
Abstract
The human solute carriers (SLCs) comprise over 400 different transporters, organized into 65 families (http://slc.bioparadigms.org/) based on their sequence homology and transport function. SLCs are responsible for transporting extraordinarily diverse solutes across biological membranes, including inorganic ions, amino acids, lipids, sugars, neurotransmitters and drugs. Most of these membrane proteins function as coupled symporters (co-transporters) utilizing downhill ion (H+ or Na+) gradients as the driving force for the transport of substrate against its concentration gradient into cells. Other members work as antiporters (exchangers) that typically contain a single substrate-binding site with an alternating access mode of transport, while a few members exhibit channel-like properties. Dysfunction of SLCs is correlated with numerous human diseases and therefore they are potential therapeutic drug targets. In this review, we identified all of the SLC crystal structures that have been determined, most of which are from prokaryotic species. We further sorted all the SLC structures into four main groups with different protein folds and further discuss the well-characterized MFS (major facilitator superfamily) and LeuT (leucine transporter) folds. This review provides a systematic analysis of the structure, molecular basis of substrate recognition and mechanism of action in different SLC family members.
Introduction
The solute carrier family (SLCs) are important membrane transport proteins that are ubiquitously distributed in different cells in both prokaryotes and eukaryotes. They transport diverse solutes such as inorganic ions, amino acids, lipids, neurotransmitters and drugs across biological membranes (Fredriksson et al., Citation2008; Hediger et al., Citation2004). The human SLCs are classified into 65 families based on sequence homology and function (http://slc.bioparadigms.org/). Dysfunction in most of SLCs (∼57 families) causes a wide variety of different diseases ranging from anemias in SLC4 to zinc transport defects in SLC30 (). However, studies of SLC family proteins are limited (Cesar-Razquin et al., Citation2015) compared with other “star” membrane proteins (G-protein-coupled receptors, ion channels, etc.) with few crystal structures to provide functional insights (). Here we summarized all the available SLC structures (examples in and all structures in ). With rare exceptions, most of the structures are from prokaryotic species. Among the structures from 23 SLC families reported by more than 70 research groups (), nine of them are from mammalian species, which are GLUT1 (Homo sapiens) (Deng et al., Citation2014), GLUT3 (Homo sapiens) (Deng et al., Citation2015), GLUT5 (Bos Taurus and Rattus norvegicus) (Nomura et al., Citation2015), Band 3 (Homo sapiens) (Arakawa et al., Citation2015), LeuT (Mus musculus) (Krishnamurthy & Gouaux, Citation2012), SERT (Homo sapiens) (Coleman et al., Citation2016), UT-B (Bos taurus) (Levin et al., Citation2012), ANT1 (Bos taurus) (Nury et al., Citation2005; Pebay-Peyroula et al., Citation2003) and RhCG (Homo sapiens) (Gruswitz et al., Citation2010), respectively (NMR/EM structures are not included). Six structures are from other species other than bacteria, which include DAT (Drosophila) (Penmatsa et al., Citation2013), PiPT (fungal) (Pedersen et al., Citation2013), YfKE (Waight et al., Citation2013), Aac2p and Aac3p (Ruprecht et al., Citation2014) from yeast and NRT1.1 (Parker & Newstead, Citation2014) from plant. Further, the SLC structures were grouped into four categories based on their protein folds, which include MFS fold (15 out of 44 proteins), LeuT fold (7/44 proteins), other antiparallel folds (5/44 proteins) and others (17/44) (). We mainly focus on the two largest folding categories-MFS and LeuT. This review provides a systematic overview of the currently available SLC structures, the molecular basis of their substrate binding specificity and their transport mechanism.
Figure 1. Representative available structures from different SLC families. Structures are downloaded from PDB (www.pdb.org) and presented using PyMOL (www.pymol.org). The helices were colored using rainbow from N to C terminal, with hues from blue through cyan closer to N terminal, while red through yellow closer to C terminal. Therefore, The TM regions are traceable in each structure by following the colors. Specifically, in MFS fold family members (e.g. LacY, GlpT, XylE, PepTso and EmrD), cool colors and warm colors are falls into two bundles, meaning that N and C terminal are separated into two bundles. This Figure is reproduced in color in Molecular Membrane Biology online.
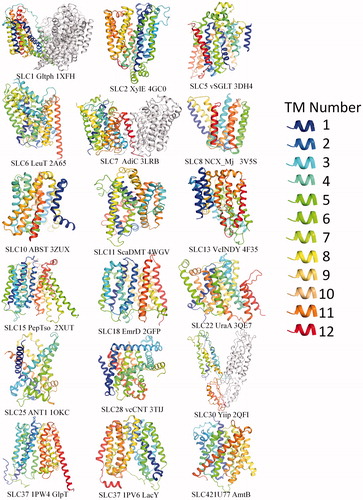
Table 1. Solute carrier family and the corresponding folds: solute carrier family and the corresponding function, folds and human diseases.
