
Abstract
We present the results of an aggregation study on the intrinsically disordered biomineralisation peptide n16N, which selects the aragonite polymorph of calcium carbonate and is expected to have aggregation-dependent structure and function. The peptide is a sub-sequence of the in vivo protein n16, with putative framework and polymorph selection roles in the nacre layer of pearl oyster (Pinctada fucata). Employing the intermediate-resolution coarse-grained protein model PLUM*, which has previously been validated with respect to n16N, we simulate assemblies of these peptide units for system sizes inaccessible to atomistic models. We use extensive conformational sampling to show that the configurational ensemble explored by n16N aggregates contains a significant proportion of ordered -structure, within which arrangement of monomers is consistent with a previous hypothesis on functionally distinct subdomains of n16N. We also study an n16N mutant which fails to aggregate in experimental studies and obtain very similar behaviour, the consequences of which are discussed.
1. Introduction
Biomineralisation refers to the formation of composite bio-inorganic materials, known as biominerals, in controlled environments by certain living organisms. Such organisms possess an ability to form hierarchically nanostructured hard tissues via biomineralisation processes which are not well understood, but have a range of potential applications [Citation1–Citation4]. In a typical scenario, biomineralisation is biologically controlled in an extracellular environment [Citation5,Citation6] by an organic matrix [Citation7–Citation9], which becomes the organic component of the final structure. The matrix is typically formed of polysaccharides, glycoproteins and proteins [Citation10] which are often members of the protein fold class known as intrinsically disordered proteins (IDPs).
IDPs are defined by their lack of a single stable tertiary structure [Citation11,Citation12], and often have a function for which their disorder is essential [Citation13]. It has been suggested that biomineralisation proteins are the most disordered functional protein class [Citation14], perhaps owing to required adaptability for multi-stage matrix assembly [Citation13], interaction with ions [Citation15] and selectivity of chemical environment in which assembly occurs [Citation16].
n16 is a biomineralisation framework protein, present in the nacre layer of the shell of pearl oyster (Pinctada fucata) [Citation17–Citation20], where it is thought to stabilise the aragonite polymorph of calcium carbonate [Citation21–Citation24]. The 30AA (amino acid chain length) N-terminal region named n16N has been used as an n16 mimic in experimental biomineralisation studies [Citation25–Citation33]. Highlights include use of n16N with a Kevlar substrate producing lamellar aragonite [Citation30], and n16N selecting aragonite when bound to -chitin [Citation29], a macromolecule with a substantial presence in the putative organic biomineralisation matrix of nacre [Citation34]. n16N has been deemed ‘the key self-assembly/aragonite forming domain’ [Citation33], and is the focus of the current work. Dynamic light scattering has confirmed that n16N oligomerises into
m-scale supramolecular assemblies [Citation32]; evidence from circular dichroism spectra and NMR show that these oligomers are composed of 54% random coil structure and 46%
-structure [Citation22,Citation32,Citation35]. Attempts have been made to converge upon a functional subdomain classification scheme for n16N, in the context of the n16N/
-chitin/calcium carbonate system. Inputs to this include bioinformatic analysis [Citation24], with the disorder-predicting algorithm GLOBPLOT2.3 [Citation36], NMR evidence [Citation22] and atomistic simulation of the monomer [Citation15]. Figure lays out the current state of these efforts, as described by Brown et al. [Citation15], on an annotated n16N chain sequence.
Figure 1. (Colour online) n16N chain sequence, labelled by putative character and function of hypothesised functional subdomain regions [Citation15]. Cationic and anionic residues are coloured in blue and red, respectively. An ellipsis indicates where the n16 parent protein’s chain continues.
![Figure 1. (Colour online) n16N chain sequence, labelled by putative character and function of hypothesised functional subdomain regions [Citation15]. Cationic and anionic residues are coloured in blue and red, respectively. An ellipsis indicates where the n16 parent protein’s chain continues.](/cms/asset/b1ec3184-1094-49fc-b473-3d33dd177909/gmos_a_1405158_f0001_oc.gif)
A fully atomistic treatment of multiple n16N peptides, their aggregation and interaction with mineral ions, remains far beyond the reach of contemporary computational capability. This is particularly true in terms of time scale and hence the ability to sample the conformation space available to large aggregates of n16N. We are therefore motivated to study the structure of n16N aggregates using lower resolution, coarse-grained models. These can provide insight into the gross structural motifs adopted by aggregates of multiple n16N units, and hence provide structural input to detailed atomistic simulations of peptide–mineral interactions at the extremities.
