

Abstract
Glanzmann thrombasthenia (GT) is a rare autosomal recessive bleeding disorder characterized by impaired platelet aggregation due to defects in integrin αIIbβ3, a fibrinogen receptor. Platelet phenotypes and allelic variations in 28 Turkish GT patients are reported. Platelets αIIbβ3 expression was evaluated by flow cytometry. Sequence analyzes of ITGA2B and ITGB3 genes allowed identifying nine variants. Non-sense variation effect on αIIbβ3 expression was studied by using transfected cell lines. 3D molecular dynamics (MDs) simulations allowed characterizing structural alterations. Five new alleles were described. αIIb:p.Gly423Asp, p.Asp560Ala and p.Tyr784Cys substitutions impaired αIIbβ3 expression. The αIIb:p.Gly128Val substitution allowed normal expression; however, the corresponding NM_000419.3:c.476G>T variation would create a cryptic donor splicing site altering mRNA processing. The β3:p.Gly540Asp substitution allowed αIIbβ3 expression in HEK-293 cells but induced its constitutive activation likely by impairing αIIb and β3 legs interaction. The substitution alters the β3 I-EGF-3 domain flexibility as shown by MDs simulations. GT variations are mostly unique although the NM_000419.3:c.1752 + 2 T > C and NM_000212.2:c.1697 G > A variations identified in 4 and 8 families, respectively, might be a current cause of GT in Turkey. MD simulations suggested how some subtle structural variations in the β3 I-EGF domains might induce constitutive activation of αIIbβ3 without altering the global domain structure.
Data availability statement
Data supporting the allelic variants described in this study are openly available in the ClinVar repository (https://www.ncbi.nlm.nih.gov/clinvar/) [Citation9] by using the accession numbers provided in and II. Assessment of the clinical signification of variants, pathogenic, likely pathogenic was performed according to the ACMG guidelines [Citation10] completed by Ross et al., [Citation11]. The criteria and classification rules used are reported in the file Supporting information 4 (Table S4-1).
Site directed mutagenesis and in-vitro expression
Site-directed mutagenesis and in vitro expression in Cos-7 / HEK-293 cells were done mainly as previously described [Citation8]. Alternatively, site-directed mutagenesis was done by using the QuikChange® II XL Site-Directed Mutagenesis Kit (Agilent Technologies, Massy, France) according to the manufacturer instructions, and by using pcDNA3.1-Zeo and pcDNA3.1-Neo (Invitrogen, Cergy Pontoise, France) to transfect αIIb and β3 cDNAs, respectively.
Intron splicing, non-synonymous variation effect, and evolutionary conservation analyses
The effect of variations on intron splicing has been predicted in silico by using the Human Splicing Finder tool [Citation12]. Polyphen-2 was used to predict potential deleterious effect of non-synonymous variations [Citation13].
To study evolutionary conservation, amino acid sequences highly similar to human αIIb and β3 protein sequences were selected by a PSI-Blast analysis (https://www.ebi.ac.uk/Tools/sss/psiblast/) [Citation14] and aligned with those of 122 species from batrachian, fish, reptile, birds, and mammal (Supporting information 1, Table S5) by using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) [Citation14]. Evolutionary conservation was calculated using the Skylign tool (http://skylign.org/).
Structural analysis
Structural analyses, in-silico mutagenesis, and molecular dynamic simulations were done as previously described in our previous works with some modifications [Citation15–16]. The β3 I-EGF-3 domain was obtained from a 2.55 Å resolution crystal structure of the αIIbβ3 integrin (PDB code 3FCS) [Citation17]. Molecular Dynamics (MD) simulations were performed using GROMACS 5.5.4 software [Citation18] with the Gromos 54a7 forcefield [Citation19]. Each system was simulated through 10 independent dynamics of 100 ns for a total of 1 µs. Molecular conformations were saved every 100 ps for downstream analysis. Protein Blocks (PBs) are a structural alphabet composed of 16 local prototypes [Citation20] that give a reasonable approximation of all local protein 3D structures [Citation21]. PB assignments were done for every residue of the β I-EGF-3 domain (positions 522–560). The equivalent number of PBs (Neq) is a statistical measurement that represents the average number of PBs a residue at a given position takes. A ΔPB value was calculated to detect changes in PBs profile between the WT and variant forms. Full description of structural analysis is presented in the Supporting information 2.