Table 2. Solute carrier family and the corresponding folds: categories of SLC structures based on folds.
MFS (6 + 6 TM) fold structures
The major facilitator superfamily (MFS) is the largest evolutionarily related super-family of secondary transporters that facilitates solute movement across the membrane (Pao et al., Citation1998). It also represents the largest fold cluster in the SLCs to date (15 proteins in ). Since 1997, MFS protein sequences shown in databases were identified based on their sequence similarity and homology (Pao et al., Citation1998). The function of MFS proteins was initially believed to transport sugars (Henderson & Maiden, Citation1990; Maiden et al., Citation1987) but following research revealed that more diverse substrates are transported by this super-family.
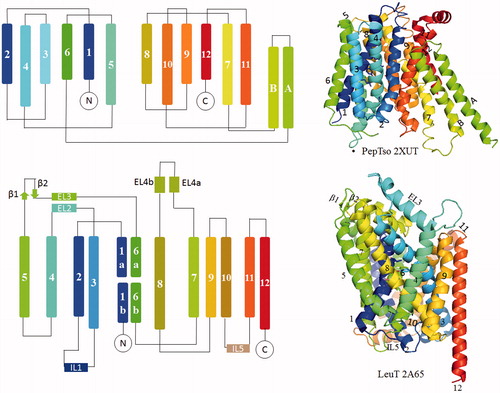
An example of the MFS fold is PepTso, which comprises 12 transmembranes (TM) segments that can be separated into two discretely folded bundles, namely, amino-(TM1-6) and carboxyl-(TM7-12) terminal bundles (domains) ( and , PepTso). Each bundle contains a “3 + 3” inverted repeats. Notably, TM helices in the amino and carboxyl-terminal bundles do not interlace with each other. The two separate bundles are usually connected by an intracellular domain or loop between TM6 and 7. In SLC families, this type of folding has been found in the glucose uniporter GLUT (SLC2 family) (Deng et al., Citation2014), D-xylose/H+ symporter XylE (SLC2 family) (Quistgaard et al., Citation2013; Sun et al., Citation2012; Wisedchaisri et al., Citation2014), Glucose/H+ symporter GlcP (SLC2 family) (Iancu et al., Citation2013), lactose/H+ symporter LacY (SLC37 family) (Abramson et al., Citation2003; Chaptal et al., Citation2011; Guan et al., Citation2007; Kumar et al. Citation2014; Mirza et al., Citation2006), phosphate/H+ symoporter PiPT (SLC22 family) (Pedersen et al., Citation2013), peptide/H+ symporter POTs (SLC15 family) (Doki et al., Citation2013; Fowler et al., Citation2015; Guettou et al., Citation2013, Citation2014; Lyons et al., Citation2014; Newstead et al., Citation2011; Solcan et al., Citation2012), nitrate transporter NRT1.1 (SLC15 family) (Parker & Newstead, Citation2014; Sun et al., Citation2014), multidrug transporter EmrD (SLC18family) (Yin et al., Citation2006) and the glycerol-3-phosphate transporter GlpT (SLC37 family) (Huang et al., Citation2003) ( and ). In addition to the TM segments, some members have their additional domains, such as the intracellular helical (ICH) domain in sugar transporters (Deng et al., Citation2014; Oka et al., Citation1990; Sun et al., Citation2012), and the two intracellular helices in POT family (Solcan et al., Citation2012).
Figure 2. Left, topology diagrams of MFS fold representative protein PepTso and LeuT fold protein LeuT. Right, structures are shown as cartoon using PyMOL (www.pymol.org). This Figure is reproduced in color in Molecular Membrane Biology online.
