
ABSTRACT
Hot water extraction of D-arabinofuranosylvitexin from the raw leaves of commercially available Basella alba “Tsurumurasaki” and subsequent acidic hydrolysis was improved to be a procedure using a high-pressure steam sterilizer to afford vitexin. The amount was estimated to be 14.1 mg from 1 g of dry weight of the raw leaves, whose recovery was calculated to be 95% based on the estimated content of D-arabinofuranosylvitexin in B. alba raw leaves. The product was dehydratively cyclized between hydroxy groups on the carbohydrate and flavone skeletons under modified Mitsunobu reaction conditions in N,N-dimethylformamide to give chafuroside B, which is known to be a bioactive Oolong tea polyphenol. Through these transformations, 10.2 mg of chafuroside B could be semisynthesized from 1 g of dry weight of the raw leaves, and the efficiency was improved compared to that from the extraction from Oolong tea (3.4 μg from 1 g of dry weight).
GRAPHICAL ABSTRACT
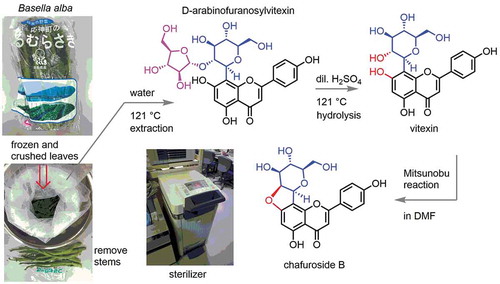
Chafuroside B, a bioactive C-glycosylated flavonoid was semisynthesized from hot water extract of Basella alba, a commercially available vegetable “Tsurumurasaki,” via vitexin.
Chafuroside B (1a, ) is a C-glycosylated flavone isolated from Oolong tea leaves, together with its regioisomer, chafuroside A [Citation1]. Although the anti-inflammatory action of 1a was less effective than that of chafuroside A, chafuroside B is promising as a photoprotective agent for skin damage induced by UV-B irradiation [Citation2]. A precursor, vitexin 2”-sulfate (2a, ) was also isolated from the same source. The cyclization with the inversion of the stereochemistry at C-2” position affords 1a. The contents of 1a (29.4 ng from 1 g of dry weight) as well as that of 2a in Oolong tea leaves are low [Citation3]. Even by an improved manufacturing procedure, only 3.4 μg of 1a could be prepared from 1 g of dry weight of Oolong tea leaves [Citation4]. To overcome such supply problems, the chemical synthesis of 1a was accomplished using vitexin (2b, ) as the precursor [Citation5] by a Mitsunobu reaction, which was synthesized through O-C glycoside rearrangement of a phloroacetophenone derivative 3 into 4 [Citation5,Citation6], the A-ring precursor of the flavone skeleton in 4–5 steps (Scheme 1).
Figure 1. Structures of C-glycosylated flavones, such as chafurosides A and B (1a) and vitexins (2ab).
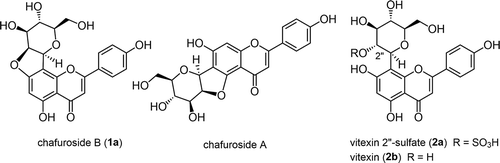
Scheme 1. So-far reported the chemical synthesis of chafuroside B (1a) [Citation5] via vitexin (2b) [Citation5,Citation6] as the key precursor.
![Scheme 1. So-far reported the chemical synthesis of chafuroside B (1a) [Citation5] via vitexin (2b) [Citation5,Citation6] as the key precursor.](/cms/asset/3ac98270-dc6b-44db-b190-5ce35a69f690/tbbb_a_1761286_sch0001_b.gif)
Based on the conversion from 2b to 1a, the large supply of 2b from natural resources facilitates the preparation of 1a. Vitexin was first isolated as a pigment in dyewood, Vitex littoralis [Citation7], and was later found in many plants, although to a lesser extent [Citation8–Citation11]. Although the most promising resource was Crataegus pinnatifida [Citation8], the concentration of 2b was estimated to be only 220 μg in 1 g of wet weight.
