
Abstract
Platelet transfusions are given to patients in hospital who have a low blood platelet count (thrombocytopenia) either because of major bleeding (following trauma or surgery) or because the bone marrow production of platelets is impaired often due to chemotherapy, infiltration with malignant cells, fibrosis or genetic disorders. We are currently entirely reliant on blood donors as a source of platelets in transfusion medicine. However, the demand for platelets continues to rise, driven by an aging population, advances in medical procedures and ever more aggressive cancer therapies, while the supply of blood donors continues to remain static.
In recent years, several groups have made major advances toward the generation of platelets in vitro for human transfusion. Recent successes include results in both generating mature human megakaryocytes as well as in developing bioreactors for extracting platelets from these megakaryocytes. Platelets made in vitro could address several issues inherent to platelets derived from blood donors – the ability to scale up/down more flexibly according to demand and therefore less precarious supply line, reduction of the risk of exposure to infectious agents and finally the possibility of engineering stem cells to reduce immunogenicity.
Here we define the quality control tools and suggest measures for implementation across the field for in vitro platelet genesis, to aid collaboration between laboratories and to aid production of the burdens of proof that will eventually be required by regulators for efficacy and biosafety. We will do this firstly, by addressing the quality control of the nucleated cells used to make the platelets with a particular emphasis to safety issues and secondly, we will look at how platelet function measurement are addressed particularly in the context of platelets derived in vitro.
Introduction
Platelet transfusions are often given to patients in hospital who have a blood platelet count below 10x109/ml (thrombocytopenia) either because of major bleeding following trauma or surgery or because the bone marrow production of platelets is impaired due to, for example, chemotherapy, infiltration with malignant cells or fibroblasts, or genetic disorders [Citation1,Citation2]. We are currently entirely reliant on blood donors as a source of platelets in transfusion medicine. However, the demand for platelets continues to rise driven by an aging population, advances in medical procedures and ever more aggressive cancer therapies, while the supply of blood donors continues to remain static ().
Figure 1. Demand for platelet units increasing over time in the UK, adapted from [Citation3].
![Figure 1. Demand for platelet units increasing over time in the UK, adapted from [Citation3].](/cms/asset/45e470ac-d4e4-45a8-8d22-b723976e2c9e/iplt_a_1769051_f0001_oc.jpg)
One unit of platelets from NHS Blood & Transplant (NHSBT, UK donor blood distributor) costs £193.15 (approx. 250 USD USD) to produce. Issuing 250,000 units for the population of the UK (66 million people) cost the NHS £52 m (approx. $67.2m USD) in 2015/16 according to NHSBT figures[Citation3]. For comparison, a unit of red blood cells cost £120.00 (approx. $155.00 USD) in 2016/17, making platelets units the most expensive of all the common blood components to be collected[Citation4].
At present, we are fully dependent on volunteer donors for our supply of platelets which can present many issues. When donor attendance drops, such as happened in North America in 2018/19 during multiple severe snowstorms, the available inventory of platelet units can become critically low, risking patients’ lives[Citation5]. Adding to this, platelets have a short shelf-life of 5–7 days compared to 35 days for red blood cells, which makes the platelet supply much more susceptible to reductions in donor attendance [Citation6,Citation7].
The use of blood donor-derived products has also been marred in recent years by incidents of infected components, particularly for components where several donations are pooled such as clotting factor concentrates. The pooling of 4 donations to make one unit of platelets remains one of the main production methods although the use of apheresis to provide single-donor platelets has markedly increased in the last two decades following the UK Bovine Spongiform Encephalopathy outbreak and the fear of transmission of variant Creutzfeldt-Jakob disease through blood transfusion (now 60–80% of all units).
For patients who receive multiple transfusions, or multiparous women, exposure to HLA Class I epitope on donor cells, or fetal cells, respectively can lead to alloimmunisation against HLA Class I. For these patients, “random” platelets that are only matched for ABO antigens have a very short life in circulation post-transfusion and therefore these patients are transfused from selected donors who have been typed based on HLA Class I and are recalled according to demand. This represents a logistical challenge and a great cost to blood providers. In the UK, HLA-matched units cost over £400.00 per unit (approx $488.00 USD).
