
Abstract
The stereochemical reaction course for the two C-3 hydrogens of leucine to produce a characteristic isoprenoidal lipid in halophilic archaea was observed using incubation experiments with whole cell Halobacterium salinarum. Deuterium-labeled (3R)- and (3S)-[3-2H]leucine were freshly prepared as substrates from 2,3-epoxy-4-methyl-1-pentanol. Incorporation of deuterium from (3S)-[3-2H]leucine and loss of deuterium from (3R)-[3-2H]leucine in the lipid-core of H. salinarum was observed. Taken together with the results of our previous report, involving the incubation of chiral-labeled [5-2H]leucine, these results strongly suggested an involvement of isovaleryl-CoA dehydrogenase in leucine conversion to isoprenoid lipid in halophilic archaea. The stereochemical course of the reaction (anti-elimination) might have been the same as that previously reported for mammalian enzyme reactions. Thus, these results suggested that branched amino acids were metabolized to mevalonate in archaea in a manner similar to other organisms.
The stereochemical reaction course for the two C-3 hydrogens of leucine to isoprenoidal lipid in halophilic archaea was observed.
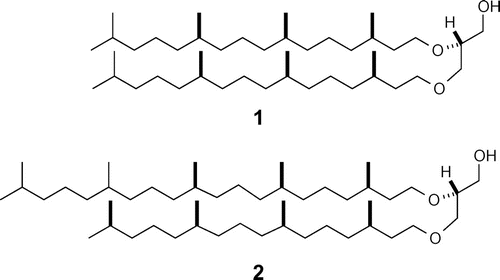
Halophilic archaea typically live in salt lakes, salt marshes, and halite rich environments, all of which represent relatively harsh conditions (high salt concentrations, ~6 M NaCl, an almost saturated solution) for any organism.Citation1,2) Based on their tolerance for high salt concentrations, there has been some work conducted with the aim of identifying halophilic archaea on Mars.Citation3,4) In terms of their characteristic features, halophilic archaea consist of a lipid-core bearing two C20 saturated isoprenoids, which are linked to a glycerol unit, known as archaeol (1). This feature contrasts well with what is typical of bacteria or eukaryotes, which normally consist of a glycerol diester lipid-core. Furthermore, one of the saturated isoprenoidal ethers is elongated to C25 (C20–C25 diether) (2) in the lipid-core of halophilic archaea (Fig. ).Citation2,5,6)
Fig. 1. Lipid-core structure of archaea.
The membrane of halophilic archaea consists of isoprenoid-based lipids. Furthermore, for unicellular organisms, characteristic isoprenoidal ether lipids might act to protect cells against harsh environments. It is, therefore, possible that efficient biosynthetic pathways could exist for isoprenoid preparation. Two biosynthetic pathways have been identified for the synthesis of the C5 unit isopentenyl diphosphate (IPP) (3), which is an important intermediate in isoprenoid biosynthesis (Fig. ). One of these pathways is the mevalonate pathway, which involves the condensation of three acetate units, followed by an appropriate functional group conversion via 3-hydroxymethylglutaryl-CoA (HMG-CoA) (4) to yield the key C6 intermediate mevalonate (5). The second pathway is the mevalonate-independent pathway. An important intermediate in the mevalonate-independent pathway is 2-C-methyl-D-erythritol-4-phosphate (MEP) (6), which is synthesized from pyruvate and glycelaldehyde-3-phosphate.Citation7) Extensive studies of the two pathways have revealed that archaea, yeast, and fungi only use the mevalonate pathway, whereas many bacteria and some protists mainly use the mevalonate-independent pathway.Citation8–11)
Fig. 2. Mevalonate pathway, MEP pathway, and leucine-to-mevalonate “shunt” pathway.
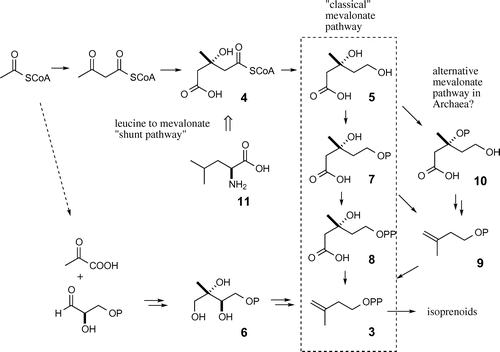
Previous labeling experiments have shown that the lipid-core of archaea is most likely biosynthesized via the mevalonate pathway.Citation12) Furthermore, HMG-CoA synthase from Haloferax volcanii has been expressed and isolated.Citation13) However, the genes of orthologs for the last two enzymes (phosphomevalonate kinase and diphosphomevalonate decarboxylase), which convert mevalonate-5-phosphate (7) to mevalonate-5-diphosphate (8) and 8 to IPP, are absent in many archaea. The alternative intermediate isopentenyl phosphate (9) from H. volcaniiCitation14) and Methanococcus jannaschiiCitation15) have been characterized. Hemmi et al.Citation16) have confirmed that mevalonate-3-phosphate (10) is formed as an intermediate by Thermoplasma acidophilum and that this material is subsequently converted to IPP. The enzymes involved in the mevalonate pathway in archaea are, therefore, different from those typically encountered in mevalonate pathways of animals and plants, but these enzymes still remain partly uncharacterized.
The relationship between branched amino acid metabolism and the mevalonate pathway is well-known. For example, it has been shown that leucine (11) is used as a substrate for the biosynthesis of isoprenoids, such as sterols and monoterpenoids, in plants,Citation17–19) animals,Citation20), and fungi.Citation21) A relationship between the shunt pathway from leucine to mevalonate has also been observed during sterol biosynthesis in some parasitic protozoa.Citation22,23) In particular, myxobacterial secondary metabolites, such as myxothiazolCitation24) and geosmin (responsible for the characteristic odor of freshly plowed soil),Citation25) are biosynthesized using leucine through the leucine-to-mevalonate shunt pathway.Citation26,27) The relationship between the life cycle of myxobacteria, such as fruiting body formation and environmental changes, and the leucine-to-mevalonate shunt pathway represents an interesting problem.