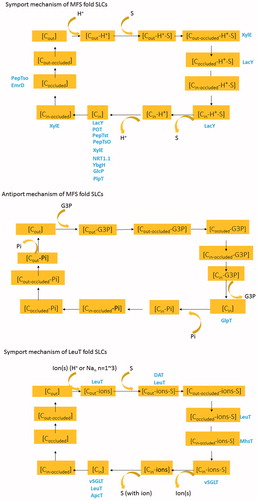
Most of the above MFS structures were captured in an inward-facing conformation (). For example, structures of POT family members POT, PepTso, PepTSt and YbgH (Zhao et al., Citation2014) were shown in inward-facing conformation either in the apo or substrate-bound state (PepTso bound with alafosfalin, PepTSt bound with Ala-Phe dipeptide and tri-Ala). The nitrate/H+ symporter NRT1.1 was crystallized facing towards the cytoplasmic side, either ligand-free or with nitrate bound. The structure of PiPT was shown in an inward-facing occluded state with a blocked entry pathway and a less occluded cytosolic side (Pedersen et al., Citation2013). The configuration of GlpT faces the cytoplasm as well. The EmrD structure represents an intermediate state (Yin et al., Citation2006). For most of this family of transporters, it seems the outward facing states represent a higher energy or a transient state within the transporting cycle, which has been harder to capture. It also suggests that the conformational change bringing the empty carrier to the outward facing state may be the rate-limiting step in the transport cycle. LacY is the most heavily studied member in MFS family, with most of the structure exhibits inward conformations (Kaback et al., Citation2011). In 2014, Kumar et al. (Citation2014)reported an almost occluded structure with galactopyranoside bound using the conformationally trapped double-Trp mutant Gly46Trp/Gly262Trp. Since the structure represents a slightly periplasmic open conformation and a tightly sealed cytoplasmic side, it suggests that intermediate conformation exists between open and closed states. In 2016, LacY was successfully crystallized in a periplasmic-open conformation using nanobody (Jiang et al., Citation2016).
Figure 3. (A) Symport model of MFSs fold SLC proteins based on structures from sugar transporters (LacY, XylE), POT family proteins (POT, PepTst, PepTso, YbgH), NRT1.1, EmrD and PipT. Specifically, sequential release in PipT seems different from the other transporters by releasing the proton first, followed by exist of substrate. C: conformation. Cout: outward conformation. Cin: inward conformation. Cin-occluded: Occluded conformation. Protein structures captured in corresponding conformations were colored blue. (B) Antiport model of MFSs fold SLC proteins based on structures from GlpT. (C) Symport model of LeuT fold SLC proteins based on structures from LeuT, DAT, MhsT, ApcT and vSGLT. This Figure is reproduced in color in Molecular Membrane Biology online.
For other sugar transporters, several members were also crystallized recently in the outward-facing state either with antibody or inhibitors (Sun et al., Citation2012). For example, the recently reported human GLUT3 was crystallized in the outward state bound with d-glucose or the exofacial inhibitor maltose (Deng et al., Citation2015), and GLUT5 was crystallized in the outward-facing state using an antibody (Nomura et al., Citation2015).
Features of substrate binding sites
Substrate binding sites are largely dependent on the properties of substrates
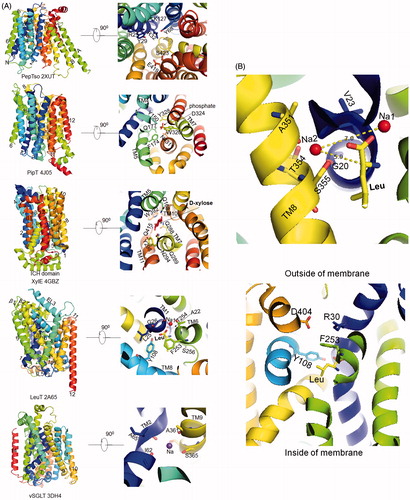
The molecular features of substrate binding sites in different families vary depending on the properties of the substrates. In the POT family, the substrate peptide is arranged within a cavity formed by opposite charges at two ends and a cluster of aromatic residues in between. In the earliest reported structure of PepTso, the substrate was supposed to be coordinated by three conserved positively charged residues Arg25, Arg32 and Lys127 on its N-terminal. Close to this are two conserved tyrosines, Tyr29 and Tyr68. The C-terminal side of the substrate binding site is the strictly conserved Glu419 and Ser423 (Newstead et al., Citation2011) (, PepTso). In the later reported alafosfalin (dipeptide analog) bound POT, the phosphonate group in the substrate is recognized by the guanidinium group of Arg43, the side-chain amide group from Gln310 and the hydroxyl groups of Tyr40 and Tyr78, whereas the N-terminal amino group is recognized by the conserved Glu413 (TM10) and Asn342 (TM8) (Doki et al. Citation2013). Similarly to the PepTso and POT, the clusters of opposite charges within the binding site were also indicated in the oligopeptide transporter YbgH (Zhao et al., Citation2014).
Figure 4. (A) Close-up view of the substrate/ion binding sites. Substrate binding sites of PepTso, PipT, XylE and LeuT and Na site of vSGLT were shown using Pymol. Bonds between atoms were not shown for clarity. For PepTso, since the presence of substrate is based on predictions from literature and map density, real substrate was not able to be shown here. (B) Top, Na2 site of LeuT. Distance between Na2 and Na1 (Leu substrate) were shown. Bottom, extracellular gate of LeuT. This Figure is reproduced in color in Molecular Membrane Biology online.