In recent years, several groups have made major advances toward the generation of platelets in vitro for human transfusion. Recent success stories include the generation of human megakaryocytes as well as the development of bioreactors for extracting platelets from these megakaryocytes [Citation8–Citation12]. Theoretically, platelets made in vitro could address several issues inherent to platelets derived from blood donors – the ability to more flexibly adjust the supply with the demand, reduction of the risk of recipient exposure to infectious agents and finally the possibility of engineering stem cells to reduce immunogenicity.
In this opinion paper, we will attempt to define the quality control tools and measures that we suggest should be implemented across the field of in vitro platelet genesis, to aid collaboration between laboratories and start to build burdens of proof that will eventually be required by regulators for efficacy and biosafety. There might be two different potential use-cases of in vitro platelets as a therapy; as prophylaxis following chemotherapy and for an acute correction of the platelet count, e.g. during trauma or cardiac surgery and different criteria may be required to assess suitability for each of these use cases. In this opinion piece, we focus on the prophylactic use-case, but many of the same lessons will likely apply to the acute use-case. We will do this firstly, by addressing the quality control of the nucleated cells used to make the platelets with a particular emphasis to safety issues and secondly, we will look at how platelet function measurement are addressed particularly in the context of platelets derived in vitro.
Stem Cells and Megakaryocytes: issues of Safety
One of the major risks associated with cellular therapies is the potential emergence of tumors following transplant/transfusion of the product into patients. Platelets and red cells have often been perceived as a “safer” cellular product with regards to tumorigenic risk because these cells do not contain a nucleus. In fact, regulators such as the European Medicine Agency do not consider platelets as an “Advanced Therapy Medicinal Product”, the umbrella designation under which cellular therapies are categorized, but as an “Investigational Medicinal Product” because they are not a cell due to the lack of a nucleus. However, this is only valid if we can remove all the nucleated precursor cells from the transfusion product. Given the number of platelets contained in one therapeutic unit (3x1011) even a 0.1% contamination with nucleated cells would still lead to 3 × 108 nucleated cells administered to the patient with each transfusion. Frequently the use of irradiation is suggested to induce apoptosis in any nucleated cells present in the final product. This procedure is routinely used to eliminate the risk of transfusion acquired “graft versus host disease” in severely immunocompromised patients where donor lymphocytes are able to engraft and mount an immune response against the recipient’s tissue. Irradiation protocols have been developed to create enough damage in donor lymphocytes to induce apoptosis. However, it remains to be seen how effective irradiation would be in reducing nucleated cell contamination from megakaryocyte cultures, and whether this degree of irradiation would affect platelet biology. We would have to demonstrate that this is equally effective against culture-derived cells that may have acquired a survival advantage from the culture process itself (or through immortalization). These mutations that provide culture cells with growth advantage will inherently be varied which may create major differences in the way each nucleated cell would respond to irradiation.
Measuring Genomic Integrity
The seed material for in vitro derived platelets are usually pluripotent stem cells or immortalized megakaryocyte progenitors derived from pluripotent stem cell lines [Citation8,Citation9]. Maintaining cell lines in culture presents major challenges with regards to genome stability. Several papers have shown how pluripotent stem cells acquire mutations in cultures and that these mutation confer a survival advantage to the stem cells so that within a few passages that whole culture is dominated by the mutant clone [Citation13,Citation14]. Immortalized cells are usually generated by manipulating the apoptosis pathway, but we need to bear in mind that apoptosis is one of the ways cells will protect themselves from acquiring mutations[Citation15]. Genomic instability is therefore even more of a concern when deriving platelets from megakaryocytes that have been generated through a process that involves immortalization at some point.