In previous work [Citation37], we explored use of the coarse-grained protein model PLUM created by Bereau and Deserno [Citation38]. PLUM is a four-bead-per-residue, implicit-solvent protein model. It includes steric backbone–backbone repulsion via Weeks–Chanlder–Anderson potentials, dipole–dipole interactions between peptide bonds, and an explicit backbone–backbone hydrogen bonding potential. Interactions between side chains are captured via further pair potentials parameterised from an analysis of square-well interaction strengths needed to reproduce structures of crystallised proteins. This analysis, which includes all possible pairs of residues, implicitly captures electrostatic interactions between charged side chains. However, the model contains no explicit electrostatic terms and is hence unsuitable for simulating the interaction of peptides with mineral ions. All other parameters, including bond and angle potentials, were fitted by Bereau and Deserno to reproduce the conformational ensemble for a number of sample protein sequences.
We demonstrated that the standard PLUM parameter set required only minor modification to produce ensembles of n16N peptide structures in agreement with structural predictions and atomistic simulations [Citation15] in the CHARMM22* model [Citation39,Citation40] with TIPS3P water [Citation41]. Specifically, a reduction in the strength of backbone–backbone hydrogen bonding produced encouraging preliminary results for both monomer and dimer, including distinguishing hypothesised subdomain regions by relative level of disorder, secondary structure manifestation, and degree and manner of involvement in dimerisation. We refer to the adjusted model as PLUM*.
This background offers the tantalising suggestion that the structure of large n16N aggregates, from which its function may derive, are accessible via simulations using PLUM-style models. The primary goal of this manuscript is therefore to build upon our one-chain and two-chain system findings by presenting results from simulations of three-, six- and eight-chain systems of n16N. These system sizes are beyond those accessible to atomistic simulations.
As a secondary objective, we also simulate a mutant of n16N called n16NN, in which the acidic residues are replaced by charge-neutral counterparts (Asp Asn, Glu
Gln, that is D
N and E
Q in Figure ). n16NN aggregates differently [Citation27,Citation30] with circular dichroism spectra suggesting a greater preference for random coil structure compared to the original peptide. It also fails to form complexes with Ca
[Citation27], likely because Asp and Glu have an active role in organic–mineral association [Citation28]. This hampers the aragonite selectivity of the peptide [Citation29,Citation31]. Our previous work studied the n16NN peptide in single- and two-chain systems, with the goals of gauging PLUM*’s sensitivity to the minor distinctions between the two chains, and forming a hypothesis about n16NN’s failure to aggregate normally. In our previous work we observed lower conformational accessibility and greater stability of aggregation-facilitating hydrogen bonds throughout the chain, including in the aggregation-driving SD2 region (see Figure ), which is identical in both systems. Here, we investigate if these distinctions persist to larger aggregates.
2. Simulation methodology
Each system simulated is referred to by the peptide name and number of chains in the system, e.g. n16N-3 for the trimer n16N system. Our aim in the present study is to establish the structure of oligomers, and the role of the three subdomains within these. Simulations of each system are initialised with the peptides in close proximity to encourage formation of oligomers. We do not sample configurations in which any of the peptides unbind. All statistics extracted from the ensemble generated should be considered conditional on oligomers of the size simulated having been already formed via aggregation. The simulation package LAMMPS [Citation42] was used with a timestep of 3 fs and a Langevin thermostat with a damping parameter of 1000 fs.
