
Abstract
The present study was designed to investigate the role of miR-30 in the development of Gastric cancer (GC). miR-30 expression was increased in GC tissues and cell lines. Downregulation of miR-30 inhibited cell proliferation and promoted apoptosis in HGC-27 cells. Upregulation of miR-30 enhanced the proliferation and inhibited apoptosis. P53 expression was decreased in GC tissues. P53 expression was correlated with miR-30 expression. Downregulation of miR-30 increased P53 expression. Knockdown of P53 inhibited miR-30-inhibitor-induced suppression of cell proliferation and increase of apoptosis. Downregulation of miR-30 increased ROS generation which was inhibited by shP53. miR-30 inhibitors induced a decrease in mitochondrial oxygen consumption, cytoplasmic release of cytochrome c, and activation of Caspase 3 and 9, activating mitochondrial apoptotic pathway. Downregulation of P53 and N-acetyl-cysteine suppressed miR-30 inhibitors-activated mitochondrial dysfunction and apoptotic events. In conclusion, we identified that miR-30 functioned as a potential oncomiR through P53/ROS-mediated regulation of mitochondrial apoptotic pathway.
Graphical abstract
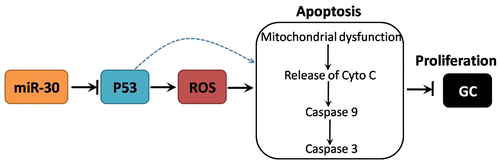
miR-30 functions as a potential oncomiR in GC through P53/ROS-mediated regulation of mitochondrial apoptotic pathway.
Gastric cancer (GC) is considered to be one of the most common malignancies and the third leading cause of cancer death worldwide.Citation1) GC is characterized by rapid tumor growth and high recurrence rate, severely limiting the survival of GC patients.Citation2) In the past few years, early diagnosis and the development of effective treatment strategies for GC, such as surgical resection and chemotherapy, has been greatly advanced and enhanced the survival rate of GC patients. However, we are still confronted with relatively short-term survival, drug resistance, metastasis, and high mortality of GC.Citation3,4) It is considered that dysregulation of oncogenes or tumor suppressor genes play important roles in the development and progression of GC.Citation5) A better understand of the molecular mechanism of GC is urgently needed.
MicroRNAs (miRNAs) are a family of small noncoding mRNA molecules with 18–25 nucleotides, which function to regulate a variety of gene expression that are involved in a battery of physiological and pathophysiological process, including development, differentiation, cell proliferation, apoptosis and many others.Citation6,7) Moreover, miRNAs also act as either oncogenes or tumor suppressor genes, playing crucial roles in the regulation of cancer development and progression in various types of cancers.Citation8) Dozens of miRNAs have been found to be dysregulated in the development and progression of GC,Citation9,10) such as miR-93,Citation11) miR-196b,Citation12) miR-203,Citation13) and miR-375,Citation14,15) etc. Dysregulation of miR-30 is associated with the development of several cancers. For instance, Tsukasa et al. found that miR-30 family promoted migratory and invasive abilities in CD133 pancreatic cancer stem-like cells.Citation16) Zhang et al. showed that miR-30a suppressed colon cancer cell growth through inhibition of insulin receptor substrate 2.Citation17) Moreover, suppression of miR-30 was found to be involved in fluorouracil-induced autophagy-related GC cell death.Citation18) However, the accurate role of miR-30 in the development of GC is not known.
The present study was designed to investigate the role of miR-30 in the development of GC and to explore the possible mechanisms that miR-30 regulates GC cell proliferation. We showed that miR-30 was highly expressed in GC tissues and cell lines and downregulation of miR-30 inhibited GC cell proliferation through enhancement of P53/reactive oxygen species (ROS)-mediated mitochondrial apoptotic pathway.
Materials and methods
Chemicals and materials
β-actin, Tubulin α, P53 and Cytochrome C (Cyto C) antibodies were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). N-acetyl cysteine (NAC) was procured from Sigma Aldrich. TRIzol Reagent and Lipofectamine 2000 were obtained from Invitrogen (Carlsbad, USA). Human gastric epithelial cell line (GES-1) and GC cell lines (HGC-27, MKN-28, MKN-45, MGC-803, SGC-7901) were purchased from American Type Culture Collection (Manassas, USA). The other chemicals used in the study were of the highest grade commercially available.