Results
Following initial GT diagnosis, αIIbβ3 surface expression on platelet of the 28 patients and their close relatives was further assessed by flow cytometry using Moabs directed to αIIb, to β3, or to the complex (respectively, M148, AP3, and AP2). Results from the patients are reported in Table S2 (Supporting information 1). Most of these patients belong to Type I GT with an αIIbβ3 expression lower than 5% of the normal. Six patients (P14, P21, P22, P26 to P28) whose platelets expressed 5% to 20% of the complex were considered as GT Type II. The Moab SZ1 revealed a normal to increased GPIbIX expression that is currently observed in GT Type I platelets [Citation22].
Sequence alterations detected in the αIIb and β3 genes
Allelic variations identified in αIIb and β3 genes are reported in and II. The variations reported herein were confirmed by new Sanger sequencings of targeted DNAs sequences from patients and their relatives (not shown). All were also confirmed by family segregation analyses. Potentially unseen large deletions were ruled out by parental analyses showing the presence of both the normal and the altered alleles (heterozygous state).
Seven homozygous ITGA2B gene alterations were identified in 11 families (patients P1 to P17). Deletions αIIb:c.240_241delGA (family F1) and αIIb:c.2975_2979delAGAGG affecting exons 4 and 29 (family F11), respectively, were predicted to lead to frame shift and erroneous synthesis of αIIb (). Four non-synonymous SNVs were detected. Three were detected in distinct families: the αIIb:c.476 G > T SNV leading to the αIIb:p.Gly128Val substitution (family F2); the SNV αIIb:c.1361 G > A resulting in the αIIb:p.Gly423Asp substitution (family F3); and the SNV αIIb:c.2444A>G leading to the αIIb:p.Tyr784Cys substitution (family F10). The SNV αIIb:c.1772A>C that induces the αIIb:p.Asp560Ala substitution was shared by families F8 and F9. The variation αIIb:c.1752 + 2 T > C affecting the donor splicing site of the exon 17 was shared by the families F4 to F7.
Only two homozygous variations affecting the β3 gene were identified in eight families (). The SNV β3:c.1641 C > A induces a stop codon and a premature termination (β3:p.Cys521Ter) in the β3 synthesis (families F12 and F13). The non-synonymous SNV β3:c.1697 G > A leads to the β3:p.Gly540Asp substitution in the I-EGF-3 domain of the β3 subunit (families F14 to F20). The patient P19 (family F13) was heterozygous for both variations.
Table II. Genomic alterations identified in the ITGB3 gene coding for the β3 subunit
SNVs effect on αIIbβ3 expression in cultured mammalian cells
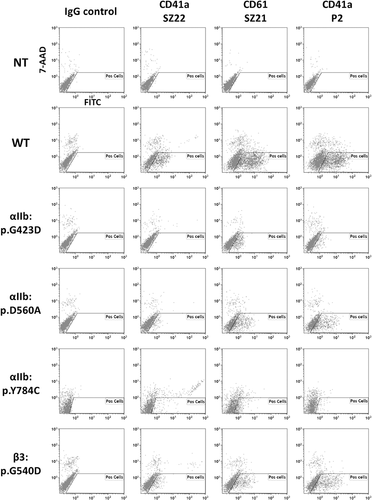
The effect of the five non-synonymous SNV was evaluated by expressing the mutated complexes in COS-7 or HEK-293 cells and flow cytometry analyses (). Compared to the WT, the expression of the αIIb:p.Asp423 and αIIb:p.Cys784 complexes was impaired whereas expression of the αIIb:p.560Ala and β3:Asp540 complexes was strongly reduced. These amounts were correlated with the phenotypes of the patients’ platelets (Supporting information 1, Table S2). However, the αIIb:p.Val128 αIIbβ3 was normally expressed at the cells surface (not shown) although the patient P2 was diagnosed as Type I GT.