This type of substrate binding arrangement has an important role in the recognition and orientation of different peptides. Peptides with various lengths, sequences and charges could be adapted appropriately within the cavity by the interactions among these hydrogen bond donors and acceptors. A detail investigation on the substrate binding site was reported in Guettou et al. (Citation2014) who defined the roles of the pockets for the recognition of peptide side chains with different subtypes. In the same year, crystal structures of di- and tripeptide-bound PepT1 exhibited a laterally orientated binding mode for dipeptide, and an alternative vertical binding mode for tripeptide (Lyons et al., Citation2014). This suggests some degree of conformation flexibility in substrate-binding site of the transport protein for binding different peptides.
The nitrate binding site of NRT1.1 is not precisely confirmed due to the resolution limit (3.25 Å) of the structure (Sun et al., Citation2014). His356 on TM7 is the only polar residue that closes to the substrate binding site and was proposed to stabilize the substrate through charge–charge interactions. Since the His356 is not conserved in other NRT (nitrate transporter) family proteins, this residue probably enabled NRT1.1 to have a high-affinity (Liu & Tsay, Citation2003) binding for nitrate.
The phosphate substrate of PipT was coordinated by Tyr150 on TM4, Gln177 and Phe174 on TM5, Trp320, Asp324, Tyr328 on TM7 and Asn431 on TM10 (, PipT) by a combination of possible hydrogen bonds and electrostatic interactions (Pedersen et al., Citation2013). Asp324 was proposed to be protonated before engaging the phosphate. Importantly, the positive charge of Lys459 is conserved at the corresponding position in organic anion transporters, such as in OAT3 (Arg454), whereas in organic cation transporters such as in OCT1 (Asp 474), the charge is reversed (Arg changed to Asp), confirming the function of the residue at this site in substrate charge specificity.
In the G3P/Pi antiporter GlpT, Arg45 from H1 (TM1) and Arg269 from H7 (TM7) most likely contribute to both the binding of the substrate G3P and Pi (Yin et al., Citation2006). EmrD was reported to export a broad spectrum of hydrophobic substrates (Pao et al., Citation1998). The substrate was proposed to be mainly stacked with two pairs of stacked aromatic groups (Tyr52/Tyr56 and Trp300/Phe249) together with other hydrophobic residues (Ile28, Ile217 and Ile253).
H-bonds are common in coordinating substrate sugars through their hydroxyl groups. Most of the substrate binding sites in sugar transporters were inferred from the substrate-free density map. In 2012, Sun et al. (Citation2012) reported that d-Xylose is bound to the outward-facing state, partly occluded XylE through polar (4 Glns and 2 Asns) and aromatic residues (Trp, Phe and Tyr) mainly from the C-terminal bundle. The hydroxyl groups of D-xylose are coordinated by eight hydrogen bonds, which are involved in the interactions with Gln168 (TM5), Gln288/Gln289/Asn294 (TM7), Trp 392 (TM10) and Gln415 (TM11) (, XylE). Besides, the substrate also indirectly connects to Tyr298 and Gln415 through water-mediated hydrogen bonds. Several aromatic residues in the vicinity of the substrate are predicted to prevent the escape of d-xylose to the extracellular environment. In 2014, a comparison of d-glucose-binding in the outward-facing XylE with the electron density of β-NG in the inward-facing human GLUT1 implied a similar substrate coordination (Deng et al., Citation2014). The only difference between the outward-facing XylE and the inward-facing GLUT1 lies in the N-terminal bundle, which partially coordinates the substrate when the protein is in the outward state but loses its binding function in the protein’s inward-facing state. The glucose binding site of GlcP is similar to those in XylE and human GLUT (Iancu et al., Citation2013), all corresponding to human GLUT1 residues: Gln161, Gln282, Gln283, Asn288 and Trp388. The recently reported GLUT3 and GLUT5 also showed similar hydrogen bond-dominated binding pattern (Deng et al., Citation2015; Nomura et al., Citation2015). The overlap of substrate binding sites in proteins’ different states suggests that a single sugar-binding site is alternately accessible from either side of the membrane, which is also indicated in other studies (Jardetzky, Citation1966; Smirnova et al., Citation2011).
