algorithm was used to numerically integrate the equations of motion in space and time for all atoms in the sheet using a time step of 0.43 fs. All simulated graphene sheets were initialised with a zero velocity applied to all atoms, and then equilibrated for a duration of at least 20 ps before loading was applied. During equilibration, the FADE algorithm [Citation24] was applied to gradually introduce the intermolecular forces on the carbon atoms using a time-weighted function with time relaxation ps. This will prevent a sudden increase in energy of the system which might otherwise result in a sudden expansion of the graphene sheet and hence a possible premature sheet fracture. To prevent the sheet from drifting within the domain, the net velocity of the sheet was set to zero during equilibration using a simple velocity constraint.[Citation25] A Langevin thermostat [Citation26] was applied to control the temperature of the sheet during relaxation and loading.
2.2. Potential functions
The net force on all atoms during the MD simulation in Equation (Equation2(2) ) is determined via the spatial derivative of four potential functions.[Citation23] These potentials are illustrated in Figure S1 (see supplementary material) and consist of: a Morse stretching potential
used to model the covalent bonds, a harmonic potential
used to model the bending angle between bonds, a twofold torsion potential
used to model out-of-plane stiffness, and a Lennard-Jones intermolecular potential
used to model van der Waals and steric bonds. These are given, respectively, by:
(3)
(4)
(5)
(6)
where the values for the constants in these equations are listed in Table , and the variables ,
and
are shown in Figure S1. The total potential energy
of the system is given as a sum of all four potentials over all carbon atoms:
(7)
where the Morse, bending angle and torsion potentials are applied to atom groups that are covalently bonded, while the Lennard-Jones potential excludes 1–2 and 1–3 interactions [Citation23] as illustrated by the shaded region in Figure S1(d) for an arbitrary atom, and excludes pair atom computations with separation greater than the cut-off radius
nm. In the initialisation stage, the atomic bonds are established and saved in a bonds list. This list is accessed throughout the simulation to avoid unnecessary computational processing required to re-establish the bonds in every time step. During loading, the Morse, angle bending and torsion bonds are then permanently broken only when the Morse bond exceeds
nm.[Citation27] In such a case, the bonds list is repopulated. Given that this usually occurs towards the end of the simulation when the sheet fractures almost instantaneously, the bonds list remains constant throughout most of the simulation. As a result of the optimised bond selection, our computational timings are approximately
where
. On the other hand, both the AIREBO and Tersoff-Brenner potentials have a reactive bond-order term, with typically either 3- or 4-body potentials. These bond order terms are also adaptive, meaning they need to access atomic position information at every time-step (in contrast to the fixed bond lists used in this work), resulting in a major computational penalty. The cost of the AIREBO and Tersoff-Brenner potentials can however be reduced, although at the expense of numerical and programming complexity. For more information on the AIREBO and Tersoff-Brenner potentials see [Citation28] and [Citation29], respectively.
Table 1. Parameter constants for the potential functions.[Citation30]
2.3. Loading method and calculations
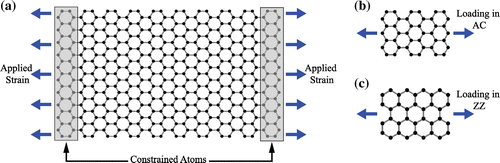
To simulate uniaxial tensile loading, hexagonal rings at opposite edges of the graphene sheet were constrained as shown in Figure . Constrained atoms were subjected to a quasistatic strain rate of in accordance with [Citation31]. We consider armchair (AC) and zigzag (ZZ) graphene sheets with an aspect ratio of approximately 1:0.3 (unless otherwise stated), which are loaded along the longitudinal axis, as indicated in Figure (b) and (c).
Figure 1. (Colour online) (a) A schematic illustration of a graphene sheet showing the edge atoms constrained (shaded in grey) for the application of the pre-defined strain rate. (b) and (c) illustrate the loading direction of an armchair (AC) sheet and zigzag (ZZ) sheet, respectively.