Ethics statement, patients and tissue samples
Tumor tissues were collected from biopsy samples that underwent resection of the primary GC in a total of 61 cases admitted in Hongqi Hospital, Mudanjiang Medical College from June 2013 to October 2015. Tumor adjacent gastric tissues were used as normal control. All the patients had provided the informed consent, and the study was approved by Hongqi Hospital, Mudanjiang Medical College and complied with the Declaration of Helsinki.
Cell culture and transfection
GES-1 and GC cells were cultured in DMEM medium supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were placed in a humidified incubator containing 5% CO2 at 37 °C. The miR-30 mimics (5′-ACAUUUGUACAUGACAAAUUAUA-3′), inhibitors (5′-UAUAAUUUGUCAUGUACAAAUGU-3′), and scramble miRNA control (5′-UUCUCCGAACGUGUCACGUTT-3′), and P53 shRNA lentivirus (5′-GACUCCAGTGGUAAUCUAC-3′) were synthesized by Genepharma (Shanghai, China). The miR30 inhibitor is siRNA. Lipofectamine 2000 was used to conduct the transient transfection according to the manufacturer’s protocols. Transfection of P53 shRNA lentivirus and stable knock down of P53 was conducted according to the manufacturer’s protocols.
Determination of cell proliferation
Measurement of cell proliferation was performed using a CCK8 assay kit according to the manufacturer’s protocols. Absorbance at 450 nm was detected using a microplate reader (BIORAD, USA).
Determination of apoptosis
To determine apoptosis, TdT-mediated dUTP nick end labeling (TUNEL) staining was performed. After the experiment, In Situ Cell Death Detection Kit (Roche) was used according to the manufacturer’s protocols. Cells were analyzed using a BD FACSCanto™ II Flow Cytometry System. Results were shown as folds of positive cells compared with control.
RNA isolation and real-time polymerase chain reaction
Total RNA was extracted from the tissue samples and cells using TRIzol reagent according to the manufacturer’s protocols. Reverse transcription of 500 ng mRNA to cDNA was performed using the First Strand cDNA synthesis kit (TaKaRa, Japan). miR-30 and P53 level was quantified by real-time polymerase chain reaction (PCR) system using SYBR Green reagents (TaKaRa, Japan) in a BIORAD Cycling Biosystem. β-actin or U6 noncoding small nuclear RNA was used as internal controls. Relative expression was analyzed using the 2−ΔΔCT method, compared with the endogenous controls. Amplification conditions were as follows: initial step at 94 °C for 5 min, followed by 40 cycles of denaturation at 94 °C for 30 s, annealing at 63 °C for 30 s, and then extension at 72 °C for 10 s. Primers used were as follows: miR-30 (Forward, 5′-P-UGAAAGCAUCCUUGACUGGAAG-3′, Reverse, 5′-UCCAGUCAAGGAUGCUUACACC-3′), p53 (Forward, 5′-TCAGATCCTAGCGTCGAGCCCCCTCT-3′ Reverse, 5′-CCCAGACGGAAACCGTAGCTGCCCT-3′), β-actin (Forward, 5′-GACCTCTATGCCAACACAGTGC-3′, Reverse, 5′-GTACTCCTGCTTGCTGATCCAC-3′), U6 (TaqMan MicroRNA Assay NR_004394).
Western blot
After the experiment, cells were lysed on ice for 30 min in RIPA Lysis buffer (Beyotime, China) supplemented with Protease and Phosphatase Inhibitor Cocktails (Thermo Scientific, USA). Cell lysates were centrifuged at 12,000 g at 4 °C for 10 min and then the supernatant was isolated. Protein concentration was determined by the BCA Assay Kit (Thermo Scientific, USA). Equal amount of proteins were subjected to SDS-PAGE and transferred to NC membranes. Then membranes were then blocked with 5% fat-free milk at room temperature for 1 h and probed with primary antibodies at 4 °C overnight. After washing for four times, the membrane was incubated with horseradish peroxidase-conjugated secondary antibodies (Thermo Scientific, USA) at 37 °C for 30 min. Bands were visualized by chemiluminescence reaction using an ECL detection system (Thermo Scientific, USA), followed by capture using BIORAD Imaging Systems (BIORAD, USA).