Therefore, it is crucial that we quantify genomic instability of both seed material or cellular intermediates as nucleated cells will inevitably find their way into the final product. Recent work has shown that cells acquire mutations directly as a result of adaptation to the tissue culture process, as there are no cell-extrinsic mechanisms (e.g. an immune system) to remove any cells that might have the potential to become dangerous. In fact, between 12.5% and 34% of all human pluripotent stem cell lines acquire specific nonrandom chromosomal abnormalities over time (particularly in chromosome structure and number in chromosomes 1, 12, 17 and 20) that were reminiscent of changes also observed in cancers[Citation16]. Additionally, immortalized cells have had their cell-intrinsic mechanisms for DNA damage control suppressed. Studies on other cell types, forced to overexpress c-Myc, show that this can directly drive the formation of tumors in vivo, making it vital that we are vigilant of this in our cell banks if we are to use these immortalized cells as a source [Citation17,Citation18].
Several approaches for the analysis of genomic integrity of cellular products have been proposed, each presenting pros and cons in terms of feasibility, sensitivity and interpretation of the results. Some methods such as karyotyping are routinely used in clinical practice but may present “low definition” results. At the other end of the spectrum whole genome sequencing would provide a comprehensive picture of the genome of the cell culture in question but is more costly than other assays and requires specialist analyses. The various methodologies to assess the culture have been reviewed elsewhere[Citation19]. Finally, the question of what genomic abnormalities are “safe” for clinical application remains unanswered.
However it remains that transparency about genomic integrity of cell lines used to produce platelets should be standardized, even when describing methodologies that are yet to leave academic laboratories as the culture methods themselves may have an influence on the “genomic profile” of the culture and therefore future safety of clinical grade products.
Measuring Cellular Product Differentiation
Pluripotent stem cells have the ability to engraft and form teratomas and in fact, this is still used as one of the measures of pluripotency. In addition, it may be that intermediate progenitors of megakaryocytes may also be able to engraft and proliferate post-transfusion. A key metric of the safety of the final product is therefore the assessment of how well differentiated and mature the megakaryocytes are, from which platelets are generated, and the purity of the culture including the identification and quantification of partially differentiated proliferative precursors.
Most laboratories use surface marker expression of major platelet receptors to assess megakaryocyte differentiation. This includes CD41 (ITGA2B) and CD61 (ITGB3). Together these form the heterodimeric integrin αIIbβ3, a fibrinogen receptor, which is expressed early during differentiation[Citation9]. Various members of the von Willebrand receptor complex GPIb-V-IX (CD42a = glycoprotein IX, CD42b = glycoprotein Ibα, CD42d = glycoprotein V) are often used as markers of megakaryocyte maturity as they are expressed later during differentiation[Citation9]. However, measuring the presence of remaining precursors in the culture is much less well established. They can be identified functionally using colony forming assays which are laborious and time-consuming and therefore poorly suited to the monitoring of culture. Flow cytometry would again be the default choice but defining which surface markers would clearly identify the progenitors in culture is at this stage unclear.
Measuring Nucleated Cell Contamination
Ideally, none of the nucleated cells would be present in the final platelet product removing all tumorigenic concerns, however in reality, this will be impossible to accomplish due to the very large numbers of platelets in the final product (see ). Essentially two different methods have been used to separate the platelet progeny from the megakaryocytes. For methods where platelets are released in liquid culture subsequent steps involve filtration and centrifugation to separate platelets from megakaryocytes based on cell-size difference[Citation11]. Other methods involve the generation of platelets using devices where the megakaryocytes are “trapped” within bespoke bioreactors or within 3-dimensional sponges and the platelets harvested in the effluent medium flown through the device [Citation10,Citation20–Citation22].
Figure 2. In vitro-produced platelet population as defined by FS/SS can also contain DAPI positive cells. Cord blood-derived megakaryocytes, permeabilised with saponin were stained for DAPI and analyzed by flow cytometry. Gates set with unstained cells.