The applied strain to the sheet at time t was found using
, where
and
are the distances between two arbitrarily pre-selected atoms from the sheet (just outside the constrained region) before and during loading, respectively. The stress
and elastic modulus E were computed using:
(8)
(9)
where is the original volume of the sheet when assuming a thickness of 0.335 nm,[Citation1] and U is the strain energy of the sheet which is measured as the difference of the total system potential energy between a time t and the unloaded sheet. To solve Equations (Equation8
(8) ) and (Equation9
(9) ) we adopt a similar method described by Le and Batra [Citation31], which involves fitting a cubic function to strain energy U against strain
, and applying straightforward differentiation.
The mdFOAM molecular dynamics code [Citation32,Citation33] was used in this work, which is available in the OpenFOAM software.[Citation34] OpenFOAM is an open-source C++ toolbox typically used for computational fluid dynamics. The mdFOAM library was extended to include the new potential functions, constraints and measurement tools described above.
3. Results and discussion
In this section, the MD method is validated by measuring the mechanical properties and investigating the fracture behaviour of pristine graphene for different orientations, sheet size and temperatures and comparing them to literature sources when available. Graphene with ordered and random vacancy defects are then investigated.
3.1. Sheet size dependence
Eleven different sheet sizes ranging from diagonal lengths of 1.35–14.51 nm (42–4760 carbon atoms) were modelled at a temperature of 0 and 300 K. Each case was run using four statistically independent realisations and an average of the mechanical properties was taken. To obtain statistically independent samples, each realisation of the same case was run for a different duration at the equilibration stage before loading was applied.
The effect of sheet size on elastic modulus, fracture stress and fracture strain is shown in Figure (a) for 0 K. The results show that the mechanical properties assessed in this work decrease with an increase in sheet size from the smallest sheets considered up to a critical length of approximately 6–7 nm, beyond which the properties stabilise to that of bulk graphene. This behaviour is especially evident in ZZ sheets whereby an increase in diagonal length from 1.42 to 5.66 nm decreases the elastic modulus, fracture stress and fracture strain by 7.2, 16.5 and 15%, respectively. On the other hand, the elastic modulus, fracture stress and fracture strain for AC graphene decrease by only 2, 1.7 and 5.8%, respectively, for the same size range.
Figure 2. (Colour online) Results for elastic modulus, fracture stress and fracture strain for AC- and ZZ-oriented graphene with increasing sheet size for two different temperatures: (a) 0 K and (b) 300 K. In all graphs, our MD results are represented by solid circles for the AC direction and inverted triangles for the ZZ direction, while dashed lines represent bulk mechanical properties.
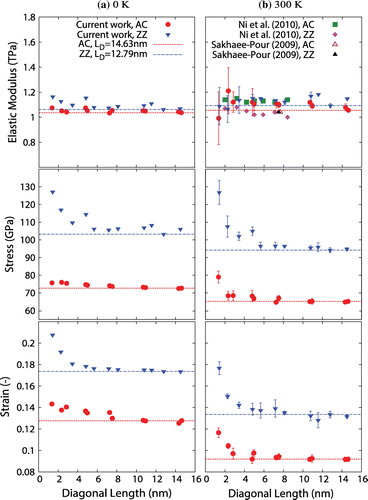
Figure 3. (Colour online) (a) Stress–strain curves of AC (red) and ZZ (blue) graphene sheets. All simulations were run at 300 K except the ones marked otherwise. We also compare in (a) our other simulations for sheets with a single vacancy (SV) defect, and nanoporous sheets with 7% defect density. In (b) and (c) we illustrate the resolved forces along selected covalent bonds during AC and ZZ loading, respectively.
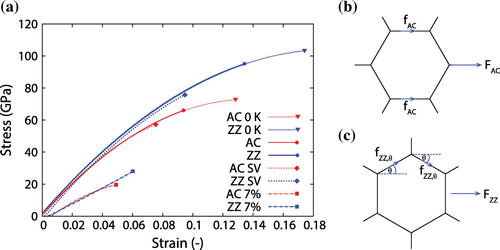
Figure 4. Fracture of: (a) a pristine AC graphene sheet with a diagonal length of 14.63 nm at 300 K; (b) a pristine ZZ graphene sheet with a diagonal length of 14.84 nm at 300 K; (c) an AC graphene sheet with a diagonal length of 14.63 nm at 300 K and a monovacancy defect at its centre; (d) a ZZ graphene sheet with a diagonal length of 14.84 nm at 300 K and a monovacancy defect at its centre; (e) a ZZ graphene sheet with a diagonal length of 14.84 nm at 300 K with 5.5% of the original atoms selected randomly and removed as vacancy defects; and (f) a ZZ graphene sheet with a diagonal length of 13.67 nm at 300 K with 5% of the original atoms being removed as vacancy defects and distributed uniformly.