In 2018, the analysis of 2b and the additionally glycosylated form, D-arabinofuranosylvitexin (5, Scheme 2) from leaves of Basella alba (Malabar spinach, “Tsurumurasaki” in Japanese) as well as the hydrolysis of 5 to 2b with hydrochloric acid was reported [Citation12]. Furthermore, the preparative isolation of 5 was intensively elaborated [Citation13]. This plant is widely cultivated and available as a vegetable mainly in Asian countries, including Japan. Arabinofuranosylated form 5 seemed attractive as the direct precursor for 2b by hydrolytic removal of D-arabinose. However, the reported procedure took operations including successive extraction with alcoholic solvents and water, chromatographic separations, and freeze drying to obtain the enriched material of 5 [Citation13]. Preparative conversion from 5 to 2b was not established, either. Here, we report the improved preparation of vitexin (2b) by hot water extraction of 5 from the raw leaves of Basella alba and subsequent acidic hydrolysis using a high-pressure steam sterilizer. The semisynthesis of chafuroside B (1a) from 2b was also achieved by modified Mitsunobu reaction conditions.
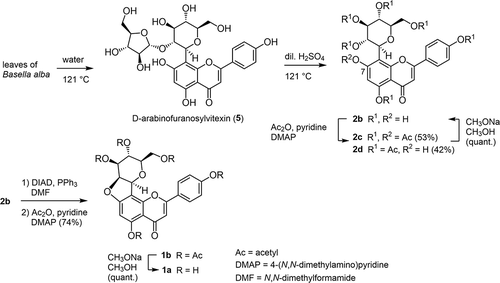
Scheme 2. Semisynthesis of chafuroside B (1a) via vitexin (2b), from hot water extract of Basella alba leaves.
Results and discussion
The “sticky” property of the aqueous exudate from this vegetable while boiling is characteristic of the favorable taste of “Tsurumurasaki” as food. To avoid vigorous foaming of the resulting solution through suction filtration and evaporation conditions under reduced pressure, alcoholic solvents have been used for the extraction of 5 in the previous reports [Citation12,Citation13]. Our purpose was not the isolation of 5 itself, but the preparation of 2b by the subsequent hydrolysis of the D-arabinose moiety in 5. Since an elevated temperature was desirable to extract both 2b and 5 from B. alba [Citation13], we chose hot water as the solvent for both the extraction and subsequent acidic hydrolysis in a high-pressure steam sterilizer at 121°C as shown in Scheme 2. In our case, instead of suction filtration to separate the water extract from the vegetable, the heated vegetable was squeezed while hot, to avoid the formation of bubbles under reduced pressure.
Next, the aqueous extract containing mainly 5 was treated in dilute sulfuric acid at 121°C again in a high-pressure steam sterilizer [Citation14–Citation16], to merge the mixture of 2b and 5 into 2b by acidic hydrolysis as shown in Scheme 2. After cooling, ammonium sulfate was dissolved in the solution to decrease the solubility of the resulting 2b. Upon standing in a refrigerator overnight, brown precipitates were easily recovered by suction filtration. After acetylation of the crude product and chromatographic purification, however, the physicochemical [Citation17] and spectral properties [Citation18] did not coincide with those reported previously. It was revealed by 1H-NMR spectroscopy that our partially purified product was a mixture of heptaacetate 2c and hexaacetate 2d in a 56:44 ratio.
For the confirmation of the structures, further separation of the above-mentioned mixture with preparative thin-layer chromatography furnished each component. The 1H-NMR spectrum of 2c in dimethyl sulfoxide (DMSO-d6) at room temperature suggested that 2c exists as two rotational isomers. Each signal collapsed to a unified broad one at an elevated temperature of 90°C (see Experimental and the spectra in supplementary materials). The existence of such rotational isomers in acetylated [Citation19] and free [Citation20] C-glycosylated flavonoids has been previously observed.