As in our previous work, we employed the replica exchange molecular dynamics (REMD) technique [Citation43,Citation44], varying replicas by thermostatted temperature and keeping the Hamiltonian of each replica the same. The replicas at the highest temperatures achieved enhanced sampling through their ability to overcome barriers on the potential energy surface. We attempted moves to swap pairs of system coordinates regularly, at fixed intervals. These proposals were accepted or rejected according to the Metropolis prescription, allowing the enhanced sampling at high temperature to propagate to the reference temperature without violating canonical ensemble statistics [Citation45].
The number of replicas required to obtain sufficiently overlapping energy histograms increases with the number ofdegrees of freedom in the system. For the smallest system considered here (3 peptides), we employ 27 replicas thermostatted at temperatures between 300 and 350 K, and for the largest (8 peptides) 84 replicas are used. We selected the temperature set for each system based on preliminary work, with the goal of achieving an acceptance rate of approximately 0.2 for swap attempts between replicas at adjacent temperatures.
Any memory of the initial configuration is very rapidly lost in the highest temperature replica. This de-correlation propagates to lower temperatures via exchanges. It is hence traversal of configurations from high to low temperature which is the limiting factor in determining the ability of our simulations to generate samples which are statistically independent of the starting configuration. To ensure the desired quality of sampling, we tracked the average time for a system to traverse the range of temperatures, and simulate for a time far in excess of this. Ordered by ascending system size, in pairs of n16N followed by n16NN systems, the simulation run times were equivalent to 6.4 and 6.4 s, 2.5 and 3.3
s, and 3.4 and 3.3
s. We stress that the concept of time in coarse-grained models such as PLUM and PLUM* is not well defined, particularly when accelerating low temperature sampling via exchange with other replicas. Times reported above should be considered an indication of sample size only.
3. Analysis methodology
We visualise the conformational ensemble for each system simulated using the standard Ramachandran plot. Briefly, this visualises the frequency at which successive peptide-bond dihedral angles ( and
measured between
and
) occur in combination over all samples from the ensemble simulated. This is typically represented as a ‘heat map’ with hotter colours indicating a higher frequency of occurrence. A high frequency of data points in the upper-left quadrant of such plots indicates a preference for
–sheet structure.
To discern the most favoured aggregate conformations in each trajectory, we use a clustering analysis based on geometric similarity. The tool g_cluster is a part of the Gromacs package [Citation46–Citation48] which performs this function. In this work, we make use of the gromos clustering algorithm [Citation49]. A pertinent detail arises from the indistinguishability of identical peptide chains. When calculating similarity to a reference structure, we must also compare to all structures resulting from permuting chain labels within that structure. For the eight-peptide system, this results in 40, 320 comparisons for each snapshot in the REMD simulation. We have modified the g_cluster tool to accommodate this, parallelising the permutations over processors using MPI.
For any clustering analysis, a root-mean-square deviation (RMSD) parameter, governing the allowable deviation of atomic coordinates of structures in the same cluster, must be set. Table shows the values we used, determined as a compromise between the level of false negative groupings and false positive groupings. Note that in general, RMSD cut-offs used in geometric clustering analysis are size-extensive; however, the cut-off distances do not follow a clear-cut, simple dependence on system size in terms of number of atoms alone due to changes in the flexibility between oligomers of differing size. The main point of our clustering analysis is not to identify a definitive set of clusters that can be compared in some absolute sense, but to highlight key structural differences between the n16N-x and n16NN-x aggregates (for a fixed value of x), and between the subdomains therein. So long as the clustering cut-off is kept the same when comparing n16N-x and n16NN-x, and for each subdomain, this remains a fair and clear way to compare the two conformational ensembles.
Table 1. Root-mean-square deviation (RMSD) values used for the geometric clustering analysis.
This clustering analysis allows comparison of flexibility between chain regions and chain types via comparison of populations in the most probable cluster, and via the discrete entropy of the ensemble. Subdomain analyses used same-length representations of each chain region: residues 1 to 8, 9 to 16 and 23 to 30 were used. For all analyses, only backbone atom positions were passed to g_cluster as input.
The VMD viewer [Citation50] was used to produce cartoon-style images of peptide aggregates. While automatic structure-identifying algorithms exist, these are not functional on the geometry of the PLUM model. Assignment was performed manually based on backbone dihedral angles and hydrogen bonding criteria.