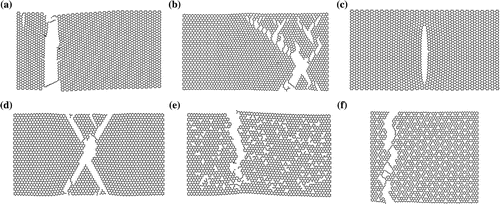
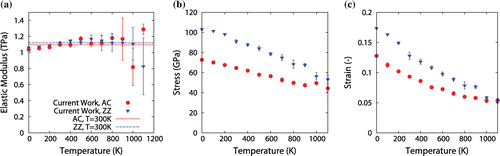
Figure 5. (Colour online) Results for (a) elastic modulus, (b) fracture stress and (c) fracture strain of AC and ZZ graphene sheets ( nm), varying with sheet temperature. In all graphs, the solid circles represent the AC direction while the inverted triangles represent the ZZ direction.
The results for sheets at 300 K are shown in Figure (b), where the critical length at which the fracture stress and fracture strain approach that of bulk graphene observed at this temperature lies between 8–10 nm; this is in good agreement with the critical length of 10 nm indicated by Zhao et al. [Citation12]. Furthermore, at 300 K a more pronounced behaviour is observed for the fracture stress and strain below this critical length. For ZZ graphene at 300 K, an increase in diagonal length from 1.42 to 5.66 nm results in a decrease of the fracture stress and fracture strain by 23.9 and 22.3%, respectively. On the other hand, the fracture stress and fracture strain for the AC graphene sheets decrease by 15.4 and 16.3%, respectively. This size dependency of fracture properties is in good agreement with Ni et al. [Citation14] (Tersoff-Brenner) and Sakhaee-Pour [Citation35] (finite-element modelling). For the smallest sheet when tested at a temperature of 300 K, we also observed a slight decrease in elastic modulus, which was not observed by Ni et al. [Citation14] and Sakhaee-Pour [Citation35], but was evident in the AIREBO MD simulations carried out by Zhao et al. [Citation12].
Following this study, we list in Table the bulk mechanical properties of graphene as measured from our MD simulations. The values for the elastic and fracture properties are in very good agreement with data found in [Citation1,Citation12,Citation35,Citation37]. Values for the elastic modulus agree very well with the experimental results of Lee et al. [Citation1] ( TPa) and the MD simulations using the AIREBO potential of Zhao et al. [Citation12] (
TPa). However, the fracture stress and fracture strain obtained from our simulations are within 30% of the experimental [Citation1] and AIREBO simulations [Citation12] results available in literature. The differences between our results and that of Zhao et al. [Citation12] may be attributed to the inability of our model to deal with changes in bond coordination which occurs just before and during fracture. In this respect, the adaptive bond-order term in the AIREBO potential performs better than our model, since it allows the bonds to change at fracture. A second possible contribution to the underestimation of fracture stress and strain values may be related to the fact that Zhao et al. [Citation12] consider an infinite sheet in the transverse direction of loading by applying periodic boundary conditions, and constrain the dynamics of atoms at a fixed pressure using the NPT ensemble. On the other hand, our simulations consider a finite graphene sheet, with dangling bonds at the transverse edge of the graphene sheet in the NVT ensemble.
Table 2. Measured mechanical properties of pristine bulk graphene.