Determination of ROS level
After the experiment, cells were incubated with 10 μM Dihydroethidium (DHE), an oxidation-sensitive probe, in serum-free medium for 20 min. After that, cells were rinsed with PBS twice and fluorescence was observed under a confocal microscopy (Olympus, Japan).
Determination of mitochondrial function
Mitochondrial function was evaluated by determination of oxygen consumption rate using a Clark Oxygen Electrode. In brief, mitochondria were isolated using a Mitochondria Isolation Kit (Thermo Scientific, USA) and then diluted into oxygen-saturated PBS. After the establishment of stable oxygen-saturated curve, mitochondria were added into the reaction container and oxygen consumption was recorded for 10 min.
Determination of Caspase activities
After the experiments, cells were homogenized using a microhomogenizer. The activities of caspase-9 and -3 in homogenates were determined using commercial assay kits (Beyotime Biotechnology, China) according to the manufacturer’s protocols.
Statistical analysis
Data in the study were shown as the mean ± SEM, and p < 0.05 was considered statistically significant. All experiments were conducted at least in triplicates. Data analysis was performed using GraphPad Software. One-way analysis of variance was used to measure the significance between more than two groups, followed by a Dunnett’s t-test for multiple comparisons. The significance of differences between two groups was analyzed by Student’s t test.
Results
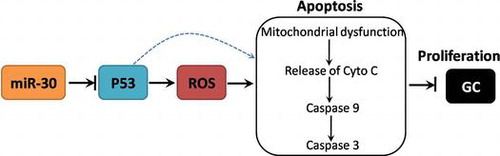
miR-30 is upregulated in GC tissues and cell lines
To assess the changes of miR-30 in the development of GC, the expression of miR-30 in GC tissues and several cell lines were determined by real-time PCR. As shown in Fig. (A) and supplemental Table 1, miR-30 expression in GC tissues was significantly higher than that of control. In Fig. (B), we showed that miR-30 expression in GC cell lines, including HGC-27, MKN-28, MKN-45, MGC-803, SGC-7901, was markedly increased, compared with that of GES-1 gastric epithelial cell line. The results demonstrated that mRNA expression of miR-30 was remarkably upregulated in GC tissues and cell lines.
Fig. 1. Expression of miR-30 in GC tissues and cell lines.
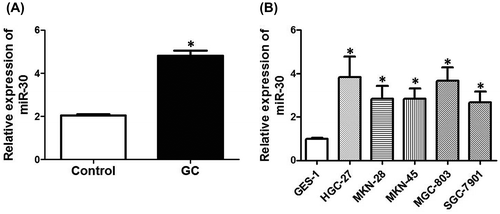
miR-30 promotes cell proliferation and inhibits apoptosis in HGC-27 cells
To examine the role of miR-30 in the proliferation of GC cells, we evaluated the effect of upregulation and downregulation of miR-30 on proliferation and apoptosis in HGC-27 cells. HGC-27 cells were transfected with miR-30 inhibitors, and the results showed that miR-30 was efficiently inhibited (Fig. (A)). Cell proliferation after the inhibition of miR-30 was determined by an assay kit. We showed that downregulation of miR-30 significantly decreased the OD values 3 days after the incubation, indicating that cell proliferation was suppressed by miR-30 downregulation (Fig. (B)). Moreover, cell apoptosis was determined by TUNEL assay and the results showed that downregulation of miR-30 markedly increased the number of apoptotic cells (Fig. (C)). Furthermore, HGC-27 cells were also transfected with miR-30 mimics to evaluate the effect of upregulation of miR-30 on HGC-27 cell proliferation and apoptosis (Fig. (D)). We showed that upregulation of miR-30 significantly promoted cell proliferation (Fig. (E)) and inhibited apoptosis (Fig. (F)) in HGC-27 cells. The results demonstrated that miR-30 promoted cell proliferation and inhibited apoptosis in GC cells.
Fig. 2. Effect of up- and down-regulation of miR-30 on cell proliferation and apoptosis in HGC-27 cells.