4. Results and discussion
Figure shows the evolution of the secondary structure of n16N chains with increasing system size. Between the n16N monomer and the n16N-6 system, these Ramachandran plots show a consistent migration of structure from the -helix peak at
, and other accessible areas, to the
-structure peak at
. We will subsequently show in detail that the n16N-6 and n16N-8 systems aggregate into full
-sheets. In the n16N-6 and n16N-8 systems, the variance of accessed
angles about the peaks is extremely low compared to the n16N-3 system.
Figure 2. (Colour online) Ramachandran heat maps of n16N systems, accompanied by difference maps between neighbouring system sizes. In the difference maps, positive values are those more favoured in the larger system.
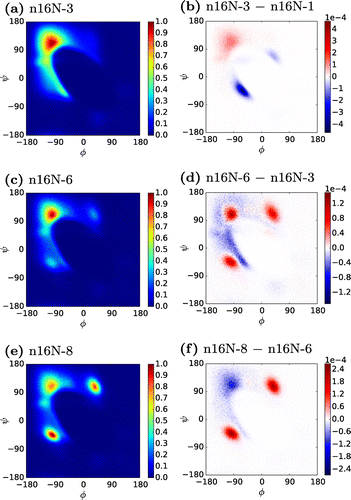
Two new peaks appear in n16N-6, Figure (c), and become dominant in n16N-8, Figure (e). The difference plots reveal that the magnitude of this change from n16N-6 to n16N-8 is approximately twice as great as any difference between the n16N-3 and n16N-6 systems. However, it will be illustrated that the n16N-8 system is in fact more strongly -sheet-dominated than any of the smaller systems. We suggest these peaks result from an artefact of the PLUM* model occurring when
-strands are tightly packed, in which strands tend to adopt alternating pairs of these
angles.
Figure is a trajectory snapshot which summarily displays the favoured aggregation behavior of the largest system of the n16N peptide, alongside the favoured structure of the monomer (inset). Geometric clustering of like conformations throughout the trajectory was used to generate the most likely structures in the conformational ensemble, and the most populous cluster is shown. This will henceforth be referred to as a system’s top cluster. In the top cluster of the n16N-8 system, the chains lie in two highly-ordered -sheets, both exhibiting a chain kink at P15 in the sequence. The smaller sheet lies at a 90
rotation about its plane to the larger sheet, optimising overlap of sheet SD2 regions (see Figure for a definition of SD2) which are hypothesised to be responsible for aggregation, without overlap of other subdomains. In stark contrast to the PLUM* predictions for the monomer system, almost no
-helical structure is present. This emergent behavior, inaccessible to atomistic approaches, demonstrates the utility of carefully parameterised intermediate resolution models, such as PLUM*.
Figure 3. (Colour online) Most populated structural group of the n16N-8 system, comprising 17.5% of trajectory snapshots. Inset: Most populated structural group of the n16N-1 system, with 4.4% [Citation37].
![Figure 3. (Colour online) Most populated structural group of the n16N-8 system, comprising 17.5% of trajectory snapshots. Inset: Most populated structural group of the n16N-1 system, with 4.4% [Citation37].](/cms/asset/a7faac67-1476-42e2-b150-24869b0c0696/gmos_a_1405158_f0003_oc.gif)
We refer readers to the supporting information for clustering results at every system size. A trend of aggregation through increasing -structure content and decreasing
-helix content as chain number increases is demonstrated.