Stress versus strain graphs for bulk graphene are also shown in Figure (a). At small strains (up to about 2%), graphene exhibits a linear behaviour, while non-linearity prevails for larger strains up to fracture, in agreement with the work in [Citation12]. While there has been some disagreement between literature sources as to which loading direction of the graphene sheet is stronger, in our simulations we find that the ZZ direction is stronger and slightly stiffer than a graphene sheet loaded from the AC direction. This anisotropic dependance agrees with [Citation12,Citation13,Citation15] which we attribute to the geometrical distribution of the covalent bonds in the hexagonal lattice, as illustrated in Figure (b) and (c). When graphene is loaded along the AC direction, roughly a third of the bonds in the sheet are perfectly aligned to the loading direction (Figure (b)), and thus the loading is directly transferred to these bonds. However, when the load is applied along the ZZ direction (Figure (c)), none of the covalent bonds are aligned to the loading direction and therefore the actual force sustained by each covalent bond is in fact a component of the force acting along the bond angle, which results in less force than those sustained by the AC sheets. As such, this allows the ZZ-loaded sheet to elongate further and hence withstand more stress and strain along the ZZ direction prior to fracture.
Based on these results, sheets with diagonal length of approximately 13 nm and aspect ratio of 1:0.3 were used in the following studies (unless otherwise stated), such that all simulations represent graphene sheets having bulk-mechanical characteristics.
3.2. Fracture behaviour of pristine graphene
We present the fracture process of pristine AC and ZZ graphene sheets at 300 K from our MD simulations in Figure (a) and (b), respectively. The fracture behaviour is mostly dependent on the alignment and position of the covalent bonds with respect to the loading direction. As such, the path of travel occurs perpendicular to aligned covalent bonds. For the AC-loaded sheet, a high percentage of covalent bonds are aligned with the loading direction and so the crack propagates perpendicular to loading. For the ZZ-loaded sheet, the covalent bonds are not oriented in the same direction as loading, and so crack propagation favours a diagonal travelling path () normal to the alignment of the bonds.
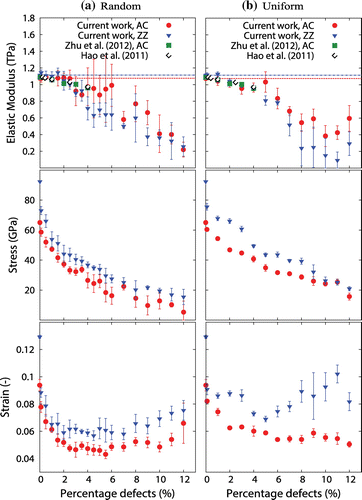
Figure 6. (Colour online) Graphs showing elastic modulus, fracture stress and fracture strain for AC- and ZZ-loaded graphene sheets at 300 K with increasing defect density distributed (a) randomly, and (b) uniformly. In all graphs, our MD results are represented by solid circles for the AC direction and inverted triangles for the ZZ direction, while dashed lines represent bulk mechanical properties.
While pristine graphene supports high fracture strains, its mode of fracture under loading is brittle since cracks rapidly propagate when they form resulting in catastrophic failure of the sheet. Similar fracture patterns to those presented in Figure (a) and (b) have been also observed with the use of the AIREBO [Citation15,Citation20,Citation22,Citation38] and Tersoff-Brenner [Citation21,Citation39] potentials. AIREBO and Tersoff-Brenner potentials are known to model the fracture behaviour accurately, thus the simulation results obtained in this study, albeit using a simpler set of potential functions, are in very good agreement with more established potentials.
3.3. Temperature dependence
Graphene is expected to be used in several different applications for which the environmental conditions vary considerably. In this study, we investigate the effect on the mechanical properties of pristine graphene at different temperatures, varying from 0 to 1100 K. For each case, five statistically independent simulations were carried out and an average was taken.
The variation of several mechanical properties with temperature is shown in Figure (a)–(c). It is evident from these graphs that the fracture stress and strain decrease substantially with an increase in temperature for both ZZ and AC loading configurations. For ZZ-loaded graphene at 1100 K, the fracture stress decreases to 53.3 GPa (48% decrease from 0 K), while the fracture strain decreases to 0.053 (70% decrease from 0 K), while for AC graphene fracture stress and strain reduce by 39 and 58%, respectively, from 0 K. The same dependance of stress and strain on temperature has been observed using the AIREBO potential in [Citation16,Citation17,Citation22,Citation37,Citation40,Citation41]. For example, Zhang et al. [Citation41] obtained a 50% decrease in intrinsic strength when the sheet temperature is increased from 0 to 1200 K.