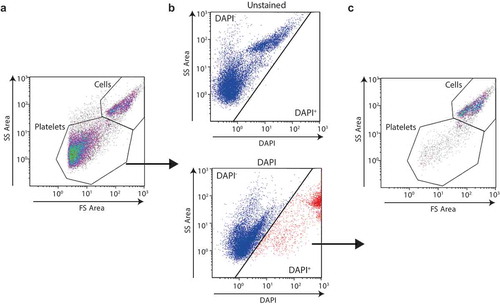
Regardless of the methodology used to generate platelets from megakaryocytes, the contamination of the final platelet concentrate with nucleated cells should be routinely disclosed not only because it would clearly be a vital part of the measurement of the product safety but also because it would allow comparison between the different methods used to release and separate platelets from the megakaryocytes.
In conclusion, with the emergence of cellular therapies, consensus around issues of safety and what testing may be required have started to emerge[Citation23]. We would advocate that this guidance is applied to platelet production in vitro. Therefore, any document pertaining to this field, be it an academic original manuscript, application for funding or regulatory approval should clearly seek to address these issues and present supportive data. We propose three steps for the quality control of the nucleated cellular product that precedes platelet production. This should include ongoing documentation of the genome integrity of stem cells/immortalized cell lines from which megakaryocytes are cultured, the documentation of the level of differentiation achieved and finally, within the final product, a measure of the number of nucleated cells that remain post-platelet release.
Assessing Potency
Defining and Counting Platelets
Addressing the “potency” of the final platelet product is not trivial. To begin with, even counting the number of platelets produced requires rigor and most importantly, consensus between laboratories. Most groups will use flow cytometry and counting beads to count platelets, usually using the forward/side scatter to identify the right size events compared to peripheral blood platelets, with the addition of cell surface markers such as CD41 or CD61, sometimes complemented by the use of other surface receptors such as CD42. However, due to their small size (<5 microns), platelets can be of a similar size to cellular debris. Such cellular debris can also nonspecifically bind antibodies, thereby overestimating the numbers of platelets produced. Some groups, including ours, have therefore additionally used metabolic viability stains such as Calcein-AM, to prove firstly that we have intact cells and not just debris and secondly that they metabolically active, therefore reducing the number of events described as “platelets” by 82% (CD41+ versus CD41+/CD42+/Calcein+ events, )[Citation22].
Figure 3. CD41 alone is not an adequate marker to define platelets. iPSc-derived megakaryocytes analyzed by flow cytometry using antibodies against CD41a, CD42a and using calcein-AM (A) gating strategy for the definition of a platelet, FS/SS, CD41/CD42+ and calcein-AM+, representative graphs (B) Flow cytometry quantification of events with the indicated gating strategies. (C) iPS cells analyzed by flow cytometry with a “platelet” staining protocol and gating strategy.
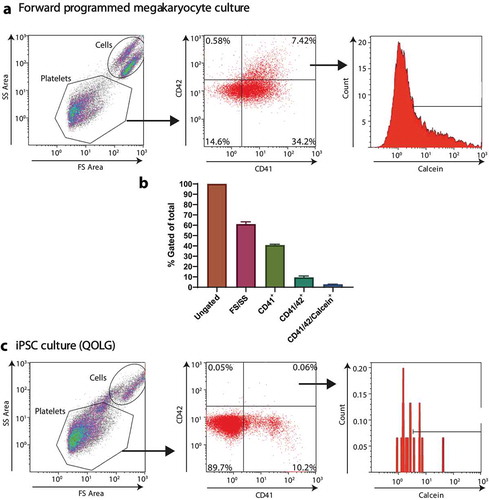
We have also shown that functionality within an in vitro-derived platelet suspension was limited to those that met all 4 the criteria described above (see ); forward and side scatter consistent with platelets, expression of both CD41 and CD42 and retention of the viability stain Calcein-AM. The lack of commonly accepted definition of what constitutes platelet when looking at flow cytometry data on cell cultures (and how to distinguish these from debris, dead platelets or intact megakaryocyte blebs) has presented a barrier preventing true comparison of the number of platelets produced by various technologies published to date.
Figure 4. Calcein-AM positive events show a marked increase in functionality. Flow cytometry analysis of iPSc-derived megakaryocytes stimulated with 10 µM TRAP6 and stained using an antibody against P-selectin (A) quantification of % P-selectin exposure in CD42+ calcein− (red) and CD42+ calcein+ (green) populations, n = 3 (B) Representative histograms of P-selectin exposure in CD42+ calcein− (red), CD42+ calcein+ (green) and CD42+ only (blue) populations.