Figure 1. Flow cytometry analysis of mutant complexes expressed in mammalian cells in culture. Transiently transfected HEK-293 cells expressing the WT or the αIIb:p.Gly423Asp, p.Tyr784Cys, p.Asp560Ala or the β3p.Gly540Asp mutant forms of αIIbβ3 were tested by flow cytometry using Moabs SZ22, SZ21 and P2 (directed to αIIb, β3 and αIIbβ3, respectively) or irrelevant mouse IgG. Untransfected cells (NT) were used as control. Following Moab incubation, bound IgGs were detected by using a polyclonal goat anti-mouse IgG conjugated to FITC. Washed cells were analyzed on a Navios flow cytometer. The results shown are representative of 3 experiments. The αIIb:p.Gly423Asp mutant form of αIIbβ3 was expressed in Cos-7 cells whereas WT and other αIIbβ3 mutants were expressed in HEK-293 cells. Untransfected or WT αIIbβ3 transfected Cos-7 cells gave similar results to those obtained with HEK-293 cells (not shown). The “Pos cell” gate allowed selecting live cells negative for the 7-AAD labeling.
αIIb:p.Ala560 and β3:p.Asp540 αIIbβ3 and fibrinogen binding
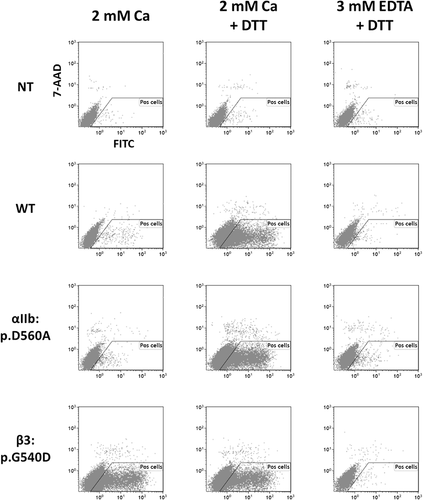
As the αIIb:p.Ala560 and β3:p.Asp540 αIIbβ3 expression was faint but significant, their functions were studied. Transiently transfected HEK-293 cells expressing either the WT or the variant forms of αIIbβ3 were incubated with fibrinogen conjugated to FITC and analyzed by flow cytometry (). The αIIb:p.Ala560 αIIbβ3 required both complex activation by 1 mM DTT and 2 mM calcium to bind fibrinogen like WT αIIbβ3. β3:p.Asp540 αIIbβ3 can bind fibrinogen in the presence of calcium but without complex activation. Untransfected cells neither bound fibrinogen whatever the experimental condition used.
Figure 2. Flow cytometry analysis of fibrinogen binding to transfected cells. Untransfected cells (NT) and transiently transfected HEK-293 cells expressing the WT or the αIIb:p.Asp560Ala or the β3:p.Gly540Asp mutant αIIbβ3 were resuspended in Tyrode buffer containing either 2 mM Calcium or 3 mM EDTA. Cell samples were treated or not by 1 mM DTT to activate the binding capacity of the αIIbβ3 for soluble fibrinogen. After incubation with FITC conjugated fibrinogen, diluted cells were analyzed for fibrinogen binding by flow cytometry. The “Pos cell” gate allowed selecting live cells negative for the 7-AAD labeling.
Variable expression of the residual β3:p.Asp540 αIIbβ3 at the platelet surface
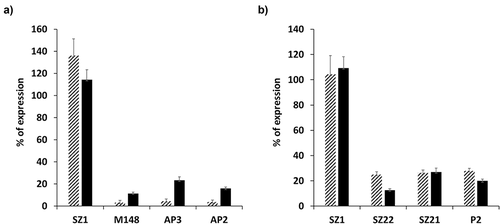
Analysis of the β3:p.Asp540 αIIbβ3 at the platelet surface revealed a first group of patients (families F15 to F17) with low residual amount whereas a second group of siblings (family F20) expressed higher residual amounts (). Membrane permeabilization revealed that both patients from the “low” or “high” group, presented similar whole platelet contents of β3:p.Asp540 αIIbβ3 (). These results suggested that the β3:p.Asp540 substitution might lead to different expressions of the residual mutant complex at the platelet surface.