The lactose permease LacY is the best-studied sugar transporter in the SLC37 family and has been considered as the model for MFS fold proteins (Kaback et al., Citation2011). The outward-occluded structure of LacY clearly defined a TDG (lactose homolog, β-d-galactopyranosyl-1-thio-β-d-galactopyranoside) binding by the hydrogen bonds with surrounding residues (Kumar et al., Citation2014), consistent with the biochemical analysis (Guan & Kaback, Citation2006; Kaback et al., Citation2001). These interactions are mainly from hydrophobic stacking between TDG galactopyranosyl ring and Trp151 indole ring, the hydrogen bonds between TDG and Glu269 (helix VIII, or TM8), Arg144 (helix V), Glu126 (helix IV), His322 (helix X) and Asn272 (helix VIII).
Most substrate binding occurs on the symmetry axis between the N and C terminal bundles
The substrate-binding sites in MFS fold transporters are primarily located at the interface between the N- and C-terminal bundles. For example, the central cavity of PepTso is mainly contributed by residues from N-terminal TM 4, 5 and C-terminal TM 10 and 11 (Newstead et al., Citation2011). The inorganic phosphate in PipT was found buried in the center of the protein at the interface (Pedersen et al., Citation2013). “An island of strong electron density” in NRT1.1 is in the middle of the transport tunnel between the N-terminal and C-terminal domains represent the substrate binding site (Sun et al., Citation2014). The putative substrate binding site in YbgH was also formed mainly by residues on the interface from N terminal TM 1, 4 and C terminal TM 8, 10 (Zhao et al., Citation2014). The substrate binding sites in the other MFS fold proteins showed similar location at the interface. This common feature indicates the relative movements between N and C terminal domains in a rocker-switch type of motion provides alternating access of the substrate binding site to the outside and inside during the transport cycles. However, the movement between the two domains is considered not rigid since most of the binding sites are unevenly distributed on either N terminal (e.g. LacY, PepTso2) or C-terminal bundle (e.g. GLUT, PipT and XylE), which is also indicated by Yan (Citation2015). In these cases, one domain provides most of the ligands for substrate binding.
Ion binding and coupling
In antiporters, substrates that are exchanged across the membrane usually compete with each other for a single binding site (Fluman et al., Citation2012), whereas in symporters, ion binding and substrate binding usually are coupled to be efficient (Jiang et al., Citation2013). In H+-coupled symporters, protonation was proposed to lower the energy barrier for substrate binding. For example, in PipT, a sequential release of protons followed by phosphate release between the N and C terminal bundles is supported by molecular dynamic simulations (Pedersen et al., Citation2013). Protonation of Asp22 in GlcP caused a disruption of a salt bridge between Asp22 (TM1) and Arg102 (TM4) and loosens the packing between TM1 and 4, enabling the subsequent glucose binding (Iancu et al., Citation2013). In NRT1.1, the protonation of His356 together with the subsequent nitrate binding were proposed to trigger the closure of the extracellular gate. Additional protonation sites in NRT1.1 were predicted to be within a conserved ExxER motif (TM1) (Parker & Newstead, Citation2014).
A detail description of the importance of the sequential binding mechanism in symport mode was raised in the protonation mimic structure GtPOT Glu310Q (Doki et al., Citation2013). The substrate peptide cannot bind to the central pocket before proton binding due to the electrostatic repulsion between the negative charge of deprotonated Glu310 and the substrate carboxylate group. An outward to inward conformation transition was supposed to occur only when both protonation and peptide binding happens sequentially, as the weak interaction between Arg43 and protonated Glu310 could not be the driving force to bring the N- and C-terminal bundles close together when the only proton is bound. A similar conclusion was also reached in the YbgH structure in which substrate binding happens after Glu21 is protonated (Zhao et al., Citation2014).
In terms of the energy landscape for transport, antiporters follow another rule completely different from symporters. In the assumed model of the antiporter GlpT (Huang et al., Citation2003), the Pi and the substrate G3P binding site were supposed to share the same ligand residues Arg45 and Arg269, implying a competing relationship between the Pi and G3P at a single binding site. Antiporters undergo a conformational change from inward- to outward-facing states in the presence of substrate, but are unable to carry out this conformational change in the absence of substrates. In contrast, symporters undergo an outward to inward conformational change in the presence of substrates but can return empty to an outward-facing state without any bound substrate. Put another way the energy barrier in antiporters for the empty carrier is high and binding substrates lowers the barrier. In symporters, the energy barrier for the movement of the empty carrier to the outward-facing state is not prohibitively high.
The gates
Transporters usually undergo local conformational changes in response to ligand binding and releasing, such as the closing of a gate on one side while opening a gate on the other side, or by forming a transient occluded state by closing both ends. During transport, the substrate is not accessible to both sides of the membrane at the same time due to the presence of a gate that transiently blocks the transport pathway. The extracellular gates of MFS fold SLCs were well studied since most of the structures were captured in inward-facing states. Many of these bacterial structures showed thick periplasmic barriers formed by clusters of different type of bonds, implying a strong prevention of the leakage or escape of ions and substrates after the transporter is fully opened to the cytoplasmic side.