The thermal fluctuations of atoms are the principal reason why graphene’s fracture stress and strain are reduced at higher temperatures. Carbon atoms possess more kinetic energy at elevated temperatures, causing significantly high vibrations and, as a result, a large out-of-plane motion (in the z-direction) in equilibrium, resulting in overall sheet rippling (see Figure S4). The distance covered by carbon atoms in the z-direction at equilibrium is shown in Figure S4 (a) as a function of temperature; assuming a sheet thickness of 0.335 nm at 0 K, the out-of-plane distance covered by molecules is 3 times larger for the 1100 K than the 0 K sheet due to thermal motion. The perturbations in the sheet are gently ironed out during loading, but since thermal fluctuations still remain, covalent bonds are more susceptible to break prematurely, producing cracks at lower strain and stress.
Conversely, the elastic modulus is largely unchanged within a large temperature range. The elastic modulus increases from 0 to 500 K by 6 and 13 % for ZZ and AC graphene, respectively, and remains relatively constant until K. At higher temperatures (
900 K), a decrease in the elastic modulus to
TPa is observed, similar to that obtained in [Citation40], but is still exceptionally high when compared to other conventional materials. The larger variation in results obtained at higher temperatures – evidenced by wider error bars – are expected due to the large thermal fluctuations.
3.4. Random and uniformly distributed vacancy defects
It is evident that the presence of atomic defects may affect the properties of graphene quite considerably. Although generally not desired, defects can originate from a fabrication process, but can also be intentionally introduced, for example, in the production of nanoporous graphene membranes for desalination [Citation32] or filtration applications. In these applications, nanoporous graphene would need to contain a high level of porosity for there to be superior permeability over conventional membranes. The porosity might, however, affect the graphene’s ability to withstand the high hydraulic pressures; MPa for typical reverse osmosis membranes. Our analysis of nanoporous graphene as a desalination/filtration application is left for future work. In this work, we simulate nanoporous graphene using MD simulations with randomly or uniformly distributed defects (single atoms removed from the sheet) varying in density from 0.1% up to 12% and measure its mechanical properties and fracture behaviour. For the following simulations, the graphene sheets were modelled with an aspect ratio that approximates a square with a diagonal length of 13.7 nm.
Figure (a) shows the elastic modulus, fracture stress and fracture strain for randomly distributed vacancy defects, while Figure (b) shows the same mechanical properties for uniformly distributed defects. In both cases, there is a considerable decrease in the calculated properties with increasing defect density. The introduction of a single vacancy defect causes a significant drop in the fracture stress and fracture strain of around 15 and 23%, respectively. This reduction can also be seen in Figure (a) and has also been reported in [Citation19,Citation42,Citation43]. We found that the elastic modulus remains relatively unaffected up to around 3–5% defects for random and uniform distributed defects, similar to the data found in [Citation44,Citation45]. The elastic modulus then decreases roughly linearly with increasing defect density. This relationship was similarly found recently in [Citation46] for nanoporous graphene with larger pore sizes. At the largest porosity we considered in this work (12%), the elastic modulus drops by around 80%. The stress–strain curve for a sheet with 7% defect density is shown in Figure (a) which is compared with that of a pristine sheet.
An important outcome from these simulations, especially for use in filtration membranes is that a sheet with uniformly distributed defects has a fracture stress of up to 197% higher than that for randomly distributed defects in the case of AC graphene sheets with 12% defects. This is caused because the latter contains a wider range of defect sizes, having pores which are larger and more susceptible to fracture as noted in [Citation46].
Another interesting outcome of these simulations is the behaviour of the fracture strain with increasing defect density above 3%. Unlike the fracture stress and elastic modulus, which continue to decrease with increasing vacancies, the fracture strain stabilises at around 3% defect density and starts to increase again above 6%. We identify this as a stage of improved ductility in graphene (above 6% defect density), and it is caused by large presence of vacancy defects that allow the sheet to stretch more under loading, and as such enabling larger fracture strains to be sustained. A similar behaviour was also observed by Xu et al. [Citation19]. For the uniform distributed vacancy defects, however, the AC-loaded sheet seems to reach a saturation phase without signs of strain improvement.