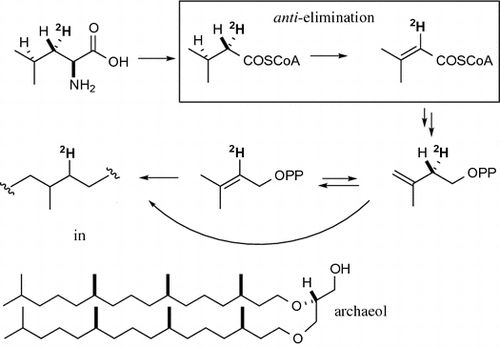
We have previously reported the stereoselective conversion of 11, which was isotopically labeled at each diasterotopic methyl group, to the hydrocarbon portions of characteristic lipid-cores in 1 and 2 via the shunt pathway for the conversion of 11 to 3.Citation28) This experiment had two important results: (i) the deuterium atoms in [Citation5,5H3]-11 were incorporated into the isoprenoidal lipid-core of archaeol and (ii) deuterium atoms in (4R)-[5-2H]leucine (12) were incorporated into archaeol efficiently (~20% monodeuterated 1 were observed compared with the non-labeled material). However, deuterium incorporation from (4S)-[5-2H]leucine was significantly low (~4% incorporation). Based on these results, it was concluded that lipid-core biosynthesis in halophilic archaea involves leucine conversion to mevalonate (Fig. ). These results also suggested that isovaleryl-CoA dehydrogenase is involved in stereospecific abstraction of a hydrogen atom in isovaleryl-CoA (13) to form 3-methylcrotonyl-CoA (14) and through the conversion from 11 to an isoprenoidal lipid-core.
Fig. 3. The result of an incorporation experiment of stereoselectively deuterated [5-2H]leucine.
![Fig. 3. The result of an incorporation experiment of stereoselectively deuterated [5-2H]leucine.](/cms/asset/fc9a41f5-8978-4d0d-bf05-6adc3fccd6b5/tbbb_a_1373588_f0003_b.gif)
Isovaleryl-CoA dehydrogenase (IVD; EC 1.3.99.10) is an enzyme involved in the conversion of 13 to 14 via an α,β-dehydrogenation step. Isovaleryl-CoA dehydrogenase deficiency in humans leads to isovaleric acidemia, which represents a congenital metabolic disorder.Citation29) This enzyme has been studied extensively using purified materials from humans, higher plants, and bacteria. Isovaleryl-CoA dehydrogenase (and its corresponding gene) is widely distributed in mammals,Citation30,31) plants,Citation32,33), and bacteria.Citation34) This enzyme belongs to the acyl-CoA dehydrogenase family, which are responsible for hydrogen transfer in flavoproteins. The stereochemical course for the enzymatic conversion of isovaleric acid has also been determined. Aberhart et al.Citation35) have demonstrated that the 2-proR hydrogen of isovaleric acid is eliminated (i.e. the 2-S hydrogen is retained) by isovaleryl-CoA dehydrogenase in biotin-deficient rats. The stereospecific elimination of the C-3 hydrogen in isovaleric acid has been confirmed using (3S)-[4-13C]isovaleric acid and purified rat liver enzyme, where the (3S)-[4-13C]isovaleric acid was converted to (3E)-[3-13C]methylcrotonyl-CoA.Citation36)
A comparison of the previous results for labeled [5-2H]leucine with that of the saturated isoprenoid in the archaeal lipid-coreCitation28) has revealed that the abstraction of a C-3 position hydrogen atom in isovaleric acid occurs with the same stereochemistry to form a diastereotopic methyl group, if isovaleryl-CoA dehydrogenase is involved in the conversion of leucine to IPP in halophilic archaea.Citation36) The corresponding acyl-CoA dehydrogenase enzymes have been observed in several archaea.Citation37) However, the purification of active forms of these enzymes from halophilic archaea remains challenging. The results from the present incorporation study of [3-2H]leucine into the archaeal lipid-core could, therefore, be used to develop a better understanding of the putative isovaleryl-CoA dehydrogenase enzyme in archaea. With this in mind, the fate of deuterium was studied following the stereoselective deuteration of leucine at its C-3 position.
Results and discussion
Synthesis of (3R)- and (3S)-[3-2H]leucine
According to Aberhart’s experiment,Citation35) it is possible to prepare (2R)- and (2S)-[2-3H]isovaleric acid from valine by the abstraction of its C-2 hydrogen atom. However, large quantities of this substrate would be required to conduct deuterium-labeling experiments in whole cell systems. Furthermore, there have been no reports in the literature pertaining to the development of preparative routes for chiral isotopic-labeling of either leucine or hydrophobic amino acids at their C-3 positions. It was, therefore, envisaged that (3R)- and (3S)-[3-2H]leucine could be prepared from other substrates. The synthesis of (3R)- and (3S)-[3-2H]leucine started from Katsuki–Sharpless chiral epoxidation of (E)-4-methyl-2-pentene-1-ol (15) was shown in Fig. . The epoxidation of 15 with a catalytic amount of diisopropyl-L-(+)-tartrate led to the enantioselective formation of the epoxide (2S,3S)-epoxy-4-methyl-1-pentanol 16.Citation38,39) The regioselective reduction of epoxide 16 with sodium bis(2-methoxyethoxy)aluminum hydride (Red-Al)Citation40) produced (3R)-4-methyl-1,3-pentanediol (17) as the sole product. The enantiomeric excess of this material was determined to be 91% by gas chromatographic analysis of the corresponding di-O-acetyl lactate derivatives.Citation41) The selective benzylation of the primary hydroxyl group in 17 gave the benzylated product 18, which was treated with p-toluenesulfonyl chloride to give the tosylate 19. The subsequent reduction of 19 with lithium aluminum deuteride occurred stereoselectively to yield the deuterated product 20, which was then converted to deuterated (3R)-[2H]leucine (21) according to a previously reported procedure.Citation42,43) The enantiomeric epoxide 22 was also prepared using diisopropyl-D-(–)-tartrate, titanium isopropoxide, and cumene hydroperoxide. This material was subsequently converted to (3R)-4-methyl-1,3-pentanediol (23) in 89% ee. Furthermore, compound 23, which is a stereoisomer of 17, was converted to 27 in the same manner. Two different isomers of C-3 deuterated leucine were then synthesized, with degrees of deuterium incorporation at 94 and 93% for the two different isomers, as measured by electrospray ionization-mass spectrographic (ESI-MS) spectroscopy. This strategy therefore, represented a convenient new route for the stereoselective deuteration of leucine at its C-3 position. This process for stereoselective isotopic-labeling could be used for structural determination of proteins by NMR.