Figure 3. Surface and whole platelet contents of the β3:pAsp540 αIIbβ3 allelic form. PFA fixed platelets expressing the β3:pAsp540 αIIbβ3 were incubated with Moabs SZ1, M148, AP3 and AP2 directed to GPIbIX, αIIb, β3 and the αIIbβ3 complex, respectively, in absence (panel A) or in presence of 1% saponin (panel B). Bound Moabs were detected by flow cytometry using a polyclonal anti-mouse IgG conjugated to FITC. The average percentage of expression ± SD calculated using MFIs obtained from the 3 platelets belonging to the “Low” (hatched bars) or “High” (plain bars) expression groups of patients is shown.
Impact of the Gly540Asp substitution on the I-EGF-3 structure
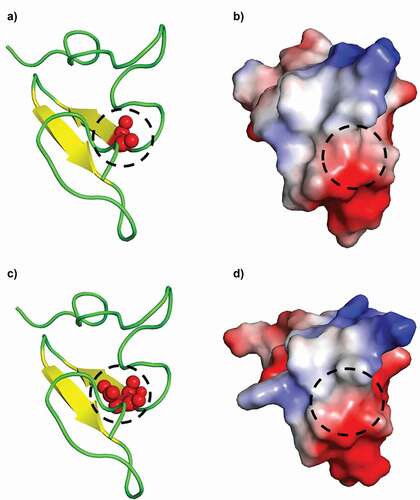
The WT I-EGF-3 and its β3:p.Asp540 variant differ by only one residue. A hydrophobic, neutral, and small amino acid glycine was changed to aspartate, a larger negatively charged amino acid (). The I-EGF-3 domain is a small globular domain mainly composed by loops with a small anti-parallel β-sheet constituted of two β-strands (positions 540–543 and 546–549). Position 540 is located at the N-terminus of first β-strand (), Gly540, and Asp540 are both well exposed. The distribution of electrostatic charges is little affected by the Gly540Asp substitution. The I-EGF-3 domain is mainly negatively charged with a large section encompassing the residue 540 (circles in ), and the only positive charged region being the loop around residue 533.
Figure 4. Visualization of the I-EGF-3 domain of β3. WT Gly540 (panels A, B) and GT variant Asp540 (panels C, D) forms of the I-EGF-3 domain are shown. Cartoons represent the peptide secondary structures (panels A, C) and its electrostatic surface (blue positive charge, red negative). Spheres (panel A) correspond to the atoms of the Glycine/Aspartic acid residues and the black dashed circles their positions (panel B).
Each system was simulated during 1 µs to ensure a correct sampling according to the size of the systems. Every simulation was carefully checked to guarantee that all classical parameters were correct. Analyses of root-mean-square deviation indicated that the WT and GT I-EGF-3 were dynamical with good flexibility (not shown).
The use of PBs allowed the precise quantification of this inner flexibility [Citation23]. For each step of the simulations, the protein structure was encoded in terms of PBs. This information provided the occurrence of each type of PB at each position as indicated in with colors coding: dark blue (PB never seen, frequency = 0) to red (this single PB is seen during dynamics, frequency = 1). These probabilities were used to compute an entropy index named Neq that ranged from 1 (a single PB is seen) to 16 (equal probability of observing every PBs). Neq can be used to define the transition from rigidity to flexibility or disorder with Neq ≥ 8 [Citation23]. Mean Neq value was 3.25 for WT I-EGF-3, i.e. close to flexible (Arrows on ), whereas it was 6.0 for GT I-EGF-3 (), meaning a huge increase in flexibility with regard to the single amino acid substitution. This result showed the impressive impact of the β3:p.Gly540Asp substitution. In addition, WT I-EGF-3 had disorder properties around residues 533 (Neq = 7.8) and 538 (Neq = 9.0), but GT I-EGF-3 changed also significantly the half C-terminus part of I-EGF-3 (e.g. Neq of position 556 increases from 1.27 [rigid] to 9.64 [disorder]).