Similar to the effect of higher thermal motion on sheet rippling in Section 3.3, we also observe in this study that the introduction of defects promotes a similar rippling behaviour, as shown in Figure S5(c)–(e) for a pristine sheet at 0 K and two sheets with 12% random and uniform defects. Figure S5(a) and (b) shows the sheet thickness in the z-direction for both random and uniform defect density for a sheet at a temperature of 300 K. Sheets with large defect densities have an effective sheet thickness of up to 18 times larger than the pristine sheet, which is similarly an indication of a decrease in fracture strain.
The fracture processes of nanoporous graphene was also analysed in this work. Figure (c) and (d) shows the fracture of AC and ZZ graphene sheets containing a monovacancy at the sheet’s centre. Their fracture behaviour is very similar to that of pristine graphene, with the only difference being that the crack nucleates at the vacancy and propagates in two opposing directions. This is in good agreement with the work in [Citation19,Citation20,Citation22]. In Figure (e), we show the fracture behaviour of a 12% randomly distributed vacancies for a ZZ graphene sheet during fracture and in a ZZ graphene sheet with uniformly distributed defects in Figure (f). Unlike the equivalent ZZ-loaded pristine graphene simulation, where the crack propagates diagonally through covalent bonds, in this case the travel path is modified by the presence of defects, as such forming an almost perpendicular crack to the loading direction. This behaviour is similar to the fracture behaviour of an AC-loaded pristine sheet. We found that this transition from a ZZ- to an AC-type fracture mode in nanoporous ZZ graphene sheets occurs at a defect density of around 4%, which is roughly equal to when the elastic modulus starts to decrease with increasing defect density. For AC graphene sheets, the crack was observed to propagate perpendicular to the loading direction, similar to what happens in pristine AC sheets, for all the defect densities investigated in this work.
4. Conclusion
In this work, we perform molecular dynamic simulations of monolayer graphene under uniaxial tensile loading using the Morse, bending angle, torsion and Lennard-Jones potentials to describe the inter- and intra-molecular carbon interactions. Our implementation of the model in OpenFOAM was found to scale with , where N is the number of carbon atoms and
. These simulations enabled the investigation of the elastic modulus, fracture stress and fracture strain of graphene with respect to the orientation, sheet size and sheet temperature. Graphene exhibits anisotropy, with the ZZ loading direction being able to withstand higher loads and strain than the AC direction, in agreement with the AIREBO force field.[Citation12,Citation15] Furthermore, size dependency is observed for graphene sheets smaller than 6–10 nm in diagonal length; the mechanical properties of larger graphene sheets rapidly approach those of bulk graphene above this size. A similar critical length had been reported in [Citation12]. We also find that higher temperatures tend to significantly decrease the fracture stress and strain almost linearly with temperature, in good agreement with [Citation41]. The elastic modulus is however only affected at system temperatures higher than approximately 900 K. The introduction of randomly and uniformly distributed vacancy defects enabled the simulation of nanoporous graphene sheets, for which it was concluded that the fracture stress decreases substantially with increasing defect density. Nanoporous graphene sheets with uniformly distributed defects are able to withstand higher loads when compared to their counterparts with random defects. Although the model implemented tends to underestimate the quantitative results of the fracture strain and stress, the qualitative fracture behaviour of pristine AC and ZZ graphene sheets was still found to be in good agreement with those obtained by the AIREBO [Citation15,Citation20,Citation22,Citation38] and Tersoff-Brenner [Citation21,Citation39] potentials. Furthermore, the fracture behaviour of nanoporous graphene sheets exhibited a ZZ- to AC-type fracture transition occurring at roughly 4% defect density. For future work, we consider investigating how this model can be improved to deal with rippling,[Citation47] and to investigate nanoporous graphene with larger pore sizes and shapes.[Citation43,Citation46]
Acknowledgements
The authors wish to thank David Willock (Cardiff University) for useful discussions.