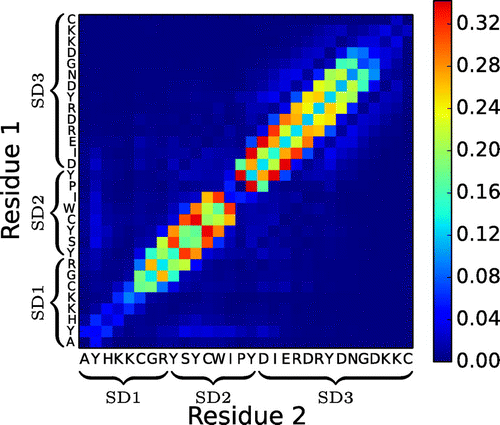
Figure shows the frequency of inter-peptide hydrogen bonds in n16N-8 over the entire ensemble. It is clear that chains exclusively undergo parallel -sheet oligomerisation, and fall into a shifted register in which inter-peptide hydrogen bonding most frequently occurs between residues i and
. This matches the staggered chains which constitute the sheets in Figure . The PLUM* model captures the fact that the proline residue is unable to form hydrogen bonds through its backbone nitrogen atom. The effect of this can be seen in the absence of hydrogen bonds from the heat map of Figure , and is likely related to the turn in each
-sheet of Figure . It is impossible for two proline residues to hydrogen bond to each other, which implies that a perfect parallel
-strand of two concurrent n16N chains cannot exist, and may account for the observation of chains preferentially aggregating out of step. Figure suggests that shifts of
, where
or 2 account for the majority of bonding, with approximate symmetry between positive and negative shifts. This leads to aggregates exhibiting a disorder in chain stacking, qualitatively distinct from amyloid-like structure. The lack of any hydrogen bonding away from the diagonal in Figure signifies a degree of specificity in aggregation behavior of n16N, suggesting that the forms of ordered structures in the conformational ensemble of n16N-8 do not deviate significantly (other than registry shifts) from the cluster in Figure .
Figure 4. (Colour online) Average number of interpeptide hydrogen bonds per residue–residue pair in the n16N-8 system.
In Figure , we present the fraction of configurations in which each residue within n16N and n16NN is involved in hydrogen bonding. Compared to Figure , information on intra-chain bonding topology is lost, masking any possible difference in aggregation behavior between the two peptides. The area under the curves can be taken as a normalised measure of total inter-peptide interaction, suggesting that hydrogen bonding in aggregates of both n16N and n16NN increases with system size, and also that differences in bonding between successive oligomers reduce with system size. We can therefore be confident that no qualitative difference in behavior will arise for larger aggregates than studied here.
Figure 5. (Colour online) Average frequency of each n16N or n16NN residue being involved in aggregation-enabling interpeptide hydrogen bonds, at each system size. For the n16NN systems, the anionic residues indicated along the axis in red are replaced in n16NN according to D N and E
Q.
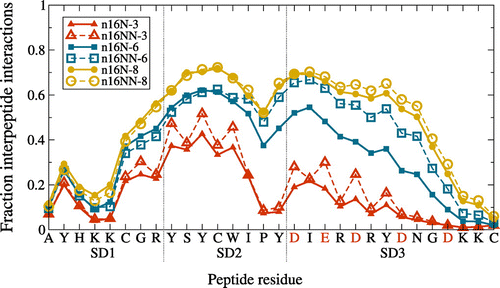
Our expectation to see SD2 as the principal driver of aggregation in n16N is largely confirmed by Figure . The residues most involved in inter-peptide hydrogen bonding are always located in SD2. However, at the largest system sizes, the N-terminal region of SD3 is also highly involved, as it is in the aggregate in Figure . The continuation of -structure well into SD3 is a result unseen at smaller system sizes. This defies the expected subdomain behavior, and is unusual given the highly charged nature of SD3. This behavior may arise from the crowding together of chains in the largest systems, coupled with the approximation inherent to the PLUM* model of describing all side-chain interactions on a simple hydrophobic scale, therefore omitting electrostatic repulsive interactions. Matching the subdomain hypothesis, SD1 remains mostly uninvolved in interpeptide hydrogen bonding for all systems.
In the two-unit systems previously studied [Citation37], a whole-chain difference was seen in measures of conformational accessibility, including the frequency of interpeptide hydrogen bonds between n16N and n16NN, with the latter being higher. This led to the hypothesis that the whole-chain conformational accessibility, which may be a prerequisite of assembly, was compromised in the mutant. However, Figure reveals that increasing system size diminishes the hydrogen bonding differences between the two peptides: the 3-chain systems are similar up to residue K5, the 6-chain ones are similar up to residue W13, and the 8-chain ones are similar throughout the entire sequence. To examine this discrepancy fully, we include an additional approach to inspecting conformational accessibility.