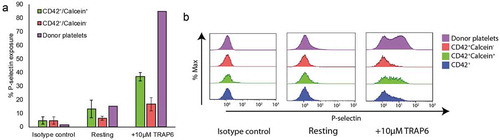
To this end, the WHO recently agreed the development of a flow cytometry standard to assess platelets preparation with the aim to provide a lyophilized preparation that will, once reconstituted, allow all users to assess their flow cytometry protocol and allow effective comparison between laboratories. Crucially, this standard will allow the calibration of instruments used for the comparison of samples using all 4 proposed markers described above.
Measuring Platelet Function
In the world of transfusion medicine, the only measure of the quality of the platelets that is required within a donated unit is the measurement of the pH[Citation24]. In contrast, when assessing platelet function for diagnostic purposes in patients or in the field of platelet research, we are offered the choice between a plethora of assays. Some of these tests only interrogates very specific platelet function such as aggregation (light transmission aggregometry), adhesion and procoagulant activities (thrombin generation) whilst others look at more complex functionality such as thrombus formation (PFA-100) or clot formation/retraction (thromboelastography). [Citation24–Citation26]
Unfortunately, applying the above assays to our in vitro produced platelets requires numbers of platelets that are unachievable using current small-scale in vitro platelet culture systems. As a result, most papers published so far have been limited to the use of low volume alternatives, often flow cytometry-based assays. The most commonly used assays analyze two different activation events; firstly the presence of fibrinogen bound to CD41/CD61 using an anti-fibrinogen antibody or PAC-1 antibody (which recognizes only the activated form of the receptor) and secondly evidence of degranulation based on the exposure of P-selectin on the platelet surface. Whilst these assays are regularly used, published results are often difficult to interpret for several reasons. Firstly, samples are often not compared to isotype controls or in the case of anti-fibrinogen antibodies, where for the polyclonal antibody there is no isotype control, an EDTA control is not used. Secondly, there is absence of comparison with donor platelets so that the amplitude of the response is not assessed against the “gold standard”. Finally, and more importantly, within the field the non-publication and comparison of resting and activated samples has become a major issue. Resting samples, especially when analyzing P-selectin exposure, can be relatively high, but through the non-disclosure of this data, agonist induced activation will be over-estimated and potentially misleading conclusions drawn. We believe that without a well-defined strategy for gating on to the “true platelet population”, as is described above, interpreting and comparing results from these flow cytometry-based assays can be difficult.
Other low volume assays have emerged such as the use of microchambers looking for thrombus formation under flow, or microchips containing pillars made of PDMS (“nanoposts”) to ascertain platelet retraction following adhesion [Citation27,Citation28]. The microchamber assay is based on coating a flow chamber with a substrate promoting platelet thrombi formation (often fibrillar collagen) into which anticoagulated whole blood is flown at a chosen shear rate. In vitro-derived platelets (or donor derived platelets) can be labeled and their incorporation into the thrombi recorded and quantified[Citation22]. The nanopost technology involves adhering platelets on top of “nano-pillars” and following activation of the platelets, recording how these pillars are “pulled” by the platelets using confocal microscopy[Citation28]. These are relatively new assays and so their quantitative value for assessing platelet functionality has not been rigorously tested, but they offer a promising addition to more traditional assays.
In vivo mouse or rabbit models are also used to quantify platelet quality by studying either recovery/survival in circulation or hemostatic effect. Due to the xenogenic barriers, platelets have a very short survival in circulation, even when using immunosuppressed animals such as NSG mice[Citation29]. Some authors have used clodronate liposomes to deplete macrophages in these animals to prolong the presence of platelets in circulation, but suppressing macrophage phagocytic activity in the mouse may obscure clinically relevant defects in the transfused platelets[Citation30]. Interpretation of the results of transfusion when using an artificial environment such as that found in animal models and, in particular, extrapolation of this data as an indication of what may happen post-transfusion to humans is unknown at this stage. The measure of hemostatic activity has usually limited itself to recording the accumulation of platelets (usually fluorescent labeling and intravital microscopy) at the site of blood vessel injury but so far only one group has produced platelets in significant enough quantities to truly assess the hemostatic power of in vitro-derived platelets in the context of bleeding in a thrombocytopenic animal model [Citation11,Citation31]. It should be noted that these functional experiments all compare in vitro generated platelets to freshly prepared donor-derived platelets as a control, as platelets from a blood bank may be several days old and this would overestimate the functionality of the in vitro platelets.