The unacetylated position in 2d was elucidated as follows. The chemical shifts of protons in the carbohydrate moieties showed no significant difference between 2c and 2d. Both the aromatic protons of H-3ʹ in 2c (δ 7.35 and 7.39 in CDCl3) and 2d (δ 7.34 in CDCl3) were downfield, which is a characteristic feature of C-4ʹ acetates. In contrast, the upfield chemical shift of H-6 in 2d (δ 6.55) compared to those in 2c (δ 6.79 and 6.88), as well as the observation of a free broadened signal of the proton on the hydroxy group (δ 8.4), indicated that the hydroxy group at C-7 was not acetylated. Generally, the reactivity of a hydroxy group at C-5 is lower toward acetylation due to hydrogen bonding with the carbonyl group at C-4. However, as shown above, the steric hindrance at the C-7 position was very high, and the reactivity at this position might lower. The absence of a hydrogen-bonded proton (δ ca. 12) in the 1H-NMR spectrum of 2d supported the presence of the acetyl group on the hydroxy group at C-5.
From 500 g of raw leaves of “Tsurumurasaki,” a total of 695 mg of 2c and 2d were obtained. Based on the previously reported content of 5 in raw leaves of “Tsurumurasaki” (1.17 mg/g) [Citation13], the recovery yields of 2c and 2d were roughly estimated to be 53% and 42%, respectively. No difference was observed based on the prefectures where the vegetables were cultivated, such as Tokushima, Fukushima, Miyagi, and Akita. We also attempted to prepare 2b from the stems of “Tsurumurasaki” after removing the leaves. The 1H-NMR spectra of the crude products after acetylation, however, only showed upfield signals, and signals attributable to either 2c or 2d were not detected.
The mixture of 2c and 2d was deacetylated under Zemplén conditions [Citation21] to give 2b in quantitative yield as shown in Scheme 2. In this way, 848 μg of 2b could be prepared from 1 g of B. alba raw leaves. Since 94% of the initial weight of B. alba raw leaves was lost by freeze drying of these leaves, the prepared amount was estimated to be 14.1 mg from 1 g of dry weight.
With the sufficient amount of 2b in hand for the semisynthesis of chafuroside B (1a), dehydrative cyclization of 2b under Mitsunobu reaction conditions was attempted, by applying tetrahydrofuran as the solvent, according to the reported procedure [Citation5]. In our hands, however, the solubility of the substrate 2b was quite low in the reaction mixture, and only a miserable yield was observed. To solve this problem, the increased solubility of the substrate 2b in N,N-dimethylformamide [Citation22] was favorable to give the desired product in an enhanced yield. The desired product 1b was purified after acetylation and obtained in 74% yield. Finally, deacetylation yielded the desired 1a in quantitative yield as shown in Scheme 2. Presently synthesized amount of 1a was estimated to be 10.2 mg from 1 g of dry weight of B. alba leaves.
Conclusion
An efficient improved procedure for the preparation of vitexin (2b) by hot water extraction of D-arabinofuranosylvitexin (5) from the commercially available vegetable “Tsurumurasaki” and subsequent acidic hydrolysis was developed. The key steps were the successive treatments for extraction and acidic hydrolysis of the materials from a vegetable, in a high-pressure steam sterilizer. In this procedure, the prepared amount of 2b was estimated to be 14.1 mg from 1 g of dry weight of the leaves. Chafuroside B was semisynthesized from 2b under modified Mitsunobu reaction conditions. Presently synthesized amount was estimated to be 10.2 mg from 1 g of dry weight of the leaves. The efficiency was higher, compared to the amount manufactured from Oolong tea (3.4 μg from 1 g of dry weight).
Experimental
General
Basella alba “Tsurumurasaki” was purchased in greengrocers in domestic supermarket stores. Diisopropyl azodicarboxylate (DIAD, A1246) was purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). Medium-pressure column chromatography was performed on Biotage IsoleraTM One flash chromatography system using Biotage SNAP Ultra silica gel cartridge FSUL-0442 (spherical and neutral; 25 μm). TLC analysis was performed using Merck TLC Silica gel 60 F254. Preparative TLC was performed with Merck Silica gel 60 F254 plates (0.5 mm thickness, No. 1.05744.0001). 1H NMR spectra were measured at 400 MHz on a VARIAN 400-MR spectrometer or at 500 MHz on a Bruker spectrometer (ascend TM), and 13C NMR spectra were measured at 100 MHz on a VARIAN 400-MR spectrometer or 125 MHz on a Bruker spectrometer (ascend TM). DMSO-d6, CD3OD, and CDCl3 were used as solvents and the residual peaks were used as internal standards (1H NMR: DMSO-d6 2.48, CD3OD 3.31, CDCl3 7.26 ppm; 13C NMR: DMSO-d6 39.9, CD3OD 49.0, CDCl3 77.0 ppm). IR spectra were measured as ATR on a Jasco FT/IR-4700 spectrometer. Optical rotation values were measured with a Jasco P-1010 polarimeter. High-resolution mass spectra were recorded on JEOL JMS-T100LP AccuTOF. TOMY LSX-500 high-pressure steam sterilizer was used for extraction and acidic hydrolysis at 121°C.