The residues forming the gates in the POT family seem controversial. Different from EmrD where hydrophobic residues form the extracellular gate (Yin et al., Citation2006), the extracellular gate in PepTso was proposed to be hydrophilic (Newstead et al., Citation2011), consistent with the role of EmrD as a hydrophobic compound transporter while PepTso is a peptide transporter. However, the structure of POT with a higher resolution at 2.4 Å seems more conceivable (Doki et al., Citation2013). The extracellular gate is tightly closed by hydrophobic and hydrogen bond interactions between the N- and C-terminal bundles in preventing the leakage of proton and substrate. The inward open state of NRT1.1 also showed an extracellular gate formed by TM1 and 2 packing against TM7 and 8 with extensive hydrophobic interactions (Parker & Newstead, Citation2014). In PipT, the entry pathway of the substrate phosphate into the transporter is blocked by a cluster of phenylalanine residues (Phe50, Phe327 and Phe369) (Pedersen et al., Citation2013). The outer cellular gate of the inward-facing apo GlpT is composed of nine aromatic side chains from TM1 and 7 which forms a 22 Å thick barrier closing the pore completely (Huang et al., Citation2003). In the inward-open conformation of XylE, the substrate is occluded from extracellular side by van der Waals and hydrophobic interactions (Quistgaard et al., Citation2013). The inward-open GLUT1 gate is dominated by a cluster of hydrogen bonds formed by residues from TM1 and 7 (Deng et al., Citation2014). The extracellular gates are diverse in different MFS proteins, and there is no evidence indicating a clear relevance between the types of substrates and the category of gates.
The intracellular gate of PepTso involves Leu427 on TM10 packing against Tyr154 and Phe150 on TM4, forming part of the highly conserved POT family PTR2_2 motif (FYxxINxG) and probably regulate the exit of peptides (Newstead et al., Citation2011). The intracellular gates of PepTst are formed by cytoplasmic ends of TM10 and 11 with a hinge region located around Gly407 (TM10) and Trp427 (TM11) (Lyons et al., Citation2014). In NRT1.1, the intracellular gate is proposed in between the Lys164 and Glu476 (Parker & Newstead, Citation2014). Interestingly, the sugar porter family showed a unique feature-the intracellular helix (ICH) domain. The ICH domain linking the N and C terminal domain in XylE and GLUT1 is proposed to be the latch that tightens the intracellular gate (Deng et al., Citation2014; Quistgaard et al., Citation2013). Numerous interactions exist on the interface between the ICH and cytosolic part of TM domain, including salt bridges from Glu153-Arg404, Arg160-Glu397 and Arg225-Glu472 and are important in the closing the door (Quistgaard et al., Citation2013). Recently in 2014, the outward-occluded state LacY was reported by Kumar et al. (Citation2014) by mutating two glycines into two tryptophans. This structure implied a tightly sealed zipper-like motif composed of Glu64 (helix II), Thr348 (helix XI), Tyr350 (helix XI), Phe334 (helix X), Phe140 (helix V), Gly141 (helix V) and Pro280 (helix VIII). However, due to the lack of enough outward-facing structures, the intracellular gates of MFS fold transporters still need investigation.
Conformational changes: alternative access mechanism
The alternating access model earlier described indicates that the central substrate binding site in the protein is accessible either from the outside or from the inside of the cell (Jardetzky, Citation1966; Vidaver, Citation1966; Widdas, Citation1952). Now that transporter structures are available, the model has been developed in greater molecular detail. In the current discussed MFS fold SLC transporters, the proposed alternating access mechanism can vary in the details.
The working mechanism of the antiporter EmrD was mostly based on comparison with previously reported structures (Yin et al., Citation2006). The substrate was proposed to enter the internal cavity either through the inner phospholipid leaflet or via the cytoplasm. However, many details including how the proton is coupled to the substrate transporting and how the alternating access is realized remain unclear.
In the antiporter GlpT, the substrate binding was proposed to lower the energy barrier to facilitate the interconversion between inward and outward state, and to allow the outward Pi gradient to drive G3P uptake (Huang et al., Citation2003).
The alternating access model reported in the uniporter hGLUT is quite unique in its “competing effect” on the affinity between N and C terminal bundle (Deng et al., Citation2014). Residues surrounding the substrate were predicted to undergo a rearrangement upon substrate binding. The protein may switch to inward conformation only when the binding affinity of the N- and C-terminal domain on the extracellular side surpasses that of the intracellular side. Also, the empty carrier must return to the outward-facing state in the absence of bound substrate.