There are other considerations that are tentatively being addressed with these in vitro generated platelets, including infectious risk management and immunogenicity Although not susceptible in the same way as donor-derived pooled platelets, the effect of an infected batch of in vitro platelets may present similar issues to that of infected donor-derived blood products. Current studies are ongoing which aim to address immunogenicity by looking at HLA knockout platelets and their effects on NK cell responses[Citation32].
At present, measurement of the half-life of these products in vivo is limited to up to 6 hours post-transfusion and does not attempt to quantify these half-lives in comparison to platelets from donors[Citation32]. Even if in vitro platelet products produced by certain methods prove to have a shorter half-life than donor-derived platelets, the resultant transfusion products could be split into two categories – those with a short half life, suitable for trauma and acute bleeding, and those with a longer half life that are suitable for prophylactic purposes. These two types of products may require different functional assessments and thus different standards.
Final Conclusions
In this paper, we have tried to give an overview of the issues faced when assessing quality control of in vitro-derived platelets. Firstly, we have particularly focused on the seed material (stem cell lines) and intermediate nucleated products (megakaryocytes) and the potential safety issues they can pose. Secondly, we looked at the challenge of assessing the potency of the product, namely how we count the produced platelets and thirdly how we measure their functionality. The required potency of the final product may depend on the intended use in patients, i.e. as a prophylactic product where consideration of half-life in circulation are more important than for therapeutic platelet transfusion in case of acute bleeding. As we near the first in human clinical studies, it is a sign of how far the field has come that we now find the need to ask ourselves these questions, notwithstanding the urgency with which these answers become apparent. It is down to the researchers themselves, peer-reviewers (and editors) of manuscripts or funding application and regulatory bodies to have the integrity to ask the hard questions and ask for standardization in the field of assessing the quality of these products. We owe it to the volunteers and patients who may one day be transfused with these novel cellular products.
Declarations Of Interests
CG is an advisory board member for Platelet Biogenesis (Boston, US), MacoPharma (Tourcoing, France) and BitBio (Cambridge, UK). SM, HRF and AKW report no conflicts of interest.
Acknowledgements
This work was supported by grants from the Rosetrees Trust (A1691), NHS Blood and Transplant and the European Union (SilkFusion: AMD-767309-3).
Additional information
Funding
References
- Williamson DR, Albert M, Heels-Ansdell D, Arnold DM, Lauzier F, Zarychanski R, Crowther M, Warkentin TE, Dodek P, Cade J, et al. Thrombocytopenia in critically ill patients receiving thromboprophylaxis: frequency, risk factors, and outcomes. Chest. 2013;144(4):1207–1215. doi:10.1378/chest.13-0121.
- Estcourt LJ, Birchall J, Lowe D, Grant-Casey J, Rowley M, Murphy MF, et al. Platelet transfusions in haematology patients: are we using them appropriately? Vox Sang. 2012;103(4):284–293. doi:10.1111/j.1423-0410.2012.01627.x.
- Cowan K. Strategies to reduce inappropriate use of platelet transfusions. Nurs Times. 2017;113: 2, 18-21.
- Blood NHS & Transplant. NHSBT Price List 2016-17. (2016). Available at: (Accessed: 4th February 2020) https://web.archive.org/web/20180603111712/http://hospital.blood.co.uk/media/28230/component-price-list-2016-2017.pdf
- American red cross. one-two punch of winter storms, canceled blood drives straining red cross blood supply. Available at: (Accessed 4 February 2020). https://www.redcrossblood.org/local-homepage/news/article/one-two-punch-of-winter-storms–canceled-blood-drives-straining-.html
- Smethurst PA. Aging of platelets stored for transfusion. Platelets. 2016;27(6):526–534. doi10.3109/09537104.2016.1171303.