4ʹ,5,7-Triacetoxy-8-(2,3,4,6-tetra-O-acetoxy-β-D-glucopyranosyl)-flavone (2c) and 4ʹ,5-Diacetoxy-8-(2,3,4,6-tetra-O-acetoxy-β-D-glucopyranosyl)-7-hydroxyflavone (2d)
Raw leaves of B. alba (ca. 200 g) were frozen and the stems were removed. The crushed leaves were wrapped doubly with nonwoven thin porous films made of polyester fabric and put in 1-L beaker. The materials in the beaker were heated in high-pressure steam sterilizer for 1 h at 121°C. After cooling to 90°C, the reaction vessel was taken out and the hot leaves were squeezed by hands, with a silicone-rubber thermal insulator. The residual leaves were soaked with water (50 mL) and the mixture was heated by microwave oven (700 W) for 2 min. Then, the leaves were squeezed again while hot. To the combined liquid extracts (ca. 200 mL) was added sulfuric acid (1 mL) and the mixture was heated in high-pressure steam sterilizer again for 2 h at 121°C. After cooling to 50°C, the reaction vessel was taken out, and ammonium sulfate (60 g) was dissolved in the mixture. The beaker was left to stand in a refrigerator overnight at 4°C. The brown precipitates were collected by filtration and washed with cold water (10 mL) and then air-dried overnight.
According to the above-mentioned procedure, the precipitates (943 mg) were obtained from leaves of B. alba (ca. 500 g) and suspended in pyridine (10 mL). To the mixture were added acetic anhydride (10 mL) and catalytic amount of 4-(N,N-dimethylamino)pyridine (DMAP, 4.2 mg, 34 μmol), and the mixture was stirred for 24 h at room temperature. The mixture was poured into ice. The precipitates were collected by filtration and dried in vacuo. The residue was purified by medium-pressure column chromatography (100 g cartridge, FSUL-0442-0100). Elution with hexane-ethyl acetate (6:1 to 0:1) furnished a mixture of 2c and 2d as yellow amorphous solid (695 mg, 2c:2d = 56:44, 95%). The ratio between 2c and 2d was estimated by the comparison of the area of signals in 1H NMR (CDCl3) [δ: 6.79 (s) and 6.88 (s) for 2c, and δ: 6.55 (s) for 2d]. The recovery yield was calculated, D-arabinofuranosyl vitexin which was so far estimated in B. alba raw leaves (1.17 mg/g) [Citation13]. The structures of 2c and 2d were determined after a small amount of the mixture was further purified by preparative TLC developed with hexane-ethyl acetate (2:1) to furnish acetates 2c and 2d.