In table , our clustering analysis is extended to include comparable, equal-length representations of subdomains SD1, SD2 and SD3, as well as the full systems. By comparing the population of every group’s top cluster, we gain a further insight into the relative conformational accessibility of each. We also compute the discrete entropy (, where
denotes the population of the ith cluster) over all clusters in the ensemble to provide an indication of the extent to which the remaining population is distributed over multiple clusters. Note that cluster population comparisons of differently-sized full systems are not meaningful. Similarly, comparison between the full chain and subdomain data is meaningless.
Table 2. Population of most populated clusters in full-system and subdomain clustering analyses, and discrete entropy of the configurational ensemble, providing an insight into the stability of the system or chain region compared to regions or systems of the same size.
By this measure, there are no significant differences in levels of disorder in a given subdomain and system size between n16N and n16NN. This includes the highly charged SD3 subdomain, where the point mutations are introduced. The population of the top cluster of SD3 increases with increasing system size, overtaking SD1 in the largest system, corroborating the evidence from the rising hydrogen bonding frequency in Figure of falling disorder.
Data in table demonstrate that SD2 consistently explores fewer conformations than the other two subdomains at all system sizes. This supports suggestions that this subdomain is key to aggregation by presenting a more consistent geometry for inter-chain hydrogen bonding.
5. Conclusions
In summary, we have presented a specific, scalable oligomer structural model for n16N peptide systems involving staggered-chain -sheets stacking at 90
rotations to each other, with overlapping SD2 regions. The structure represents an emergence of order entirely unseen in the monomer state, matching expectations of disordered framework proteins [Citation13]. The structure conforms in the main to existing hypotheses about the peptide’s three subdomains [Citation15], with aggregation being centred on SD2. We observe the maximum propensity for inter-chain hydrogen bonding between SD2 subdomains, particularly in the smaller aggregates, coupled with the most consistent subdomain structure. Results from this aggregation study do not highlight a clear role for SD1 and SD3. However these two regions are less involved in aggregation-enabling inter-peptide hydrogen bonds, which leaves them available to perform other roles, such as the ‘fly-casting’ mechanism proposed previously. Given our validation of the n16N PLUM* model against atomistic simulations, we expect this conclusion to be qualitatively robust. We caution that inclusion of side-chain interactions with atomistic detail could feasibly result in different quantitative conformational flexibility in this region.
We believe that the approach adopted here for simulating IDPs, i.e. validation of a PLUM-style model against atomistic simulations via the conformational ensemble for small numbers of peptides, followed by scale-up to larger aggregates, is a viable route to identifying representative structures for use in detailed atomistic simulations.
As a secondary objective, we have compared properties of the structural ensemble of n16N with those of n16NN in an attempt to understand their differing ability to facilitate biomineralisation. The resulting data do not demonstrate any significant differences in the ensemble statistics of the two peptides when formed into oligomers. Structural differences reported previously [Citation37] for the dimer do not persist into these larger aggregates. Conclusions regarding n16NN are somewhat more tentative than for n16N, as PLUM* data have not been validated against atomistic simulations for the mutant. However, even if the PLUM* model is transferable to n16NN, there are a number of potential reasons why no difference in aggregation behaviour was observed. Firstly, we stress that our sampling does not consider unbinding of peptide chains and hence does not inform on the relative stability of bound versus unbound configurations. Detailed binding free energy calculations may reveal that n16NN structures are less stable than their n16N equivalents due to the weaker side-chain interactions. Secondly, the inability of the n16NN mutant to function correctly in biomineralisation processes may lie in kinetic effects (such as the mobility in solution of individual peptides), or interactions with ions during aggregation rather than in relative stabilities of different aggregate structures. Neither of these possibilities can be captured in the present model, motivating future studies into how such details can be captured in tractable models.
Supplementary_Material.pdf
Download PDF (1.9 MB)Acknowledgements
This work used the ARCHER UK National Supercomputing Service (http://www.archer.ac.uk) and facilities provided by the Research Technology Platform for Scientific Computing at the University of Warwick. MPA and GOR acknowledge helpful discussions with Tristan Bereau.