Figure 5. Analysis of I-EGF-3 dynamics. WT Gly540 (panels A, B) and GT variant Asp540 (panels C, D) MDs are analyzed with Protein Blocks though PB maps (panels A, C) and Neq (panels B, D). Panel E shows ΔPB plot. Arrows underlined important values on Neq and ΔPB plot.
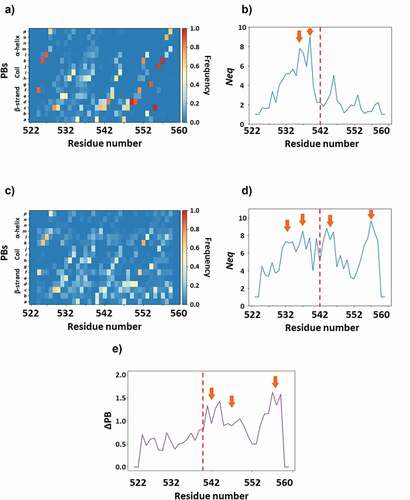
It was possible to compute a direct difference between PB distributions. shows the ΔPB, namely the sum of these differences at each position. This last plot shows an average value higher than 1 that reflects impressive changes even for a very flexible domain. Arrows in ) highlight regions with ΔPB close to 1.5 that had totally changed their dynamics. All these results showed that position 540 was originally slightly rigid with a glycine and became flexible with an aspartate. The Asp540 increased drastically the dynamics of the I-EGF-3 domain that may be considered as highly flexible with some disordered regions (arrows, ) and near complete disruption of the small anti-parallel β-sheet. It should be notice that the impact remains highly local with no change of the complete protein fold. Nonetheless, in terms of structures, the impact of β3:p.Gly540Asp substitution may be considered significant and correlated well with the platelet phenotype: The substitution alters both the expression of the complex at the cell surface (platelets and transfected cells), and its function (constitutive activation).
Discussion
Flow cytometry analyses done on patients’ platelets confirmed GT diagnosis made in Turkey. Nine variations were identified in both αIIb and β3 genes. All variations were confirmed by a second sequence analysis but also by sequencing of DNAs from their close relatives (parents and siblings). Non-synonymous and nonsense variations, deletion, and SNV affecting splice sites were identified. The αIIb:c.240_241delGA was predicted to lead to frame-shift, erroneous sequence synthesis of 21 residues following the Glu79, resulting in the deletion of most of the αIIb. The αIIb:c.2975_2979delAGAGG induced a frame-shift predicted to cause the replacement of the αIIb transmembrane and cytoplasmic domains by an erroneous sequence of 42 amino acids. The β3:c.1641 C > A led to a nonsense codon. This variation results in a premature synthesis termination in the middle of β3 (p.Cys521Ter) and the loss of all domains downstream the I-EGF-2 domain. Such variations leading to major abnormality in the primary peptide sequence of αIIb or β3 were considered as a current cause of GT and were not studied further.
The five identified non-synonymous SNVs affected highly conserved amino acids in vertebrates and mammals and were predicted to be deleterious by using Polyphen-2 (Supporting information 1, Table S3). Their effects were studied by expressing the mutant complexes in Cos-7 or HEK-293 cells (). The expression of αIIb:p.Asp423 and αIIb:p.Cys784 αIIbβ3 complexes was impaired, correlating with the platelet GT Type I phenotype. The αIIb:p.Ala560 αIIbβ3 expression in cultured cells was only reduced but platelets from patient P14 (Supporting information 1, Table S2) expressed 10% of the complex. Located in a highly conserved amino acid sequence of the Thigh domain, this substitution may affect the stability of the complex, preventing its normal expression. Polyphen-2 predicted a strong deleterious effect (1/0,999). This variation referenced in the NCBI dbSNP database (rs778608263, allelic frequency 1/250606), was discovered during whole genome studies without any reported pathology. Although the mutant complex retained the ability to bind fibrinogen (), residual amount and possible altered stability might prevent its ability to support platelet aggregation. The αIIb:p.Gly128Val substitution allowed normal complex expression in transfected Cos-7 cells (not shown) although this substitution of a highly conserved amino acid was expected to be deleterious (Supporting information 1, Table S5 and Figure S1). To explain this discrepancy, the Human Splice Finder tool predicted that the αIIb:c.476 G > T SNV might create a cryptic donor splicing site in exon 4 leading to a frame shift (αIIb:p.Gly128Alafs). The predicted erroneous αIIb synthesis may be responsible for type I GT (not shown). Whereas the αIIb:Gly128Ser substitution led to an impaired processing of the complex that resulted in Type I GT [Citation24], the αIIb:c.476 G > T SNV might rather impair the transcript synthesis.