2c: Rf value: 0.53 [developed with chloroform-ethyl acetate (1:3)]. 1H-NMR (a 2:3 mixture of rotational isomers, 400 MHz, CDCl3) δ: 1.69 (1.2H, s), 1.74 (1.8H, s), 1.90 (1.8H, s), 1.99 (1,8H, s), 2.04 (1.2H, s), 2.06 (1.2H, s), 2.07 (1.2H, s), 2.10 (1.8H, s), 2.35 (1.8H, s), 2.37 (1.2H, s), 2.42 (4.8H, s), 2.44 (1.2H, s), 3.84–3.92 (1H, m), 4.00 (0.4H, br), 4.19 (0.6H, dd, J = 2.2 Hz, 12.5 Hz), 4.29 (0.6H, dd, J = 4.1 Hz, 12.5 Hz), 4.44 (0.4H, dd, J = 4.1 Hz, 12.5 Hz), 4.94 (0.6H, d, J = 10.4Hz), 5.23 (0.4H, t, J = 9.6 Hz), 5.32 (0.4H, d, J = 10.2 Hz), 5.38–5.50 (1.6H, m), 5.69 (0.4H, t, J = 9.6 Hz), 5.83 (0.6H, dd, J = 9.0 Hz, 10.4 Hz), 6.61 (0.4H, s), 6.69 (0.6H, s), 6.79 (0.6H, s), 6.88 (0.4H, s), 7.35 (0.8H, d, J = 8.6Hz), 7.39 (1,2H, d, J = 8.6 Hz), 7.87 (0.8H, d, J = 8.6 Hz), 8.07 (1.2H, d, J = 8.6 Hz). 1H-NMR (the same sample as above, 400 MHz, DMSO-d6) δ: 1.54 (0.6H, s), 1.64 (2.4H, s), 1.79 (2.4H, s), 1.91 (2.4H, s), 1.95 (0.6H, s), 1.97 (0.6H, s), 2.01 (0.6H, s), 2.05 (2.4H, s), 2.31 (0.6H, s), 2.31 (2.4H, s), 2.32 (0.6H, s), 2.32 (2.4H, s), 2.36 (0.6H, s), 2.44 (2.4H, s), 3.92 (0.2H, br), 4.11 (0.8H, dd, J = 2.4 Hz, 12.7 Hz), 4.21 (0.8H, dd, J = 4.0 Hz, 12.7 Hz), 4.27 (0.2H, dd, J = 4.7 Hz, 12.6 Hz), 4.37 (0.8H, ddd, J = 2.4 Hz, 4.0 Hz, 9.8 Hz), 4.42 (0.2H, br), 5.08 (0.2H, t, J = 9.6 Hz), 5.25 (0.8H, d, J = 9.8 Hz), 5.33 (0.8H, t, J = 9.8 Hz), 5.43–5.58 (0.6H, m), 5.62 (0.8 H, t, J = 9.8Hz), 5.69 (0.8H, t, 9.8 Hz), 6.96 (0.2H, s), 7.02 (0.8H, s), 7.12 (0.2H, s), 7.12 (0.8H, s), 7.38 (0.4H, d, J = 9.0 Hz), 7.41 (1.6H, d, J = 8.8 Hz), 8.20 (0.4H, d, J = 9.0 Hz), 8.28 (1.6H, d, J = 8.8 Hz). 1H-NMR (500 MHz, DMSO-d6, 90°C) δ: 1.66 (3H, br), 1.85 (3H, br), 1.86 (3H, s), 2.06 (3H, s), 2.33 (3H, s), 2,34 (3H, s), 2.43 (3H, br), 4.15 (1H, br), 4.24 (1H, dd, J = 4.5 Hz, 12.4 Hz), 4,30–4.34 (1H, m), 5.29 (2H, br), 5.53 (1H, t, J = 9.3Hz), 5.72 (1H, br), 6.89 (1H, s), 7.06 (1H, s), 7.42 (2H, d, J = 8.8 Hz), 8.21 (2H, dd, J = 8.8 Hz). IR νmax cm–1: 1739, 1649, 1366, 1182, 1167, 1016. HR-MS [ESI+, (M+ Na)+]: calculated for C35H34NaO17, 749.1694; found, 749.1665.