For the PipT and POT family symporters, substrate binding after protonation was proposed to trigger the closure of the extracellular gates in NRT1.1 (Parker & Newstead, Citation2014; Sun et al., Citation2014), PipT (Pedersen et al., Citation2013) and PepTst (Lyons et al., Citation2014) and the gate might even be strengthened by a salt bridge between Arg33 and Glu300 (Lyons et al., Citation2014). Upon transition to the occluded state, the intracellular gate opens to facilitate the substrate/ion the access to the cytoplasm. The opening of the gate seems commonly accomplished by breaking salt bridges in several transporters (Lyons et al., Citation2014; Parker & Newstead, Citation2014). Interestingly, PiPT seems different from others (Pedersen et al., Citation2013). It was proposed that exposure of the negatively charged cytosolic tunnel in PiPT pulls a proton before the substrate release, and the resulting electrostatic repulsion between the deprotonated binding site and phosphate force the phosphate to exit to the cytoplasm.
In GlcP, the effect of protonation or deprotonation of Asp22 on the salt bridge between Asp22 and Arg102 appears to be key for the transporter to overcome the energy barrier between outward and inward conformation. Binding of glucose after protonation was proposed to bring the N- (helix 5) and C-terminal (helices 7 and 10) domains closer together to close the periplasmic gate (Iancu et al., Citation2013).
The alternating access to the substrate binding site was speculated to be achieved through the rotation of N and C terminal bundles pivoting around the bound substrate (Guan & Kaback, Citation2006; Law et al., Citation2008). However, with the existence of several intermediate structural snapshots, including the occluded EmrD (Yin et al., Citation2006) and PepTso (Newstead et al., Citation2011), the inward occluded GlcP (Iancu et al., Citation2013) and PiPT (Pedersen et al., Citation2013) with phosphate, the outward partially occluded XylE, and the occluded LacY bound to sugar (Kumar et al., Citation2014) all suggest that more than two intermediate states may exist beside the typical inward and outward conformations. This, on the other hand, rejects the model of a simple rigid body rotation between N and C terminal bundles in MFS fold proteins. What's more, the recently reported outward facing GLUT3 (Deng et al., Citation2015) and GLUT5 (Nomura et al., Citation2015) structures further indicate that at least sugar porters may rotate mainly through their C-terminal bundle while transferring between inward or outward states.
A large number of structures captured in the inward-facing state indicates that this state may represent a low free energy conformation in MFS fold proteins as indicated previously (Forrest et al., Citation2011), even when the ligands are bound. However, sugar porters seem to be an exception. XylE bound to its substrate was captured in an outward occluded state, as was also seen in the substrate-fully ligated LacY (Kumar et al., Citation2014), GLUT3 (Deng et al., Citation2015), GLUT5 (Nomura et al., Citation2015) and MFS transporter FucP (Dang et al., Citation2010), whereas other MFS transporters were found either in the occluded state (Newstead et al., Citation2011) or inward occluded state (PipT with phosphate) (Pedersen et al., Citation2013) when bound to substrate. The phenomenon indicates a possible larger energy barrier that exists in sugar porter than other proteins (POT/PiPT proteins, etc.) during the outward-to-inward conformation transitions. So Iwata et al. suggested that in GLUT members, those with salt bridges at the cytosolic side may favor the outside facing state (Nomura et al., Citation2015), which provides an explanation for the conformations of GLUTs. Finally, the preferred conformations in different structures seem to have no correlation with the classification of the transporters on whether it is an antiporter, uniporter or a symporter.
Relationship with diseases and drugs
Many of current available SLC homolog or paralog structures that are in MFS fold have medical or pharmacological relevance (). For example, the POT transporters which are in the intestinal track provide a major route for the uptake of orally administered drugs such as β-lactam antibiotics (Faria et al., Citation2004; Smith et al., Citation2013; Tamai et al., Citation1997; Wenzel et al., Citation1995). Therefore, a structural investigation of this family is essential to understand the molecular basis of their substrate recognition and substrate transport. This knowledge may also help to improve the pharmacokinetic properties of peptide prodrugs such as valacyclovir (Ganapathy et al., Citation1998) and val-val-lopinavir (Brandsch, Citation2009).