The β3:c.1697 G > A SNV identified in this study was previously reported in two GT Type I patients [Citation25,Citation26]. To our surprise, patients P20 to P28 homozygous for this variation expressed from 2% to about 25% (Family F20, P26, 27, and 28) of residual αIIbβ3 at the surface of their platelets. They were classified accordingly as Type I or II GT (See Supporting information 1, Table S2). Furthermore, our results indicated that the β3:p.Asp540 αIIbβ3 was expressed at the surface of HEK-293 cells in low amount (). This reduced expression correlated with the GT Type II patients’ platelet phenotype (Supporting information 1, Table S2, patients P20 to P28). Nurden et al. [Citation25] only reported on the Type I GT status (<5%) of the patient without giving further detail on a possible detection of residual amount. Wihadmadyatami et al [Citation26]. used a single Moab Gi9 to evaluate the effect of this substitution on the complex expression. However, this substitution might impair the epitope of the Moab Gi9, leading to a lack of sensitivity and a failure to detect residual Asp540 β3, both in platelets and transfected CHO. Our results showed that different amounts of the β3:p.Asp540 αIIbβ3 complex could be expressed at the platelet surface (Supporting information 1, Table S2). Indeed, platelets from patients P20 to P23 expressed very low complex amount whereas patients P26 to P28 had clearly detectable amounts of mutated αIIbβ3 at the surface of their platelets (). Systematic calibration of the flow cytometer ruled out a possible experimental bias and allowed comparing the results (see the Material and methods section). The analyses of both surface and whole-platelet β3:p.Asp540 αIIbβ3 contents showed that similar internal pools () led to different surface expressions () depending on the patient family. Taken together, these observations suggested that factors other than the β3:p.Gly540Asp substitution might affect the expression of the mutated complex at the membrane. Genetic differences linked to the family would explain this apparent discrepancy of a single variation leading to Type I or II GT. Up to our knowledge such an observation is novel in GT and deserves to be further studied.
The β3:p.Asp540 αIIbβ3 expressed at the surface of HEK-293 cells can bind soluble fibrinogen in absence of complex activation (). Several substitutions in β3 I-EGF-3 or −4 domains led to a reduced expression (Type II GT) and a constitutive activation of αIIbβ3 in transfected cells [Citation27–32]. The β3:p.Gly540Asp substitution located in I-EGF-3 affects a highly conserved amino acid (Supporting information 1, Table S5 and Figure S1). Structural analyses revealed that the β3:p.Gly540Asp substitution neither altered the global structure of the domain nor its surface electrostatic charge repartition (). However, MD simulations indicated that the β3:p.Gly540Asp substitution strongly modified local flexibility. From a nearly flexible domain (average Neq of 3.25), it became highly flexible (average Neq of 6) with even disordered structure in some region (Neq > 8). Furthermore, different PBs patterns indicated strong alterations of local conformations (). In the closed bent form of the complex, the I-EGF-3 domain interacts with both αIIb Calf-2 and the β3 Thigh domains [Citation17]. The β3:p.Gly540Asp substitution likely deconstructs the interaction surfaces by modifying local flexibility and conformations. Isolation of the I-EGF-3 domain to perform structural analyzes might bias MD simulations results by introducing artificial high flexibility. Few MD simulations with a polypeptide encompassing the I-EGF-2, -3 and -4 domains indicated that the I-EGF-3 domain isolation did not impact the results reported herein (not shown). The β3:p.Asp540 αIIbβ3 synthesis occurred likely because the substitution did not alter the global I-EGF-3 structure. Nonetheless, its capacity to destabilize αIIb and β3 legs interactions might alter cell surface expression and lead to constitutive activation regarding the “Clasp” model of activation [Citation33]. The β3:p.Gly540Asp substitution like several GT Type I variations