Fig. 4. Synthesis of (3R)- and (3S)-[3-2H]leucine.
![Fig. 4. Synthesis of (3R)- and (3S)-[3-2H]leucine.](/cms/asset/b9d10e82-f49d-4cb2-96fa-096aa2fcaeb8/tbbb_a_1373588_f0004_b.gif)
Incorporation experiments
Incorporation experiments were performed according to the procedures described in our previous report.Citation28) Labeled amino acid (200 mg) was added to medium for Halobacterium salinarum, which was then inoculated with a preculture of H. salinarum and incubated at 37 °C for 7 d. No significant inhibition of growth was observed by turbidity (A650) compared with an unlabeled standard. The cells were collected and lyophilized, and the lipids were extracted. The lipid mixture was then hydrolyzed successively with acid and base, and the resulting lipid-cores purified by silica gel chromatography. The deuterium content of the lipid-core was analyzed using two different methods, including (i) direct analysis of the lipid-core by ESI-MS in a CHCl3-MeOH solution and (ii) conversion of the hydrophobic portion of the lipid-core to a hydrocarbon through an HI degradation reaction, followed by reduction of the iodide to a hydrocarbon with lithium aluminum hydride. The hydrocarbon product was subsequently analyzed by gas chromatography (GC)-MS to estimate its deuterium content, and the results for the incorporation experiments (run 1 and 2) from these two methods are shown in Tables and . The results revealed considerable levels of deuterium incorporation following cell incubations with (3S)-[3-2H]leucine. In contrast, incubation experiments involving (3R)-[3-2H]leucine resulted in much lower levels of deuterium incorporation. These results therefore, confirmed that a stereospecific and efficient conversion had been achieved.
Table 1. Incorporation of the deuterium of (3S)- and (3R)-[3-2H]leucine estimated by ESI-MS.
Table 2. Incorporation of the deuterium of (3S)- and (3R)-[3-2H]leucine estimated by GC-MS.
The experiments involving (3R)-[3-2H]leucine resulted in <2% incorporation, according to GC-MS analyses, which was within the experimental error, suggesting that this substrate most likely underwent non-specific incorporation with the complete degradation and reincorporation of the deuterium. However, deuterium incorporation observed in the current study following cell incubation with (3S)-[3-2H]leucine (~10%) was lower than that observed in our previous study, in which (4R)-[5-2H]leucine was incubated under the same conditions (~20%).Citation28) The incorporation process would have proceeded through multiple steps (involving different enzymes) to give the (saturated) isoprenoid in the lipid-core of halophilic archaea. Each step in this conversion process must, therefore, be considered to determine the fate of the hydrogen atom at the C-2 position of 3-methylcrotonyl-CoA. If the enzymes involved from leucine to IPP and DMAPP via mevalonate were similar to the leucine-to-mevalonate shunt pathway in myxobacteria,Citation26,27) the steps described below are critical. Notably, there is no evidence that leucine to IPP goes via mevalonate in archaea; reduction of 3-methylcrotonyl-CoA to dimethylallyl alcohol might be one of a simpler option.
The conversion of 3-methylglutaconic acid to HMG-CoA has been shown to proceed via the syn-addition of water by 3-methylglutaconyl-CoA hydratase (Fig. , step (A).Citation44,45) The C-2 hydrogen in 3-methylcrotonyl-CoA would then be converted to the pro-S hydrogen in IPP. The key step in isoprenoid elongation involves the intermediate IPP, which exists in equilibrium with dimethylallyl pyrophosphate (DMAPP). The well-known stereochemistry of IPP isomerase leads to isomerization of the pro-R hydrogen atom in IPP to a one hydrogen at the methyl group of DMAPP (Fig. , step (B).Citation46,47) The deuterium of (2S)-[2-2H]IPP, which would be derived from (3S)-[3-2H]leucine, retained at the C-2 of DMAPP. The chain elongation step is catalyzed by a trans-prenyltransferase (Fig. , step (C) with the loss of pro-R hydrogen atom from the C-2 position of IPP.Citation48,49) The deuterium of (2S)-[2-2H]IPP would be retained in the isoprenoidal portion. Therefore, the deuterium of [2-2H]-3-methylcrotonyl-CoA would be at least almost incorporated into the lipid-core’s isoprenoidal portion.
Fig. 5. The fate of hydrogen from (3S)-[3-2H]leucine and the stereochemical course of whole reaction series from leucine to isoprenoidal lipid-core of halophilic archaea.
![Fig. 5. The fate of hydrogen from (3S)-[3-2H]leucine and the stereochemical course of whole reaction series from leucine to isoprenoidal lipid-core of halophilic archaea.](/cms/asset/6150fe2c-89bb-4d2d-822f-e6d35f7dba94/tbbb_a_1373588_f0005_b.gif)
The concerning enzymes in this archaeon would proceed via the same stereochemical course as in known enzymes of bacterial and archaeal origin. In this course, the deuterium in (3S)-[3-2H]leucine was incorporated into the lipid-core’s isoprenoidal portion in halophilic archaea. So, the fate of deuterium in (3R)- and (3S)-[3-2H]leucine into the lipid-core would have been driven by the dehydrogenation reaction catalyzed by the putative isovaleryl-CoA dehydrogenase.