- García-Roa M, Del Carmen Vicente-Ayuso M, Bobes AM, Pedraza AC, González-Fernández A, Martín MP, Sáez I, Seghatchian J, Gutiérrez L, et al. Red blood cell storage time & transfusion: current practice, concerns & future perspectives. Blood Transfusion. 2017;15(3):222–231. doi:10.2450/2017.0345-16.
- Nakamura S, Takayama N, Hirata S, Seo H, Endo H, Ochi K, Fujita K-I, Koike T, Harimoto K-I, Dohda T, et al. Expandable megakaryocyte cell lines enable clinically applicable generation of platelets from human induced pluripotent stem cells. Cell Stem Cell. 2014;14(4):535–548. doi:10.1016/j.stem.2014.01.011.
- Moreau T, Evans AL, Vasquez L, Tijssen MR, Yan Y, Trotter MW, Howard D, Colzani M, Arumugam M, Wu WH, Dalby A, et al. Large-scale production of megakaryocytes from human pluripotent stem cells by chemically defined forward programming. Nat Commun. 2016;7:11208doi:10.1038/ncomms11208.
- Thon JN, Mazutis L, Wu S, Sylman JL, Ehrlicher A, Machlus KR, Feng Q, Lu S, Lanza R, Neeves KB, et al. Platelet bioreactor-on-a-chip. Blood. 2014;124(12):1857–1867. doi:10.1182/blood-2014-05-574913.
- Ito Y, Nakamura S, Sugimoto N, Shigemori T, Kato Y, Ohno M, Sakuma S, Ito K, Kumon H, Hirose H, et al. Turbulence activates platelet biogenesis to enable clinical scale ex vivo production. Cell. 2018;174(3):636–648.e18. doi:10.1016/j.cell.2018.06.011.
- Tozzi L, Laurent P-A, Di Buduo CA, Mu X, Massaro A, Bretherton R, Stoppel W, Kaplan DL, Balduini A, et al. Multi-channel silk sponge mimicking bone marrow vascular niche for platelet production. Biomaterials. 2018;178:122–133doi:10.1016/j.biomaterials.2018.06.018.
- Merkle FT, Ghosh S, Kamitaki N, Mitchell J, Avior Y, Mello C, Kashin S, Mekhoubad S, Ilic D, Charlton M, et al. Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature. 2017;545(7653):229–233. doi:10.1038/nature22312.
- Baker D, Hirst A, Gokhale P, Juarez M, Williams S, Wheeler M, Bean K, Allison T, Moore H, Andrews P, et al. Detecting genetic mosaicism in cultures of human pluripotent stem cells. Stem Cell Rep. 2016;7(5):998–1012. doi:10.1016/j.stemcr.2016.10.003.
- Zhang J, Hirst AJ, Duan F, Qiu H, Huang R, Ji Y, Bai L, Zhang F, Robinson D, Jones M, et al. Anti-apoptotic mutations desensitize human pluripotent stem cells to mitotic stress and enable aneuploid cell survival. Stem Cell Rep. 2019;12(3):557–571. doi:10.1016/j.stemcr.2019.01.013.
- Na J, Baker D, Zhang J, Andrews PW, Barbaric I. Aneuploidy in pluripotent stem cells and implications for cancerous transformation. Protein Cell. 2014;5(8):569–579. doi10.1007/s13238-014-0073-9.
- Merve A, et al. c-MYC overexpression induces choroid plexus papillomas through a T-cell mediated inflammatory mechanism. Acta Neuropathol Commun. 2019;7:2.
- Soucek L, Evan GI. The ups and downs of Myc biology. Curr Opin Genet Dev. 2010;20(1):91–95. doi10.1016/j.gde.2009.11.001.