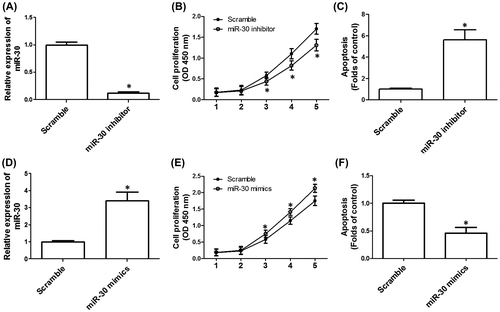
Regulation of P53 was involved in miR-30-induced regulation of cell proliferation and apoptosis in HGC-27 cells
Previous studies have shown that P53 is a potential target of miR-30. In the current study, we next examined the possible role of P53 in miR-30-exhibited regulation of cell proliferation and apoptosis in HGC-27 cells. The expression of P53 in GC tissues was significantly lower than that of control tissues (Fig. (A)). In GC tissues, the expression of P53 was significantly negatively correlated with the expression of miR-30 (Fig. (B)). However, in normal tissues, there was no significant correlation between P53 and miR-30 levels (figure not shown). In Fig. (C) and (D), we found that downregulation of miR-30 significantly increased the mRNA and protein expression of P53, indicating that P53 may be a target of miR-30. Moreover, we constructed lentivirus carrying shRNA of P53 and transfected HGC-27 cells to build a cell line with stable knockdown of P53. Knockdown of P53 significantly enhanced cell proliferation (Fig. (E)). Moreover, knockdown of P53 remarkably inhibited miR-30-inhibitor-induced suppression of cell proliferation in HGC-27 cells (Fig. (E)). Downregulation of P53 reduced apoptosis, compared with that of scramble (Fig. (F)). Furthermore, miR-30-inhibitor-induced increase in apoptosis was suppressed by shP53 (Fig. (F)). The results indicated that upregulation of P53 was involved in downregulation of miR-30-induced inhibition of cell proliferation and promotion of apoptosis in HGC-27 cells.
Fig. 3. Role of P53 in the effect of down-regulation of miR-30 on cell proliferation and apoptosis in HGC-27 cells.
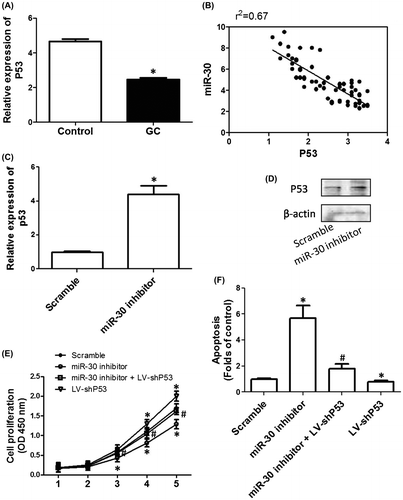
P53-mediated mitochondrial apoptotic pathway was involved in miR-30-induced regulation of cell proliferation and apoptosis in HGC-27 cells
To explore the molecular mechanism of P53-mediated regulation of cell proliferation and apoptosis induced by downregulation of miR-30, ROS generation and mitochondrial apoptotic pathway were evaluated. As shown in Fig. (A), knockdown of P53 markedly decreased DHE fluorescence, but transfection of miR-30 inhibitors significantly increased DHE fluorescence. shP53 notably inhibited the increase in DHE fluorescence induced by miR-30 inhibitors, indicating that P53 was required for miR-30 inhibitors-induced ROS generation. In the next step, we evaluated the mechanism of ROS-mediated inhibition of cell proliferation induced by miR-30 inhibitors. In Fig. (B), we showed that miR-30 inhibitors markedly decreased oxygen consumption rate in HGC-27 cells, indicating the induction of mitochondrial dysfunction. shP53 and NAC, a potent antioxidant, notably suppressed miR-30 inhibitors-induced decrease in oxygen consumption rate, indicating that P53 activation and the subsequent ROS generation were involved in miR-30 inhibitors-induced mitochondrial dysfunction.
Fig. 4. Role of P53/ROS-mediated mitochondrial pathway in the effect of downregulation of miR-30 on cell proliferation and apoptosis in HGC-27 cells.