2d: Rf value: 0.23 [developed with chloroform-ethyl acetate (1:3)]. 1H-NMR (500 MHz, CDCl3) δ: 1.61 (3H, br), 2.03 (3H, s), 2.10 (3H, s), 2.14 (3H, br), 2.36 (3H, s), 2.41 (3H, s), 4.03 (1H, br), 4.20 (1H, dd, J = 2.0 Hz, 12.5 Hz), 4.38 (1H, dd, J = 3.0 Hz, 12.5 Hz), 5.35 (1H, t, J = 9.0 Hz), 5.45–5.48 (3H, m), 6.55 (1H, s), 6.64 (1H, s), 7.34 (2H, d, J = 8.5 Hz), 7.89 (2H, d, J = 8.5 Hz), 8.36 (1H, br). 13C-NMR (125 MHz, CDCl3) δ: 20.0, 20.5, 20.6, 20.6, 21.1, 21.1, 61.2, 67.6, 70.5, 73.4, 74.5, 77.6, 106.3, 108.5, 110.6, 111.0, 122.8, 127.5, 128.9, 151.2, 153.4, 155.1, 160.9, 161.0, 168.4, 168.9, 169.3, 169.3, 170.2, 170.5, 176.0. IR νmax cm–1: 3144, 2888, 1642, 1605, 1506, 1415, 1188, 1168, 1033. HR-MS [ESI+, (M+ Na)+]: calculated for C33 H32NaO16, 707.1588; found, 707.1588. [α]D22 + 66.9° (c 0.38, chloroform).
8-(β-D-Glucopyranosyl)-4ʹ,5,7-trihydroxyflavone (vitexin, 2b)
To a suspension of 2c and 2d (695 mg, 2c:2d = 56:44, 0.981 mmol) in anhydrous methanol (30 mL), was added sodium methoxide (435 mg, 8.05 mmol) under argon atmosphere and the mixture was stirred for 3 h at room temperature. The reaction was neutralized with DOWEX 50 W-X8 (H+ form) and the resulting mixture was filtered to remove insoluble materials. The solid residue was washed with methanol, and the combined filtrate and washings were concentrated in vacuo to furnish 2b as yellow amorphous solid (424 mg, quantitative yield). Rf value: 0.48 [developed with chloroform-methanol (2:1)]. 1H-NMR (400 MHz, DMSO-d6) δ: 3.17–3.28 (3H, m), 3.45–3.56 (1H, m), 3.73–3.87 (2H, m), 4.62 (1H, t, J = 5.6 Hz), 4.68 (1H, d, J = 9.8 Hz), 4.72 (1H, d, J = 5.3 Hz), 5.00 (1H, d, J = 6.0 Hz), 5.01 (1H, d, J = 7.4 Hz), 6.27 (1H, s), 6.79 (1H, s), 6.89 (2H, d, J = 8.8 Hz), 8.03 (2H, d, J = 8.8 Hz), 10.36 (1H, s), 10.85 (1H, s), 13.17 (1H, s). 13C-NMR (100 MHz, DMSO-d6) δ: 62.2, 71.5, 71.8, 74.3, 79.6, 82.8, 99.1, 103.4, 105.0, 105.6, 116.8, 122.6, 129.9, 157.0, 161.3, 162.1, 163.5, 164.9, 183.1. IR νmax cm–1: 3154, 2925, 1751, 1650, 1366, 1177. HR-MS [ESI+, (M+ Na)+]: calculated for C21 H22NaO10, 455.0954; found, 455.0924. [α]D22 – 9.5° (c 1.0, DMSO).
2”,7-Anhydro-4ʹ,5-diacetoxy-8-(3,4,6-tri-O-acetoxy-β-D-glucopyranosyl)flavone (1b)
To a solution of 2b (19.7 mg. 45.6 mol) in anhydrous N,N-dimethylformamide (DMF, mL) were added triphenylphosphine (30.3 mg, 0.115 mmol) as solid and diisopropyl azodicarboxylate (40% in toluene, 60 μL, 0.11 mmol) at 0°C under argon atmosphere and the mixture was stirred for 4 h at room temperature. After the consumption of 2b judged by TLC analysis, to the mixture were directly added acetic anhydride (1 mL), pyridine (1 mL) and catalytic amount of DMAP (0.9 mg, 7 μmol) and the resulted mixture was further stirred for 19 h at room temperature. Then, the mixture was poured into ice. The precipitates were collected by filtration and dried in vacuo. The residual solids were purified by medium-pressure column chromatography (10 g cartridge, FSUL-0442-0010). Elution with hexane-ethyl acetate (4:1 to 1:0) furnished 1b as white amorphous solid (21.1 mg, 74%). Rf value: 0.51 [developed with hexane-ethyl acetate (1:2)]. 