- Allison TF, Andrews PW, Avior Y, Barbaric I, Yamanaka S. Assessment of established techniques to determine developmental and malignant potential of human pluripotent stem cells. Nat Commun. 2018;9:1925
- Blin A, Le Goff A, Magniez A, Poirault-Chassac S, Teste B, Sicot G, Nguyen KA, Hamdi FS, Reyssat M, Baruch D, et al. Microfluidic model of the platelet-generating organ: beyond bone marrow biomimetics. Sci. Rep. 2016;6(1). doi:10.1038/srep21700
- Pallotta I, Lovett M, Kaplan DL, Balduini A. Three-dimensional system for the in vitro study of megakaryocytes and functional platelet production using silk-based vascular tubes. Tissue Eng. 2011;17(12):1223–1232. doi10.1089/ten.tec.2011.0134.
- Shepherd JH, Howard D, Waller AK, Foster HR, Mueller A, Moreau T, Evans AL, Arumugam M, Bouët Chalon G, Vriend E, et al. Structurally graduated collagen scaffolds applied to the ex vivo generation of platelets from human pluripotent stem cell-derived megakaryocytes: enhancing production and purity. Biomaterials. 2018;182:135–144doi:10.1016/j.biomaterials.2018.08.019.
- Stacey GN, Andrews PW, Barbaric I, Boiers C, Chandra A, Cossu G, Csontos L, Frith TJ, Halliwell JA, Hewitt Z, et al. Stem cell culture conditions and stability: A joint workshop of the plurimes consortium and pluripotent stem cell platform. Regen. Med. 2019;14(3):243–255. doi:10.2217/rme-2019-0001.
- Cardigan R, Turner C, Harrison P. Current methods of assessing platelet function: relevance to transfusion medicine. Vox Sang. 2005;88(3):153–163. doi10.1111/j.1423-0410.2005.00618.x.
- Born GV. Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature. 1962;194(4832):927–929. doi10.1038/194927b0.
- Finkenstaedt-Quinn SA, Ge S, Haynes CL. Cytoskeleton dynamics in drug-treated platelets. Anal. Bioanal. Chem. 2015;407(10):2803–2809. doi10.1007/s00216-015-8523-7.
- Roest M, Reininger A, Zwaginga JJ, King MR, Heemskerk JWM. Flow chamber-based assays to measure thrombus formation in vitro: requirements for standardization. J Thrombosis Haemostasis. 2011;9(11):2322–2324. doi10.1111/j.1538-7836.2011.04492.x.
- Feghhi S, Munday AD, Tooley WW, Rajsekar S, Fura AM, Kulman JD, López JA, Sniadecki NJ, et al. Glycoprotein Ib-IX-V complex transmits cytoskeletal forces that enhance platelet adhesion. Biophys. J. 2016;111(3):601–608. doi:10.1016/j.bpj.2016.06.023.
- Newman PJ, Aster R, Boylan B. Human platelets circulating in mice: applications for interrogating platelet function and survival, the efficacy of antiplatelet therapeutics, and the molecular basis of platelet immunological disorders. J Thromb Haemost. 2007;5:305–309. doi10.1111/j.1538-7836.2007.02466.x.
- Hu Z, Yang YG. Full reconstitution of human platelets in humanized mice after macrophage depletion. Blood. 2012;120(8):1713–1716. doi10.1182/blood-2012-01-407890.
- Rosen ED, Raymond S, Zollman A, Noria F, Sandoval-Cooper M, Shulman A, Merz JL, Castellino FJ, et al. Laser-induced noninvasive vascular injury models in mice generate platelet- and coagulationdependent thrombi. Am. J. Pathol. 2001;158(5):1613–1622. doi:10.1016/S0002-9440(10)64117-X.
- Suzuki D, Flahou C, Yoshikawa N, Stirblyte I, Hayashi Y, Sawaguchi A, Akasaka M, Nakamura S, Higashi N, Xu H, et al. iPSC-derived platelets depleted of HLA class I are inert to anti-HLA class I and natural killer cell immunity. Stem Cell Rep. 2020;14(1):49–59. doi:10.1016/j.stemcr.2019.11.011.