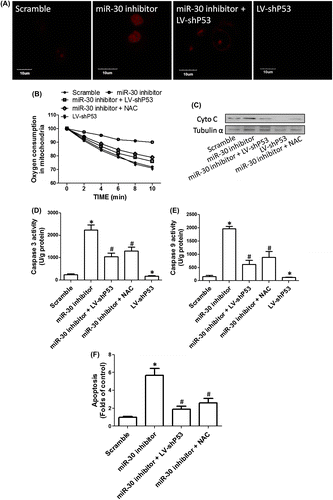
In Fig. (C)–(E), we showed knockdown of P53 notably decreased Cyto C expression in cytoplasm and the activities of Caspase 3 and 9. We also found that miR-30 inhibitors significantly increased the cytoplasmic expression of Cyto C and the activities of Caspase 3 and 9, implicating that inhibition of miR-30 activated mitochondrial apoptotic pathway. Moreover, the release of Cyto C and the activation of Caspase 3 and 9 induced by miR-30 inhibitors were suppressed by shP53 and NAC, indicating the involvement of P53 activation and the subsequent ROS generation in miR-30 inhibitors-induced mitochondrial apoptotic pathway (Fig. (C)–(E)). As respected, miR-30 inhibitors-induced apoptosis was inhibited by both shP53 and NAC. The results indicated that P53/ROS-mediated mitochondrial apoptotic pathway was involved in miR-30-induced regulation of cell proliferation and apoptosis in HGC-27 cells (Fig. (F)).
Discussion
Dysregulation of miR-30 is associated with the development of several cancers, such as pancreatic cancer and colon cancer.Citation16,17,19) Yang et al. found that fluorouracil could suppress miR-30 to upregulate Beclin-1 to induce autophagic cell death and cell proliferation arrest in GC cells.Citation18) However, the mechanism of miR-30-mediated regulation of GC cell proliferation is far from completely understood. Enhanced cell proliferation is a hallmark of cancer cell and the regulation of cell proliferation is usually used to evaluate the oncogenic activity of microRNAs.Citation20–22) In the current study, we explored the role of miR-30 in GC cell proliferation and elucidated the possible molecular mechanisms. We found that miR-30 expression was significantly increased in GC tissues and cell lines. Downregulation of miR-30 inhibited cell proliferation and promoted apoptosis in HGC-27 cells. In contrast, upregulation of miR-30 enhanced the proliferation and inhibited the apoptosis of HGC-27 cells. The results demonstrated that miR-30 was involved in GC cell proliferation.
Previous literature has supported that P53 is a potential target of miR-30. For instance, Li et al. found that miR-30 family members inhibited mitochondrial fission through suppressing the expression of p53 and its downstream target Drp1.Citation23) miR30a/p53 axis was found to be involved in triiodothyronine-induced prevention against cardiac ischemia/reperfusion mitochondrial impairment and cell loss.Citation24) In the study, we examined the possible role of P53 in the regulation of cell proliferation and apoptosis in GC cells. Consistently, we found that downregulation of miR-30 enhanced the expression of P53 and inhibition of P53 significantly reversed the decrease of cell proliferation and increase of apoptosis induced by downregulation of miR-30. The results demonstrated that miR-30 regulate GC cell proliferation and apoptosis via modulation of P53.
The P53 functions as a well-known tumor suppressor which is involved in various forms of cell death including apoptosis, necrosis, and ferroptosis.Citation25,26) The mitochondrial apoptotic pathway is main pathway that mediates the anti-tumor effect of P53.Citation27,28) In response to death stimuli, p53 mediates mitochondrial outer membrane permeabilization and permeability transition pore, followed by release of apoptogenic proteins, such as cytochrome c, are released to the cytosol from the mitochondrial inter-membrane space.Citation29,30) Release of apoptogenic proteins results in the activation of caspase cascades, leading to final apoptotic cell death.Citation31,32) ROS participates in P53-mediated mitochondrial apoptosis through regulating mitochondrial membrane potential and inducing Cyto C release and activation of caspase cascades.Citation33–35) In addition to ROS, P53 could activate mitochondrial pathway through regulation of Bax and Bcl-2 family members.Citation36–38) The results in the current study showed that downregulation of miR-30 promoted ROS generation through activation of P53. Moreover, P53 activation and the subsequent ROS generation mediated downregulation of miR-30-resulted activation of mitochondrial apoptotic pathway and final apoptosis and inhibition of cell proliferation. The data do not preclude the possibility that other mechanism besides ROS generation participates in P53-mediated activation of mitochondrial apoptosis induced by downregulation of miR-30. Further studies are needed to clarify this mechanism.