Additional information
Funding
Notes
No potential conflict of interest was reported by the authors.
References
- Lee C, Wei X, Kysar JW, et al. Measurement of the elastic properties and intrinsic strength of monolayer graphene. Science. 2008;321:385–388.
- Novoselov KS, Geim AK, Morozov SV, et al. Electric field effect in atomically thin carbon films. Science. 2004;306:666–669.
- Boukhvalov DW, Katsnelson MI. Chemical functionalization of graphene with defects. Nano Lett. 2008;8:4373–4379.
- Cohen-Tanugi D, Grossman JC. Water desalination across nanoporous graphene. Nano Lett. 2012;12:3602–3608.
- Jiang D, Cooper VR, Dai S. Porous graphene as the ultimate membrane for gas separation. Nano Lett. 2009;9:4019–4024.
- Lee JH, Loya PE, Lou J, et al. Dynamic mechanical behavior of multilayer graphene via supersonic projectile penetration. Science. 2014;346:1092–1096.
- Chen R, Zhao T, Wu W, et al. Free-standing hierarchically sandwich-type tungsten disulfide nanotubes/graphene anode for lithium-ion batteries. Nano Lett. 2014;14:5899-5904.
- Stankovich S, Dikin DA, Piner RD, et al. Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide. Carbon. 2007;45:1558–1565.
- Gomez-Navarro C, Weitz RT, Bittner AM, et al. Electronic transport properties of individual chemically reduced graphene oxide sheets. Nano Lett. 2007;7:3499–3503.
- Blakslee OL, Proctor DG, Seldin EJ, et al. Elastic constants of compression annealed pyrolytic graphite. J. Appl. Phys. 1970;41:3373–3382.
- Bunch JS, Verbridge SS, Alden JS, et al. Impermeable atomic membranes from graphene sheets. Nano Lett. 2008;8:2458–2462.
- Zhao H, Min K, Aluru NR. Size and chirality dependent elastic properties of graphene nanoribbons under uniaxial tension. Nano Lett. 2009;9:3012–3015.
- Wong C, Vijayaraghavan V. Nanomechanics of free form and water submerged single layer graphene sheet under axial tension by using molecular dynamics simulation. Mater. Sci. Eng.: A. 2012;556:420–428.
- Ni Z, Bu H, Zou M, et al. Anisotropic mechanical properties of graphene sheets from molecular dynamics. Physica B. 2010;405:1301–1306.
- Jhon YI, Jhon YM, Yeom GY, et al. Orientation dependence of the fracture behavior of graphene. Carbon. 2014;66:619–628.
- Dewapriya M, Phani AS, Rajapakse R. Influence of temperature and free edges on the mechanical properties of graphene. Modell. Simul. Mater. Sci. Eng. 2013;21:065017.
- Zhao H, Aluru N. Temperature and strain-rate dependent fracture strength of graphene. J. Appl. Phys. 2010;108:064321.
- Sun Y, Ma F, Ma D, et al. Stress-induced annihilation of stone-wales defects in graphene nanoribbons. J. Phys. D: Appl. Phys. 2012;45:305303–305307.
- Xu L, Wei N, Zheng Y. Mechanical properties of highly defective graphene: from brittle rupture to ductile fracture. Nanotechnology. 2013;24:505703–505709.
- Ito A, Okamoto S. Molecular dynamics analysis on effects of vacancies upon mechanical properties of graphene and graphite. Eng. Lett. 2012;20:271–278.
- Ansari R, Motevalli B, Montazeri A, et al. Fracture analysis of monolayer graphene sheets with double vacancy defects via md simulation. Solid State Commun. 2011;151:1141–1146.
- Dewapriya M, Rajapakse R, Phani A. Atomistic and continuum modelling of temperature-dependent fracture of graphene. Int. J. Fract. 2014;187:199–212.
- Walther JH, Jaffe RL, Halicioglu T, et al. Carbon nanotubes in water: structural characteristics and energetics. J. Phys. Chem. B. 2001;105:9980–9987.