Then, these results strongly suggested the involvement of the leucine-to-mevalonate pathway, especially isovaleryl-CoA dehydrogenase, in the production of 3-methylcrotonyl-CoA. Notably, this result proceeded along the same stereochemical course as Aberhart’s experiment involving the corresponding rat enzyme.Citation35) Mechanistically, the results of this study suggested that the abstraction of a hydrogen atom from isovaleryl-CoA occurred via the anti-elimination pathway. The stereochemical course of this process was, therefore, consistent with the known acyl-CoA dehydrogenase and FAD cofactor-dehydrogenase.
Conclusion
The stereospecific conversion of leucine (stereospecifically labeled at its C-3 position) to the lipid-core of halophilic archaea was studied in detail. Taken together with our previous results, the findings of this study strongly suggested that a leucine-to-mevalonate “shunt” pathway was involved in lipid-core biosynthesis in halophilic archaea. This stereospecific conversion was attributed to a putative isovaleryl-CoA dehydrogenase, which catalyzed the anti-elimination of a hydrogen atom from isovaleryl-CoA to give methylcrotonyl-CoA. It is noteworthy that this process followed the same stereochemical course as enzymes belonging to the acyl-CoA dehydrogenase family in all other organisms.
Experimental
Infrared spectra were obtained using a PerkinElmer Spectrum One FT-IR spectrometer (PerkinElmer, Inc., Waltham, MA, USA). Samples were dissolved in CHCl3 and measured in a NaCl cell. Optical rotations were recorded on a JASCO DIP 1000 digital polarimeter (JASCO, Inc. Tokyo, Japan). 1H-NMR and 13C-NMR spectra were recorded on a JEOL ECX-500 spectrometer (JEOL Ltd., Tokyo, Japan). Tetramethylsilane and sodium 3-trimethylsilyl-1-propanate (0 ppm) were used as internal standards for 1H-NMR spectra recorded in CDCl3 and D2O, respectively. ESI-MS was recorded using an ABSciex Mariner spectrometer (SCIEX, Framingham, MA, USA). GC-MS analyses were conducted on a Shimadzu QP-5000 spectrometer (Shimadzu Corp., Kyoto, Japan). Chromatographic separations were carried out over silica gel (Merck Kieselgel 60, 70–230 mesh, Merck KGaA, Darmstadt, Germany).
Preparation of (3R)- and (3S)-[3-2H]Leucine
(2S,3S)-Epoxy-4-methyl-1-pentanol (16) was prepared according to the procedure reported by Sunazuka et al.Citation39) = −22.4 (c = 1.32), (lit. −32.5).Citation38) (2R,3R)-Epoxy-4-methyl-1-pentanol (22) was also prepared in this way using diisopropyl-D-(–)-tartrate as a reagent.
= + 24.7 (c = 4.14).
(3R)-4-Methyl-1,3-pentanediol (17)
A solution of 16 (2.81 g, 24.2 mmol) in toluene (90 mL) was cooled to −20 °C and Red-Al solution was added (3.6 M in toluene, 25 mL, 90 mmol) via a syringe under a N2 atmosphere. The resulting mixture was stirred at −20 °C for 30 min and then warmed to room temperature with stirring for 1 h. The mixture was diluted with hexane (90 mL) and slowly treated with a 3 mol/L solution of aqueous HCl. The resulting biphasic mixture was agitated for 5 min and allowed to settle. The water layer was collected and thrice extracted with 100 mL volumes of EtOAc. The combined EtOAc layers were then washed with brine, dried over Na2SO4, filtered, and evaporated to dryness to yield a residue, which was purified by chromatography over silica gel (hexane/EtOAc, 1/1 to 1/2, v/v) to give the desired product 17 (2.35 g; 82.2%). = + 10.5 (c = 2.16, CHCl3); (lit. + 13.5, CHCl3).Citation50) The enantiomeric excess of this material was evaluated based on chromatograms of the corresponding di-O-acetyl lactate derivative, which gave a value of 91% enantiomeric excess (ee).
(3R)-4-Methyl-1-O-benzyl-1,3-pentanediol (18)
To a solution of 17 (5.30 g, 44.9 mmol) in tetrahydrofuran (50 mL), n-Bu4NI (4.1 g, 11.1 mmol) was added, followed by NaH (60% oil dispersion, 1.3 g, 90.4 mmol, 2.0 eq.), and the resulting mixture stirred under an atmosphere of N2 at ambient temperature for 1 h. Benzyl bromide (5.4 ml, 7.7 g, 45.0 mmol) was added dropwise, and the resulting mixture stirred at ambient temperature for 2 h. The mixture was then quenched with water (50 mL) and stirred for 1 h before being extracted twice with 100 mL volumes of EtOAc. The organic layers were combined and washed sequentially with 1 M HCl, sat. NaHCO3, and brine before being dried over Na2SO4, filtered, and evaporated to dryness to provide a residue. Purification by column chromatography over silica gel (hexane/EtOAc, 20/1 to 4/1, v/v) produced the desired product as a liquid (8.71 g, 93.3%). = −2.70 (c = 2.43, CHCl3); 1H-NMR (500 MHz, CDCl3): δ 0.91 (3H, d, J = 6.9 Hz), 0.93 (3H, d, J = 6.9 Hz), 1.67 (1H, dq, J = 5.5 and 6.9 Hz), 1.73 (2H, q, J = 6.5 Hz), 2.37 (1H, broad, OH), 3.56 (1H, q, J = 6.0 Hz), 3.66 (1H, dt, J = 6.0 and 9.6 Hz), 3.74 (1H, dt, J = 5.5 and 9.6 Hz), 4.56 (2H, t, J = 9.5 Hz), and 7.26–7.37 (5H, m). IR (CHCl3): 3500 (broad), 3010, 2962, 2871, 1373, 1248, and 1094 cm−1. High resolution (HR)MS (ESI, positive, [M + H]+): Calcd. for 12C141H2116O2: 209.1543, Found: 209.1543.