In conclusion, we identified miR-30 as a potential oncomiR in GC patients and found that miR-30 regulated GC cell proliferation through P53/ROS-mediated regulation of mitochondrial apoptotic pathway (Fig. ). Further studies are needed to explore the mechanism of P53-mediated ROS generation induced by downregulation of miR-30. Overall, the results suggest that miR-30/P53/ROS-induced mitochondrial apoptotic pathway may serve as novel therapeutic targets and prognostic markers in GC patients.
Fig. 5. Schematic figure of the mechanism of miR-30-mediated regulation of GC cell proliferation.
Disclosure statement
No potential conflict of interest was reported by the authors.
Author Contributions
J.W. conducted the experiments and analyzed the data; Y.J. designed the experiments, analyzed the data and wrote the paper; L.C. conducted the experiments; L.J. conducted the experiments.
Supplementary material
The supplemental material for this paper is available at http://dx.doi.org/10.1080/09168451.2016.1238294.
TBBB_1238294_Supplementary_Material.docx
Download MS Word (16.4 KB)Related Research Data
References
- Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J. Clin. 2015;65:87–108.10.3322/caac.21262
- Choi YY, Noh SH, Cheong JH. Molecular dimensions of gastric cancer: translational and clinical perspectives. J. Pathol. Transl. Med. 2016;50:1–9.10.4132/jptm.2015.09.10
- Marano L, Polom K, Patriti A, et al. Surgical management of advanced gastric cancer: an evolving issue. Eur. J. Surg. Oncol. 2016;42:18–27.10.1016/j.ejso.2015.10.016
- Wu J, Qin H, Li T, et al. Characterization of site-specific glycosylation of secreted proteins associated with multi-drug resistance of gastric cancer. Oncotarget. 2016;7:25315–25327.
- Irmak-Yazicioglu MB. Mechanisms of microRNA deregulation and microRNA targets in gastric cancer. Oncol. Res. Treat. 2016;39:136–139.10.1159/000443224
- Kang C, Song JJ, Lee J, et al. Epigenetics: an emerging player in gastric cancer. World J. Gastroenterol. 2014;20:6433–6447.10.3748/wjg.v20.i21.6433
- Juzenas S, Salteniene V, Kupcinskas J, et al. Analysis of deregulated microRNAs and their target genes in gastric cancer. PLoS ONE. 2015;10:e132327.
- Ishiguro H, Kimura M, Takeyama H. Role of microRNAs in gastric cancer. World J. Gastroenterol. 2014;20:5694–5699.10.3748/wjg.v20.i19.5694
- Yang G, Gong Y, Wang Q, et al. The role of miR-100-mediated Notch pathway in apoptosis of gastric tumor cells. Cell. Signal. 2015;27:1087–1101.10.1016/j.cellsig.2015.02.013
- Li Z, Li D, Zhang G, et al. Methylation-associated silencing of MicroRNA-335 contributes tumor cell invasion and migration by interacting with RASA1 in gastric cancer. Am. J. Cancer Res. 2014;4:648–662.
- Liang H, Wang F, Chu D, et al. miR-93 functions as an oncomiR for the downregulation of PDCD4 in gastric carcinoma. Sci. Rep. 2016;6:23772.10.1038/srep23772
- Li NA, Wang W, Xu B, et al. miR-196b regulates gastric cancer cell proliferation and invasion via PI3K/AKT/mTOR signaling pathway. Oncol. Lett. 2016;11:1745–1749.
- Chu SJ, Wang G, Zhang PF, et al. MicroRNA-203 suppresses gastric cancer growth by targeting PIBF1/Akt signaling. J. Exp. Clin. Cancer Res. 2016;35:87.10.1186/s13046-016-0323-1
- Xu Y, Jin J, Liu Y, et al. Snail-regulated MiR-375 inhibits migration and invasion of gastric cancer cells by targeting JAK2. PLoS ONE. 2014;9:e99516.10.1371/journal.pone.0099516
- Shen ZY, Zhang ZZ, Liu H, et al. miR-375 inhibits the proliferation of gastric cancer cells by repressing ERBB2 expression. Exp. Ther. Med. 2014;7:1757–1761.