- Borg MK, Lockerby DA, Reese JM. The FADE mass-stat: A technique for inserting or deleting particles in molecular dynamics simulations. J. Chem. Phys. 2014;140:074110.
- Borg MK, Macpherson GB, Reese JM. Controllers for imposing continuum-to-molecular boundary conditions in arbitrary fluid flow geometries. Mol. Simul. 2010;36:745–757.
- Izaguirre JA. Langevin stabilisation of multiscale mollified molecular dynamics. In: Brandt A, Bernholc J, Binder K,editors. Multiscale computational methods in chemistry and physics. Vol. 117, NATO science series: series III. Amsterdam: IOS Press; 2001. p. 34–47.
- Jin Y, Yuan F. Atomistic simulations of J-integral in 2D graphene nanosystems. J. Nanosci. Nanotechnol. 2005;5:2099–2107.
- Stuart SJ, Tutein AB, Harrison JA. A reactive potential for hydrocarbons with intermolecular interactions. J. Chem. Phys. 2000;112:6472–6486.
- Griebel M, Knapek S, Zumbusch G. Numerical simulation in molecular dynamics: numerics, algorithms, parallelization, applications. Vol. 5, Springer science and business media. 2007.
- Walther JH, Jaffe RL, Kotsalis EM, et al. Hydrophobic hydration of c60 and carbon nanotubes in water. Carbon. 2004;42:1185–1194.
- Le MQ, Batra RC. Single-edge crack growth in graphene sheets under tension. Comput. Mater. Sci. 2013;69:381–388.
- Nicholls W, Borg MK, Lockerby DA, et al. Water transport through carbon nanotubes with defects. Mol. Simul. 2012;38:781–785.
- Ritos K, Dongari N, Borg MK, et al. Dynamics of nanoscale droplets on moving surfaces. Langmuir. 2013;29:6936–6943.
- The OpenFOAM Foundation Ltd. Available from: http://www.openfoam.org. 2016.
- Sakhaee-Pour A. Elastic properties of single-layered graphene sheet. Solid State Commun. 2009;149:91–95.
- Shokrieh MM, Rafiee R. Prediction of young’s modulus of graphene sheets and carbon nanotubes using nanoscale continuum mechanics approach. Mater. Des. 2010;31:790–795.
- Wang MC, Yan C, Ma L, et al. Effect of defects on fracture strength of graphene sheets. Comput. Mater. Sci. 2012;54:236–239.
- Lu Q, Gao W, Huang R. Atomistic simulation and continuum modeling of graphene nanoribbons under uniaxial tension. Modell. Simul. Mater. Sci. Eng. 2011;19:054006.
- Terdalkar SS, Huang S, Yuan H, et al. Nanoscale fracture in graphene. Chem. Phys. Lett. 2010;494:218–222.
- Zhang YY, Gu Y. Mechanical properties of graphene: effects of layer number, temperature and isotope. Comput. Mater. Sci. 2013;71:197–200.
- Zhang H, Duan Z, Zhang X, et al. Strength and fracture behavior of graphene grain boundaries: effects of temperature, inflection, and symmetry from molecular dynamics. Phys. Chem. Chem. Phys. 2013;15:11794–11799.
- Dewapriya MAN, Rajapakse RKND, Phani A. Molecular dynamics simulation of fracture of graphene. In: 13th international conference on fracture; 2013; Beijing
- Liu Y, Chen X. Mechanical properties of nanoporous graphene membrane. J. Appl. Phys. 2014;115:034303.
- Zhu J, He M, Qiu F. Effect of vacancy defects on the young’s modulus and fracture strength of graphene: A molecular dynamics study. Chin. J. Chem. 2012;30:1399–1404.
- Hao F, Fang D, Xu Z. Mechanical and thermal transport properties of graphene with defects. Appl. Phys. Lett. 2011;99:041901.
- Cohen-Tanugi D, Grossman JC. Mechanical strength of nanoporous graphene as a desalination membrane. Nano Lett. 2014;14:6171–6178.
- Shenoy VB, Reddy CD, Ramasubramaniam A, et al. Edge-stress-induced warping of graphene sheets and nanoribbons. Phys. Rev. Lett. 2008;101:245501–245504.