1H-NMR (400 MHz, CDCl3) δ: 2.00 (3H, s), 2.10 (3H, s), 2.17 (3H, s), 2.35 (3H, s), 2.45 (3H, s), 3.90 (1H, ddd, J = 2.5 Hz, 5.5 Hz, 8.8 Hz), 4.12 (1H, dd, J = 2.5 Hz, 12.3 Hz), 4.28 (1H, dd, J = 5.5 Hz, 12.3 Hz), 4.97 (1H, dd, J = 3.8 Hz, 4.9 Hz), 5.27 (1H, t, J = 8.8 Hz), 5.46 (1H, dd, J = 4.9 Hz, 8.8 Hz), 5.47 (1H, d, J = 3.8 Hz) 6.57 (1H, s), 6.69 (1H, s), 7.25 (2H, d, J = 9.0 Hz), 7.84 (2H, d, J = 9.0 Hz). 13C-NMR (125 MHz, CDCl3) δ: 20.7, 20.7, 20.7, 21.1, 21.1, 62.7, 66.3, 69.9, 74.0, 74.6, 83.0, 104.6, 108.8, 112.0, 112.2, 122.4, 127.5, 128.8, 153.2, 153.2, 154.3, 160.7, 164.4, 168.9, 169.4, 169.4, 170.3, 170.6, 176.0. IR νmax cm–1: 1738, 1644, 1634, 1608, 1370, 1205, 1047, 907. HR-MS [ESI+, (M+ Na)+]: calculated for C31 H28NaO14, 647.1377; found, 647.1376. [α]D22 – 174.1° (c 1.0, chloroform).
2”,7-Anhydro-8-(β-D-glucopyranosyl)-4ʹ,5-dihydroxyflavone (chafuroside B, 1a)
The deacetylation of 1b (15.6 mg, 25.0 μmol) was carried out in a similar manner with that for 2c and 2d. After workup, 1a was obtained as yellow amorphous solid (10.4 mg, quantitative yield). Rf value: 0.72 [developed with chloroform-methanol (2:1)]. 1H-NMR (400 MHz, CD3OD) δ: 3.43–3.48 (1 H, m), 3.61 (1H, t, J = 9.4 Hz), 3.63 (1H, dd, J = 5.9 Hz, 12.1 Hz), 3.86 (1H, dd, J = 2.5 Hz, 12.1 Hz), 4.03 (1H, dd, J = 4.7 Hz, 9.4 Hz), 4.76 (1H, dd, J = 4.7 Hz, 3.3 Hz), 5.45 (1H, d, J = 3.3 Hz), 6.37 (1H, s), 6.66 (1H, s), 6.94 (2H, d, J = 8.8 Hz), 7.90 (2H, d, J = 8.8 Hz). 13C-NMR (125 MHz, CD3OD) δ: 63.0, 69.3, 73.3, 74.6, 80.1, 88.6, 95.9, 104.3, 106.3, 108.0, 117.1, 123.1, 129.7, 154.6, 163.0, 165.7, 166.1, 168.5, 184.0. IR νmax cm–1: 3464, 3333, 3071, 1660, 1573, 1428, 1079, 831. HR-MS [ESI+, (M+ Na)+]: calculated for C21H18NaO9, 437.0849; found, 437.0857. [α]D22 – 140.8° (c 0.17, methanol). [lit [Citation5]. [α]D23 − 169.3° (c 0.17, methanol)].
Author contributions
K.K. and T.S. designed this study; K.K. and T.S. carried out the experiments; K.H. contributed to analytical works; K.K. wrote the manuscript with assistance from all authors; and S.H. and T.S. supervised the research.
ChafurosideSugaiSM.pdf
Download PDF (1.9 MB)Acknowledgments
K.K. and T.S. deeply thank Professor Fumiyuki Kiuchi of this faculty, for his valuable advices.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental material for this article can be accessed here.
Additional information
Funding
References
- Ishikura Y, Tsuji K, Nukaya H Novel derivative of flavone C-glycoside and composition containing the same. WO Patent, 2004, 005296.
- Hasegawa T, Shimada S, Ishida H, et al. an Oolong tea polyphenol, ameliorates UVB-induced DNA damage and generation of photoimmunosuppression related mediators in human keratinocytes. Plos One. 2013;8:e77308.