Additional information
Funding
Notes
No potential conflict of interest was reported by the authors.
Supplemental material for this article can be accessed https://doi.org/10.1080/08927022.2017.1405158.
Related Research Data
References
- Evans JS. ‘Tuning in’ to mollusk shell nacre- and prismatic-associated protein terminal sequences. implications for biomineralization and the construction of high performance inorganic--organic composites. Chem Rev. 2008;108:4455–4462.
- Aizenberg J. New nanofabrication strategies: inspired by biomineralization. MRS Bull. 2010;4(35):323–330.
- Kim S, Park CB. Bio-inspired synthesis of minerals for energy, environment, and medicinal applications. Adv Funct Mater. 2013;23:10–25.
- Cheng Q, Jiang L, Tang Z. Bioinspired layered materials with superior mechanical performance. Acc Chem Res. 2014;47:1256–1266.
- Lowenstam H. Minerals formed by organisms. Science. 1981;211:1126–1131.
- Mann S. Mineralization in biological systems. In: Inorganic elements in biochemistry. Vol. 54, Structure and bonding. Berlin, Heidelberg: Springer; 1983. p. 125–174.
- Weiner S, Dove PM. An overview of biomineralization processes and the problem of the vital effect. Rev Mineral Geochem. 2003;54:1–29.
- He G, Dahl T, Veis A, et al. Nucleation of apatite crystals in vitro by self-assembled dentin matrix protein 1. Nat Mater. 2003;2:552–558.
- Bettencourt V, Guerra A. Growth increments and biomineralization process in cephalopod statoliths. J Exp Mar Biol Ecol. 2000;248:191–205.
- Lowenstam HA, Weiner S. On biomineralization. New York (NY): Oxford University Press; 1989.
- Dunker AK, Babu MM, Barbar E, et al. What’s in a name? Why these proteins are intrinsically disordered. Intrinsically Disord Proteins. 2013;1. DOI:10.4161/idp.24157
- Ball KA, Wemmer DE, Head-Gordon T. Comparison of structure determination methods for intrinsically disordered amyloid-β peptides. J Phys Chem B. 2014;118:6405–6416. DOI:10.1021/jp410275y
- Dunker AK, Brown CJ, Lawson JD, et al. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–6582.
- Kalmar L, Homola D, Varga G, et al. Structural disorder in proteins brings order to crystal growth in biomineralization. Bone. 2012;51:528–534.
- Brown AH, Rodger PM, Evans JS, et al. Equilibrium conformational ensemble of the intrinsically disordered peptide n16N: linking subdomain structures and function in nacre. Biomacromolecules. 2014;15:4467–4479.
- Namba K. Roles of partly unfolded conformations in macromolecular self-assembly. Genes Cells. 2001;6:1–12.
- Nogawa C, Baba H, Masaoka T, et al. Genetic structure and polymorphisms of the N16 gene in pinctada fucata. Gene. 2012;504:84–91.
- Gardner L, Mills D, Wiegand A, et al. Spatial analysis of biomineralization associated gene expression from the mantle organ of the pearl oyster pinctada maxima. BMC Genomics. 2011;12:455–469.
- Marie B, Joubert C, Tayalé A, et al. Different secretory repertoires control the biomineralization processes of prism and nacre deposition of the pearl oyster shell. Proc Natl Acad Sci USA. 2012;109:20986–20991.
- Montagnani C, Marie B, Marin F, et al. Pmarg-pearlin is a matrix protein involved in nacre framework formation in the pearl oyster Pinctada margaritifera. ChemBioChem. 2011;12:2033–2043.
- Samata T, Hayashi N, Kono M, et al. A new matrix protein family related to the nacreous layer formation of pinctada fucata. FEBS Lett. 1999;462:225–229.
- Collino S, Evans JS. Molecular specifications of a mineral modulation sequence derived from the aragonite-promoting protein n16. Biomacromolecules. 2008;9:1909–1918. pMID: 18558739.
- Perovic I, Chang EP, Lui M, et al. A nacre protein, n16.3, self-assembles to form protein oligomers that dimensionally limit and organize mineral deposits. Biochemistry. 2014;53:2739–2748.