(3R)-4-Methyl-1-O-benzyl-3-O-p-toluenesulfonyl-1,3-pentanediol (19)
TsCl (11.0 g, 58.2 mmol) was added to a solution of 18 (8.07 g, 38.8 mmol) in pyridine (50 mL) and the resulting mixture stirred under a N2 atmosphere at ambient temperature for 12 h. The reaction was quenched by the addition of water (50 mL), and the resulting mixture stirred for 1 h. The mixture was then extracted twice with 100 mL volumes of EtOAc, and the combined organics washed sequentially with 1 M HCl, sat. NaHCO3, and brine before being dried over Na2SO4, filtered, and evaporated to dryness. The resulting residue was purified by column chromatography over silica gel (hexane/EtOAc, 20/1 to 4/1, v/v) to give the desired product as a liquid (12.20 g, 87.1%). = +12.9 (c = 2.35, CHCl3); 1H-NMR (500 MHz, CDCl3): δ 0.86 (3H, d, J = 6.8 Hz), 0.87 (3H, d, J = 6.8 Hz), 1.85 (2H, dd, J = 5.5 and 6.6 Hz), 1.97 (2H, dq, J = 4.8 and 6.8 Hz), 3.35 (1H, dt, J = 6.3 and 10.0 Hz), 3.45 (1H, dt, J = 6.3 and 9.0 Hz), 4.33 (2H, t, J = 12.0 Hz), 4.68 (1H, dt, J = 6.4 and 10.0 Hz), 7.28–7.36 (5H, m), 7.29 (2H, d, J = 8.0 Hz), and 7.78 (2H, d, J = 8.0 Hz). IR (CHCl3) 3031, 3010, 2968, 2935, 2875, 1599, 1361, 1175, 1097, and 910 cm−1. HRMS (ESI, negative, [M + Cl + CH3OH]−) Calcd. for 12C211H3016O532S35Cl: 429.1494, Found: 429.1481.
(3S)-[3-2H]-4-Methyl-1-O-benzylpentanol (20)
Lithium aluminum deuteride (500 mg, 11.9 mmol) was added to a solution of 19 (5.40 g, 14.9 mmol) in tetrahydrofuran (50 mL), and the resulting mixture stirred at ambient temperature for 30 min before being refluxed for 3 h. The mixture was then cooled to ambient temperature and slowly treated with sat. potassium sodium tartrate solution (20 mL). The mixture was extracted twice with 50 mL volumes of EtOAc, and the combined extracts washed with brine, dried over Na2SO4, filtered, and evaporated to dryness to produce a residue. Purification by column chromatography over silica gel (hexane/EtOAc, 40/1 to 10/1, v/v) gave the desired product as a liquid (2.53 g, 89.0%). 1H-NMR (500 MHz, CDCl3): δ 0.88 (3H, d, J = 6.2 Hz), 1.22 (1H, broad q, J = 6.6 Hz), 1.50–1.64 (3H, m), 3.44 (2H, t, J = 6.8 Hz), 4.50 (2H, s), and 7.26–7.35 (5H, m). IR (CHCl3): 3010, 2957, 2869, 1466, 1454, 1094, and 910 cm−1. Ex. Anal., Calcd for C13H192HO: C 80.77, H(+2H) 10.41, Found: C 80.74, H 10.11.
(3R)-[3-2H]Leucine 21
This material was prepared according to a previously reported method,Citation45) as well as the method reported by Hill,Citation44) with minor modifications. Iodotrimethylsilane (2.8 g, 14 mmol) was added dropwise to a solution of compound 20 (2.53 g, 13.2 mol) in CH2Cl2 (15 mL), and the resulting mixture stirred for 1 h at ambient temperature. The solvent was removed to yield a residue, which was purified by column chromatography over silica gel (hexane/EtOAc, 1/1, v/v) to obtain the alcohol product (893 mg). Without further purification, the alcohol was subjected to a Jones oxidation in acetone to produce the corresponding carboxylic acid, which was brominated with Br2 in the presence of small pieces of I2 and 1 drop of PCl3 to provide the α-brominated carboxylate. This material was dissolved in 28% aqueous ammonium hydroxide solution (20 mL) and held at ambient temperature for 9 d. The mixture was subsequently evaporated to dryness to obtain a residue, which was passed through an Amberlite IRA 120 (H+) resin. The resulting leucine hydrochloride was eluted with 1 M HCl and the eluent evaporated to dryness to produce a residue, which was passed through Amberlite IRA 410 (HCOO−) resin to yield free leucine (845 mg, 48.5% for 4 steps). 1H-NMR (500 MHz, D2O): δ 0.95 (6H, t, J = 6.5 Hz), 1.65 (1H, m), 1.68 (1H, m), 3.69 (1H, d, J = 8.0 Hz). 13C-NMR (125.7 MHz, D2O): δ 21.87, 23.03, 25.08, 40.44 (t, J = 21 Hz), 54.38, and 176.61. HRMS (ESI, positive, [M + H]+) Calcd. for 12C61H1314N116O22H1: 133.1088, Found: 133.1088, Relative int. 132.1 (6.02), 133.1 (100).
(3S)-4-Methyl-1,3-pentanediol (23)
This compound was prepared according to the same procedure as that for preparation of 16. Compound 22 (5.04 g, 43.4 mmol) was selectively reduced with Red-Al (3.6 mol/L in toluene, 12 mL) to give the desired product as a liquid (5.00 g, 97.5%). = −10.5 (c = 3.84, CHCl3); (lit. + 13.5, CHCl3 antipode).Citation50) The enantiomeric excess was evaluated by GCMS analysis of the corresponding di-O-acetyl lactate derivative and determined to be 89% ee.
(3S)-4-Methyl-1-O-benzyl-1,3-pentanediol (24)
This compound was prepared according to the same procedure as that used for preparation of 18. The selective benzylation of 23 (5.14 g, 43.6 mmol) with benzyl bromide (5.2 ml, 7.5 g, 44.0 mmol) yielded the desired product as a liquid (8.41 g, 91.9%). = +2.63 (c = 2.78, CHCl3). HRMS (ESI, positive, [M + H]+): Calcd. for 12C141H2116O2: 209.1543, Found: 209.1530.