- Ishida H, Wakimoto T, Kitao Y, et al. Quantitation of chafurosides A and B in tea leaves and isolation of prechafurosides A and B from Oolong tea leaves. J Agric Food Chem. 2009;57:6779–6786.
- Ishida H. Sulfated C-glycoside, method for isolating same and method for synthesizing same. WO Patent. 2010, 076879.
- Furuta T, Nakayama M, Suzuki H, et al. Concise synthesis of chafurosides A and B. Org Lett. 2009;11:2233–2236.
- Mahling J-A, Jung K-H, Schmidt RR. Synthesis of flavone C-glycosides vitexin, isovitexin, and isoembigenin. Liebigs Ann Chem. 1995;3:461–466. .
- Perkin AG. Colouring matters of the New Zealand dyewood Puriri, Vitex littoralis. Part I. J Chem Soc. 1898;73:1019–1031.
- Liu W, Chen G, Cui T. Determination of flavones in Crataegus pinnatifida by capillary zone electrophoresis. J Chromatogr Sci. 2003;41:87–91.
- Zhang Y, Jiao J, Liu C, et al. Isolation and purification of four flavone C-glycosides from antioxidant of bamboo leaves by macroporous resin column chromatography and preparative high-performance liquid chromatography. Food Chem. 2008;107:1326–1336.
- Sharma KK, Sharma AK, Sharma MC, et al. Isolation of orientin and vitexin from stem bark of Parkinsonia aculeata (Caesalpiniaceae) and their successive blending on sheep wool fiber. Int J Pharmacogn Phytochem Res. 2014;6:557–561.
- Shin H, Park Y, Jeon YH, et al. Identification of Polygonum orientale constituents using high-performance liquid chromatography high-resolution tandem mass spectrometry. Biosci Biotechnol Biochem. 2018;82:15–21.
- Aboshi T, Ishiguri S, Shiono Y, et al. Flavonoid glycosides in Malabar spinach Basella alba inhibit the growth of Spodoptera litura larvae. Biosci Biotechnol Biochem. 2018;82:9–14.
- Kusano S, Okazaki K. Simple method for preparing flavonoid-C-glycoside, arabinofuranosyl-vitexin, from Basella alba leaves and application of it to functional food materials. Jpn J Food Eng. 2018;19:131–136.
- Kurahayashi K, Hanaya K, Higashibayashi S, et al. Synthesis of trilobatin from naringin via prunin as the key intermediate: acidic hydrolysis of the α-rhamnosidic linkage in naringin under improved conditions. Biosci Biotechnol Biochem. 2018;82:1463–1467.
- Tsunekawa R, Katayama K, Hanaya K, et al. Synthesis of 5-hydroxy-3ʹ,4ʹ,7-trimethoxyflavone (HTMF) and related compounds and elucidation of their reversal effects on BCRP/ABCG2-mediated anticancer drug resistance. ChemBioChem. 2019;20:210–220.
- Fujita R, Hanaya K, Higashibayashi S, et al. Synthesis of oroxylin A starting from naturally abundant baicalin. Heterocycles. 2018;97:1165–1174.
- Péteri E. Experiments on vitexin. J Chem Soc. 1939:1635–1637.
- Gentili B, Horowitz RM. Flavonoids of citrus. IX. Some new C-glycosylflavones and a nuclear magnetic resonance method for differentiating 6- and 8-C-glycosyl isomers. J Org Chem. 1968;33:1571–1577.
- Shie -J-J, Chen C-A, Lin -C-C, et al. Regioselective synthesis of di-C-glycosylflavones possessing anti-inflammation activities. Org Biomol Chem. 2010;8:4451–4462.
- Frank JH, Powder-George YL, Ramsewak RS, et al. Variable-temperature 1H-NMR studies on two C-glycosylflavones. Molecules. 2012;17:7914–7926.
- Ren B, Wang M, Liu J, et al. Zemplén Transesterification: a name reaction that has misled us for 90 years. Green Chem. 2015;17:1390–1394.
- Camp D, Harvey PJ, Jenkins ID. The effect of solvent polarity on the rate of the Mitsunobu esterification reaction. Tetrahedron. 2015;71:3932–3938.