- Ponce CB, Evans JS. Polymorph crystal selection by n16, an intrinsically disordered nacre framework protein. Crystal Growth Des. 2011;11:4690–4696.
- Kim IW, DiMasi E, Evans JS. Identification of mineral modulation sequences within the nacre-associated oyster shell protein, n16. Crystal Growth Des. 2004;4:1113–1118.
- Kim I, Darragh M, Orme C, et al. Molecular ‘tuning’ of crystal growth by nacre-associated polypeptides. Crystal Growth Des. 2006;6:5–10.
- Delak K, Collino S, Evans JS. Expected and unexpected effects of amino acid substitutions on polypeptide-directed crystal growth. Langmuir. 2007;23:11951–11955.
- Metzler RA, Kim IW, Delak K, et al. Probing the organic-mineral interface at the molecular level in model biominerals. Langmuir. 2008;24:2680–2687.
- Keene EC, Evans JS, Estroff LA. Matrix interactions in biomineralization: Aragonite nucleation by an intrinsically disordered nacre polypeptide, n16N, associated with a β-chitin substrate. Crystal Growth Des. 2010;10:1383–1389.
- Metzler RA, Evans JS, Killian CE, et al. Nacre protein fragment templates lamellar aragonite growth. J Am Chem Soc. 2010;132:6329–6334.
- Keene EC, Evans JS, Estroff LA. Silk fibroin hydrogels coupled with the n16N-β-chitin complex: An in vitro organic matrix for controlling calcium carbonate mineralization. Crystal Growth Des. 2010;10:5169–5175.
- Amos FF, Ponce CB, Evans JS. Formation of framework nacre polypeptide supramolecular assemblies that nucleate polymorphs. Biomacromolecules. 2011;12:1883–1890.
- Seto J, Picker A, Chen Y, et al. Nacre protein sequence compartmentalizes mineral polymorphs in solution. Crystal Growth Des. 2014;14:1501–1505.
- Levi-Kalisman Y, Falini G, Addadi L, et al. Structure of the nacreous organic matrix of a bivalve mollusk shell examined in the hydrated state using cryo-TEM. J Struct Biol. 2001;135:8–17.
- Kim J, Straub JE, Keyes T. Statistical-temperature monte carlo and molecular dynamics algorithms. Phys Rev Lett. 2006;97:050601.
- Linding R, Russell RB, Neduva V, et al. Globplot: exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003;31:3701–3708.
- Rutter GO, Brown AH, Quigley D, et al. Testing the transferability of a coarse-grained model to intrinsically disordered proteins. Phys Chem Chem Phys. 2015;17:31741–31749.
- Bereau T, Deserno M. Generic coarse-grained model for protein folding and aggregation. J Chem Phys. 2009;130. DOI:10.1063/1.3152842
- Piana S, Lindorff-Larsen K, Shaw DE. How robust are protein folding simulations with respect to force field parameterization? Biophys J. 2011;100:L47–L49.
- MacKerell AD, Bashford D, Bellott M, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616.
- Jorgensen WL, Chandrasekhar J, Madura JD, et al. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935.
- Plimpton S. Fast parallel algorithms for short-range molecular dynamics. J Comp Phys. 1995;117:1–19.
- Sugita Y, Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem Phys Lett. 1999;314:141–151.
- Swendsen RH, Wang JS. Replica monte carlo simulation of spin-glasses. Phys Rev Lett. 1986;57:2607–2609.
- Frenkel D, Smit B. Understanding molecular simulation: from algorithms to applications. London: Academic Pr; 2002.
- Berendsen H, van der Spoel D, van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput Phys Commun. 1995;91:43–56.
- Hess B, Kutzner C, van der Spoel D, et al. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–447.
- Pronk S, Páll S, Schulz R, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854.
- Daura X, Gademann K, Jaun B, et al. Peptide folding: when simulation meets experiment. Angew Chem Int Ed. 1999;38:236–240.
- Humphrey W, Dalke A, Schulten K. VMD -- visual molecular dynamics. J Mol Graph. 1996;14:33–38.