(3S)-4-Methyl-1-O-benzyl-3-O-p-toluenesulfonyl-1,3-pentanediol (24)
This compound was prepared according to the same procedure as that used for preparation of 19. Compound 24 (793 mg, 3.8 mmol) was tosylated with p-toluenesulfonyl chloride (2.17 g, 11.4 mmol, 3 eq.) in pyridine (4 mL) to give the desired product as a liquid (1.14 g, 82.6%). = −18.9 (c = 3.20, CHCl3). HRMS (ESI, positive, [M + H]+): Calcd. for 12C201H2616O432S1: 363.1631, Found: 363.1643.
(3R)-[3-2H]-4-Methyl-1-O-benzylpentanol (26)
This compound was prepared according to the same procedure as that used for preparation of 20. Compound 25 (12.0 g, 33 mmol) was reduced with lithium aluminum deuteride (1.07 g, 25 mmol) in tetrahydrofuran (THF) to give the desired product as a liquid (5.45 g, 86.7%). Ex. Anal., Calcd for C13H192HO: C 80.77, H(+2H) 10.41, Found: C 80.95, H 10.13.
(3S)-[3-2H]Leucine (27)
This compound was prepared according to the same procedure as that used for preparation of 21. Compound 26 (2.31 g, 11.9 mmol) was used as a substrate, affording the desired product as a white solid (1.07 g, 68.1%). HRMS (ESI, positive, [M + H]+) Calcd. for 12C61H1314N116O22H1:133.1088, Found: 133.1093. Relative intensities, 132.1 (7.30) and 133.1 (100).
Archaeal culture and lipid extraction
This procedure was conducted in a similar manner to that of a previously reported procedure.Citation28) H. salinarum (JCM 9210) was obtained from the RIKEN BioResource Center, Ibarabki, Japan. This material was cultivated at 37 °C with shaking for 7 d in 100 mL of medium containing casamino acids (0.75 g), yeast extract (1.0 g), sodium citrate (0.25 g), NaCl (25 g), KCl (0.2 g), MgSO4·7H2O (2.0 g), and labeled leucine (200 mg) at pH 7.4. The cells were then harvested by centrifugation (3000 × g, 15 min), washed with water, and lyophilized. The lipids were extracted according to the method by Bligh and Dyer,Citation51,52) and total lipids hydrolyzed directly (no acetone precipitation) in 10 mL of 3% MeOH-water under reflux for 3 h. The mixture was then treated with 2 mL of a 7 M NaOH solution and the resulting mixture heated at reflux for 6 h. The lipid-cores were thrice extracted with 10 mL volumes of hexane. The combined hexane extracts were dried over Na2SO4, filtered, and concentrated to dryness. The resulting residue was purified by column chromatography over silica gel eluted with hexane, followed by hexane/EtOAc (10/1, v/v), with core lipid eluted in the latter solvent (typically 0.9 mg). Half of the purified lipid-core was treated with concentrated HI (2 mL) and the resulting mixture heated at reflux for 6 h. The mixture was then treated with water and the organic layer extracted with hexane. The combined hexane extracts were dried and evaporated to yield a residue. The residue was treated with THF (3 mL) and LiAlH4 (50 mL), and the resulting mixture heated to 80 °C (bath temp.) with stirring for 2 h. The reaction mixture was quenched by the addition of a small amount of water, and the hydrocarbon extracted with diethyl ether. The purified lipid-core was dissolved in CHCl3/methanol (1/3, v/v) at a concentration of 0.1 mg/mL and analyzed by ESI-MS (negative ion mode, nozzle temp. 140 °C, nozzle potential 90 V, and spray tip potential 3000 V). The hydrocarbon fraction containing phytane (C20H42) was analyzed directly by GC-MS (30 m InertCap 5, OV-5 equivalent; He 50 mL/min; injector at 260 °C, column maintained for 3 min at 120 °C, and then increased to 280 °C at 5 °C/min).Citation30)
Author Contributions
N.Y. conceived and designed the study. N.Y. performed the synthesis of labeled leucine. R.T. performed archaeal culture, lipid extraction, analysis of labeled lipid-core, and data analysis. N. Y. performed the incubation experiments involving (3R)-[3-2H]leucine in part. The results were well discussed with the two authors. The draft manuscript was written by R.T and N.Y. and N.Y reviewed and revised the final manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Acknowledgments
NMR measurements were conducted as the Centre of Advanced Instrumental Analysis, Kyushu University. Elemental analyses were measured at the Service Centre for the Elementary Analysis of Organic Compounds, Faculty of sciences, Kyushu University. We are also grateful to two anonymous reviewers for valuable and thoughtful review comments, which are very much helpful for improving the manuscript.
References
- Kates M. Membrane-Lipids of extreme halophiles–biosynthesis, function and evolutionary significance. Experientia. 1993;49(12):1027–1036.10.1007/BF01929909
- Kates M. Diether and tetraether phospholipids and glycolipids as molecular markers for archaebacteria (archaea). In: Eganhouse RP, editor. ACS symposium series 671 molecular markers in environmental geochemistry. Washington (DC): American Chemical Society; 1977. p. 35–51.
- Foster IS, King PL, Hyde BC, et al. Characterization of halophiles in natural MgSO4 salts and laboratory enrichment samples: astrobiological implications for Mars. Planet Space Sci. 2010;58(4):599–615.10.1016/j.pss.2009.08.009
- Fendrihan S, Musso M, Stan-Lotter H. Raman spectroscopy as a potential method for the detection of extremely halophilic archaea embedded in halite in terrestrial and possibly extra-terrestrial samples. J Raman Spectrosc. 2009;40(12):1996–2003.10.1002/jrs.v40:12
- Morita M, Yamauchi N, Eguchi T, et al. Structural diversity of the membrane core lipid of extreme halophiles. Biosci Biotechnol Bioch. 1998;62(3):596–598.10.1271/bbb.62.596
- Kamekura M, Kates M. Structural diversity of membrane lipids in members of Halobacteriaceae. Biosci Biotechnol Bioch. 1999;63(6):969–972.10.1271/bbb.63.969
- Fresch G, Rohmer M. Prokaryotic hopanoids: the biosynthesis of the bacteriohopane skeleton. Formation of isoprenic units from two distinct acetate pools and a novel type of carbon/carbon linkage between a triterpene and D-ribose. Eur J Biochem. 1988;175(2):405–411.