affecting the αIIb Calf-1 domain [Citation34], confirmed that amino acid substitutions may be deleterious by altering the dynamic structure of distant residues. According to the literature and our results, the β3:p.Asp540 form of αIIbβ3 was responsible of the observed GT type I/II phenotypes. Despite the ability of the constitutively activated αIIbβ3 mutants to bind fibrinogen, the residual amount of complex expressed at the platelet surface might not be sufficient to support aggregation[Citation30]. Furthermore, variations in the I-EGF-3 and I-EGF-4 domains resulting in αIIbβ3 constitutive activation led to a fibrinogen saturation of non-activated platelets that failed to aggregate upon stimulation [Citation35]. Constitutive complex activation in autosomal dominant macrothrombocytopenia can lead from mild to severe bleeding. However, no bleeding phenotype has been so far reported for the heterozygous relatives of the patients, suggesting that the decreased expression of αIIbβ3 is rather responsible for the GT phenotype than its constitutive activation.
Excepted patient P19 who was heterozygous for both deleterious β3:c.1641 C > A and β3:c.1697 G > A variations, all patients studied herein presented with homozygous GT variation. As GT is an autosomal recessive hereditary disorder, the results of this study verified the high prevalence of consanguinity (22%) described in Turkey [Citation36]. Several families sharing the same variations presented few polymorphisms already reported in the NCBI dbSNP (see rs codes in Supporting information 1, Table S3). However, the polymorphisms patterns were different confirming that these families were unrelated. The SNV αIIb:c.1752 + 2 T > C identified in 4 families (F4 to F7) was previously reported in Turkey in 6 of the 12 studied patients [Citation37]. These observations suggested that the αIIb:c.1752 + 2 T > C variation may be frequently encountered in Turkey. In the present study, most of the families inhabited around the Lake Van suggesting a possible founder effect in the East part of Turkey. Pillitteri et al. also identified this variation in a German GT patient who might have a Turkish origin [Citation38]. The β3:c.1697 G > A variation identified in 8 families (F13 to F20) may also be frequent in Turkey.
In conclusion, nine variations were identified in 28 GT patients (20 families) and five were unreported. Cell expression models allowed showing that all non-synonymous SNV but one were responsible for the Type I or II GT phenotype. The non-synonymous c.476 G > T SNV might affect mRNA splicing and lead to erroneous αIIb synthesis. All studied patients but one, were homozygous for the GT variation correlating with the high prevalence of consanguineous marriages in Turkey. The αIIb:c.1752 + 2 T > C and β3:c.1697 G > A variations identified in several families might be common in patients with GT in this country.
Notes on contributors
VJ and AGdeB designed the experiments; VJ, CC, FB and AGdeBr, conducted the experiments; MYK, NS, CA, BZ, EZ, BS, DA, BK, HA, MK, contributed for the patients; VJ and AGdeB analyzed the data; VJ, AGdeB, RP, and MYK supervised the research and wrote the manuscript; All authors approved the final version of the manuscript.
Acknowledgements
The authors are grateful to Dr. William Vainchencker for helping in the manuscript redaction. This work was supported by a grant n° 96 (2014) from the “Association Recherche et Transfusion” (ART) Paris, AGdB acknowledges 1) the French National Research Agency with grant ANR-19-CE17-0021 (BASIN); 2) to Indo-French Centre for the Promotion of Advanced Research/CEFIPRA for collaborative grant (number 5302-2); and 3) granted access to high-performance computing (HPC) resources at the French National Computing Centre CINES under grants no. A0040710426 and no. A0070710961 funded by the GENCI (Grand Equipement National de Calcul Intensif). Calculations were also performed on an SGI cluster granted by Conseil Régional Ile de France and INTS (SESAME Grant), France (n° 96, 2014).
Disclosure statement
The authors report no conflicts of interest.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.