- Tsukasa K, Ding Q, Miyazaki Y, et al. miR-30 family promotes migratory and invasive abilities in CD133 pancreatic cancer stem-like cells. Hum. Cell. 2016;29:130–137.
- Zhang Q, Tang Q, Qin D, et al. Role of microRNA 30a targeting insulin receptor substrate 2 in colorectal tumorigenesis. Mol. Cell. Biol. 2015;35:988–1000.10.1128/MCB.01242-14
- Yang C, Pan Y. Fluorouracil induces autophagy-related gastric carcinoma cell death through Beclin-1 upregulation by miR-30 suppression. Tumour Biol. 2015. doi:10.1007/s13277-015-3775-6
- Che S, Sun T, Wang J, et al. miR-30 overexpression promotes glioma stem cells by regulating Jak/STAT3 signaling pathway. Tumour Biol. 2015;36:6805–6811.10.1007/s13277-015-3400-8
- Yin G, Zhou H, Xue Y, et al. MicroRNA-340 promotes the tumor growth of human gastric cancer by inhibiting cyclin G2. Oncol. Rep. 2016;36:1111–1118.
- Lin J, Xu K, Wei J, et al. MicroRNA-124 suppresses tumor cell proliferation and invasion by targeting CD164 signaling pathway in non-small cell lung cancer. J. Gene. Ther 2016; 2. pii: 6.
- Zhou C, Tan W, Lv H, et al. Hypoxia-inducible microRNA-488 regulates apoptosis by targeting Bim in osteosarcoma. Cell. Oncol. (Dordr) 2016;39:463–471.
- Li J, Donath S, Li Y, et al. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. 2010;6:e1000795.10.1371/journal.pgen.1000795
- Forini F, Kusmic C, Nicolini G, et al. Triiodothyronine prevents cardiac ischemia/reperfusion mitochondrial impairment and cell loss by regulating miR30a/p53 axis. Endocrinology. 2014;155:4581–4590.10.1210/en.2014-1106
- Jiang L, Kon N, Li T, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62.10.1038/nature14344
- Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell. 2001;7:683–694.10.1016/S1097-2765(01)00214-3
- Liu J, Zhang C, Hu W, et al. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer Lett. 2015;356:197–203.10.1016/j.canlet.2013.12.025
- Chi SW. Structural insights into the transcription-independent apoptotic pathway of p53. BMB Rep. 2014;47:167–172.10.5483/BMBRep.2014.47.3.261
- Saleem A, Iqbal S, Zhang Y, et al. Effect of p53 on mitochondrial morphology, import, and assembly in skeletal muscle. Am. J. Physiol. Cell Physiol. 2015;308:C319–C329.10.1152/ajpcell.00253.2014
- Mihara M, Erster S, Zaika A, et al. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell. 2003;11:577–590.10.1016/S1097-2765(03)00050-9
- Kadenbach B, Arnold S, Lee I, et al. The possible role of cytochrome c oxidase in stress-induced apoptosis and degenerative diseases. Biochim. Biophys. Acta. 2004;1655:400–408.10.1016/j.bbabio.2003.06.005
- Haupt S, Berger M, Goldberg Z, et al. Apoptosis—the p53 network. J. Cell Sci. 2003;116:4077–4085.10.1242/jcs.00739
- Li PF, Dietz R, von Harsdorf R. p53 regulates mitochondrial membrane potential through reactive oxygen species and induces cytochrome c-independent apoptosis blocked by Bcl-2. EMBO J. 1999;18:6027–6036.10.1093/emboj/18.21.6027
- Wang L, Hu T, Shen J, et al. Miltirone induced mitochondrial dysfunction and ROS-dependent apoptosis in colon cancer cells. Life Sci. 2016;151:224–234.
- Fallahian F, Aghaei M, Abdolmohammadi MH, et al. Molecular mechanism of apoptosis induction by Gaillardin, a sesquiterpene lactone, in breast cancer cell lines: Gaillardin-induced apoptosis in breast cancer cell lines. Cell Biol. Toxicol. 2015;31:295–305.10.1007/s10565-016-9312-6
- Leu JI, Dumont P, Hafey M, et al. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat. Cell Biol. 2004;6:443–450.10.1038/ncb1123
- Oda E, Ohki R, Murasawa H, et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–1058.10.1126/science.288.5468.1053
- Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–299.