- Bach TJ. Some new aspects of isoprenoid biosynthesis in plants—A review. Lipids. 1995;30(3):191–202.10.1007/BF02537822
- Rohmer M. Mevalonate-independent methylerythritol phosphate pathway for isoprenoid biosynthesis. Elucidation and distribution. Pure Appl Chem. 2003;75(2–3):375–387.
- Kuzuyama T, Seto H. Diversity of the biosynthesis of the isoprene units. Nat Prod Rep. 2003;20(2):171–183.10.1039/b109860 h
- Kuzuyama T, Hemmi H, Takahashi S. Mevalonate pathway in bacteria and archaea. In: Townsend CA, Ebizuka Y, editors. Comprehensive natural products II. Chemistry and biology. Vol. 1, Natural products structural diversity-I. Secondary metabolites; Organization and biosynthesis. Oxford:Elsevier; 2010. p. 493–516.10.1016/B978-008045382-8.00014-9
- De Rosa M, Gambacorta A, Gliozzi A. Structure, biosynthesis, and physicochemical properties of archaebacterial lipids. Microbiol Rev. 1986;50(1):70–80.
- VanNice JC, Skaff DA, Wyckoff GJ, et al. Expression in Haloferax volcanii of 3-hydroxy-3-methylglutaryl coenzyme A synthase facilitates isolation and characterization of the active form of a key enzyme required for polyisoprenoid cell membrane biosynthesis in halophilic archaea. J Bacteriol. 2013;195(17):3854–3862.10.1128/JB.00485-13
- VanNice JC, Skaff DA, Keightley A, et al. Identification in Haloferax volcanii of phosphomevalonate decarboxylase and isopentenyl phosphate kinase as catalysts of terminal enzyme reactions in an archaeal alternate mevalonate pathway. J Bacteriol. 2014;196(5):1055–1063.10.1128/JB.01230-13
- Grochowski LL, Xu H, White RH. Methanocaldococcus jannaschii uses a modified mevalonate pathway for biosynthesis of isopentenyl diphosphate. J Bacteriol. 2006;188(9):3192–3198.10.1128/JB.188.9.3192-3198.2006
- Azami Y, Hattori A, Nishimura H, et al. (R)-Mevalonate 3-phosphate is an intermediate of the mevalonate pathway in Thermoplasma acidophilum. J Biol Chem. 2014;289(23):15957–15967.10.1074/jbc.M114.562686
- Suga T, Hirata T, Shishibori T, et al. The first proof of the biosynthesis of isoprenoid from amino acid in higher plant. The incorporation of L-Leucine into linalool. Chem Lett. 1974;3(2):189–192.10.1246/cl.1974.189
- Suga T, Hirata T, Tange K. Biosynthesis of isoprenoid from amino acid in higher plant. Incorporation of L-leucine and L-valine into geraniol and citronellol. Chem Lett. 1975;4(3):243–246.
- Anastasis P, Freer I, Overton K, et al. The role of leucine in isoprenoid metabolism-incorporation of [3-13C]leucine and of [2-3H,4-14C]-β,β-dimethylacrylic acid into phytosterols by tissue-cultures of Andrographis paniculata. Chem Commun. 1985;(3):148–149.10.1039/C39850000148
- Stillway LW, Weigand DA, Riefler JF. Leucine and isoleucine as in vitro precursors for lipid biosynthesis by rat aorta. Lipids. 1977;12:1012–1016.10.1007/BF02533327
- Domenech CE, Giordano W, Avalos J, et al. Separate compartments for the production of sterols, carotenoids and gibberellins in Gibberella fujikuroi. Eur J Biochem. 1996;239(3):720–725.10.1111/ejb.1996.239.issue-3
- Ginger ML, Prescott MC, Reynolds DG, et al. Utilization of leucine and acetate as carbon source for sterol and fatty acid biosynthesis by old and new world Leishmania species, Endotrypanum monterogeii and Trypanosoma cruzi. Eur J Biochem. 2000;267(9):2555–2566.10.1046/j.1432-1327.2000.01261.x
- Ginger ML, Chance ML, Sadler IH, et al. The biosynthetic incorporation of the intact leucine skeleton into sterol by the Trypanosomatid Leishmania mexicana. J Biol Chem. 2001;276(15):11674–11682.10.1074/jbc.M006850200
- Mahmud T, Bode HB, Silakowski B, et al. A novel biosynthetic pathway providing precursors for fatty acid biosynthesis and secondary metabolite formation in myxobacteria. J Biol Chem. 2002;277(36):32768–32774.10.1074/jbc.M205222200
- Dickschat JS, Bode HB, Mahmud T, et al. A novel type of geosmin biosynthesis in myxobacteria. J Org Chem. 2005;70(13):5174–5182.10.1021/jo050449 g
- Bode HB, Ring MW, Schwar G, et al. 3-Hydroxy-3-methylglutaryl-coenzyme A (CoA) synthase in involved in biosynthesis of isovaleryl-CoA in the Myxobacterium Myxococcus xanthus during fruiting body formation. J Bacteriol. 2006;188(18):6524–6528.10.1128/JB.00825-06
- Li Y, Luxenburger E, et al. An alternative isovaleryl CoA biosynthetic pathway involving a previously unknown 3-methylglutaconyl CoA decarboxylase. Angew Chem Int Ed. 2013;52(4):1304–1308.10.1002/anie.201207984
- Yamauchi N. The pathway of leucine to mevalonate in halophilic archaea: efficient incorporation of leucine into isoprenoidal lipid with the involvement of isovaleryl-CoA dehydrogenase in Halobacterium salinarum. Biosci Biotech Bioch. 2010;74(2):443–446.10.1271/bbb.90814
- Ikeda Y, Tanaka K. Mutant isovaleryl-CoA dehydrogenase in isovaleric acidemia cells: assay of activity and molecular characterization. Methods Enzymol. 1988;166:155–166.10.1016/S0076-6879(88)66022-8
- Ikeda Y, Tanaka K. Purification and characterization of isovaleryl coenzyme A dehydrogenase from rat liver mitochondria. J Biol Chem. 1983;258:1077–1085.
- Willard JM, Reinard T, Mohsen AW, et al. Cloning of genomic and cDNA for mouse isovaleryl-CoA dehydrogenase (IVD) and evolutionary comparison to other known IVDs. Gene. 2001;270(1–2):253–257.10.1016/S0378-1119(01)00466-8
- Reinard T, Janke V, Willard J, et al. Cloning of a gene for an acyl-CoA dehydrogenase from Pisum sativum L. and purification and characterization of its product as an isovaleryl-CoA dehydrogenase. J Biol Chem. 2000;275(43):33738–33743.10.1074/jbc.M004178200
- Faivre-Nitschke SE, Couée I, Vermel M, et al. Purification, characterization and cloning of isovaleryl-CoA dehydrogenase from higher plant mitochondria. Eur J Biochem. 2001;268(5):1332–1339.10.1046/j.1432-1327.2001.01999.x
- Förster-Fromme K, Jendrossek D. Biochemical characterization of isovaleryl-CoA dehydrogenase (LiuA) of Pseudomonas aeruginosa and the importance of liu genes for a functional catabolic pathway of methyl-branched compounds. FEMS Microbiol Lett. 2008;286(1):78–84.10.1111/fml.2008.286.issue-1
- Aberhart DJ, Tann C-H. Substrate stereochemistry of isovaleryl-CoA dehydrogenase elimination of the 2-pro-R hydrogen in biotin-deficient rats. Bioorg Chem. 1981;10(2):200–205.10.1016/0045-2068(81)90023-7
- Aberhart DJ, Finocchiaro G, Ikeda Y, et al. Substrate stereochemistry of isovaleryl-CoA dehydrogenase. II. Steric course of C-3 hydrogen elimination. Bioorg Chem. 1986;14(2):170–175.10.1016/0045-2068(86)90027-1
- Swigoňová Z, Mohsen A-W, Vockley J. Acyl-CoA dehydrogenases: dynamic history of protein family evolution. J Mol Evol. 2009;69(2):176–193.10.1007/s00239-009-9263-0
- Caldwell CG, Bondy SS. A convenient synthesis of enantiomerically pure (2S, 3S)- or (2R,3R)-3-hydroxyleucine. Synthesis. 1990;1990(1):34–36.
- Sunazuka T, Nagamitsu T, Tanaka H, et al. An efficient asymmetric synthesis of the four stereoisomers of 3-hydroxyleucine. Tetrahedron Lett. 1993;34(28):4447–4448.10.1016/0040-4039(93)88055-N
- Ishikawa H, Sone H, Kigoshi H, et al. Enantioselective total synthesis of doliculide, a potent cytotoxic cyclodepsipeptide of marine origin and structure-cytotoxicity relationships of synthetic doliculide congeners. Tetrahedron. 1994;50(45):12853–12882.
- Modandi A, Gessner M, Günther C, et al. (S)-O-Acetyllactyl chloride – a versatile chiral auxiliary in stereodifferentiation of enantiomeric flavor components. J. High Resol Chrom. 1987;10:67–70.
- Hill RK, Abächerli C, Hagishita S. Synthesis of (2S, 4S)- and (2S, 4R)-[5,5,5-2H3]leucine from (R)-pulegone. Can J Chem. 1993;72:110–113.
- Yamauchi N, Endoh S. Improved isotopic deuterium labeling at the diastereotopic methyl group of leucine: a synthetic route to (4S)-and (4R)-[5-2H1]leucine. Biosci Biotech Bioch. 2006;70(1):276–278.10.1271/bbb.70.276
- Messner B, Eggerer H, Cornforth JW, et al. Substrate stereochemistry of the hydroxymethylglutaryl-CoA lyase and methylglutaconyl-CoA hydratase reactions. Eur J Biochem. 1975;53(1):255–264.10.1111/ejb.1975.53.issue-1
- Mack M, Liesert M, Zschocke J, et al. 3-Methylglutaconyl-CoA hydratase from Acinetobacter sp. Arch Microbiol. 2006;185(4):297–306.10.1007/s00203-006-0095-7
- Barkley SJ, Desai SB, Poulter CD. Proton exchange in type II isopentenyl diphosphate isomerase. Org Lett. 2004;6(26):5019–5021.10.1021/ol0477273
- Kao C, Kittleman W, Zhang H, et al. Stereochemical analysis of isopentenyl diphosphate isomerase type II from Staphylococcus aureus using chemically synthesized (S)- and (R)-[2-2H]isopentenyl diphosphates. Org Lett. 2005;7(25):5677–5680.10.1021/ol0524050
- Cornforth JW, Cornforth RH, Donninger C, et al. Studies on the biosynthesis of cholesterol. XIX. Steric course of hydrogen eliminations and of C-C bond formations in squalene biosynthesis. Proc R Soc Lond B Biol Sci. 1966;163:492–514.10.1098/rspb.1966.0004
- Cornforth JW, Cornforth RH, Popjác G, et al. Studies on the biosynthesis of cholesterol XX. Steric course of decarboxylation of 5-pyrophosphomevalonate and of the carbon to carbon bond formation in the biosynthesis of farnesyl pyrophosphate. J Biol Chem. 1966;241(17):3970–3987.
- Clark JK, Jones PS, Palin R, et al. Asymmetric synthesis of N-3 substituted phenoxypropyl piperidine benzimidazol-2-one derivatives, potent and selective NOP agonists. Tetrahedron. 2008;64(14):3119–3126.10.1016/j.tet.2008.01.125
- Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can. J Biochem Physiol. 1959;37(8):911–917.10.1139/o59-099
- Tornabene TG, Kates M, Gelpi E, et al. Occurrence of squalene, di- and tetrahydrosqualenes, and vitamin MK8 in an extremely halophilic bacterium. Halobacterium cutirubrum. J Lipid Res. 1969;10(3):294–303.