For sugar transporters, dysfunction of mammalian glucose transporter GLUT4 causes type II diabetes (Kahn, Citation1992), and a GLUT1 mutation is correlated with glucose transporter 1 deficiency syndrome, including infantile seizures and developmental delay (Escary et al., Citation2000). Mutations of human glucose-6-phosphate transporter (G6PT) generate glycogen storage disease type 1b (Chen et al., Citation2002). The first human MFS fold structure is from glucose uniporter GLUT1, which is presented by Deng et al. (Citation2014). In this study, the disease-causing mutants were mapped onto the 3D structure and were divided into three clusters. Cluster I contains residues surrounding substrate binding, while cluster II is at the interface between the TM domain and the intracellular ICH domain and Cluster III is situated mainly on the extracellular side. This atomic resolution mapping confers a potential interpretation that the disease may be caused either by a failure of substrate binding, conformational changes associated with transport, or more general defect in protein folding. However, since most MFS structures are from prokaryotic species, more human structures and models are needed to provide the precise molecular mechanism that leads to various diseases.
LeuT (5 + 5) fold transporters
In addition to the MFS fold, a strikingly similar LeuT (Leucine Transporter) fold was also observed in several seemingly unrelated SLC structures ( and ), including the neurotransmitter/Na+ symporter LeuT (Kantcheva et al., Citation2013; Krishnamurthy & Gouaux, Citation2012; Kroncke et al., Citation2010; Malinauskaite et al., Citation2016; Piscitelli & Gouaux, Citation2012; Quick et al., Citation2009; Singh et al., Citation2007, Citation2008; Yamashita et al., Citation2005; Zhou et al., Citation2007, Citation2009; Wang & Gouaux, Citation2012; Wang et al., Citation2012, Citation2013 ), the amino acid/H+ symporter ApcT (Shaffer et al., Citation2009), the dopamine transport/Na+ symporter DAT (Penmatsa et al., Citation2013), the neurotransmitter/Na+ symporter MhsT (Malinauskaite et al., Citation2014) and serotonin/Na+ symporter SERT (Coleman et al., Citation2016), the divalent metal cation/H+ symporter scaDMT (Ehrnstorfer et al., Citation2014) and the Galactose/Na+ symporter vSGLT. The common fold usually features a “5 + 5” inverted repeat with an anti-parallel orientation and a pseudo-symmetry axis in the membrane plane (, LeuT). The first two helices within each repeat form a four-helix bundle (core domain, TMs 1, 2, 6 and 7). This bundle is surrounded by a second bundle (also called scaffold hash domain) contributed by the third and fourth helices from each repeat (TMs 3, 4, 8 and 9). Notably, the first TM of each repeat usually contains an unwound region involved in ligand binding and transport.
In spite of the similarity of the overall fold, different transporters have their own specific structural modifications. For example, the Galactose/Na+ symporter vSGLT contains 14 TMs with a central “5 + 5” inverted repeat and extracellular amino- and carboxyl-termini (Faham et al., Citation2008; Watanabe et al., Citation2010). The structure of DAT was “modified” by including an additional kink in TM12 halfway across the membrane, a cholesterol molecule wedged within a groove formed by TM1a, 5 and 7, and a C-terminal latch which caps the cytoplasmic gate that was proposed to be involved in protein kinase C-mediated endocytic trafficking (Holton et al., Citation2005; Kristensen et al., Citation2011; Penmatsa et al., Citation2013), as well as the interactions with IL1 and TM1a (Penmatsa et al., Citation2013). The Neurotransmitter/Na+ symporter MhsT (Malinauskaite et al., Citation2014) and metal/H+ symporter ScaDMT (Ehrnstorfer et al., Citation2014) have 11 TMs. An unwound region was also observed in MhsT (Malinauskaite et al., Citation2014), which was proposed to create a solvent pathway to the Na2 site from the cytoplasmic side.
Features of substrate binding sites
Substrate binding sites are located within non-α-helical regions of TMs and are usually connected with ion binding sites
The substrate binding site in LeuT is formed by residues mainly from TM1, 6 and TM3, 8 (Yamashita et al., Citation2005) (, LeuT). Among all the interactions, polar residues in TM1 and 6 make most of the contacts to the bound leucine. The amino group of leucine interacts with the main-chain carbonyl oxygens of Ala22 (TM1), Phe253 and Thr254 (TM6) and a side-chain hydroxyl from Ser256 (TM6). The carboxyl group of leucine is connected directly with Na+, the amide nitrogens of Leu25 and Gly26 (TM1), and the hydroxyl from Tyr108 (TM3) (, LeuT). On the other hand, the side chain of the leucine is cradled within a hydrophobic pocket contributed by Val104 and Tyr108 (TM3), Phe253, Ser 256 and Phe259 (TM6), Ser355 and Ile359 (TM8). Clearly many interactions, both side-chain and back-bone, contribute to the specificity of substrate recognition by LeuT.
In addition to the primary Na+ binding site (S1 site) in LeuT, a second binding site (S2) was proposed and supported in different experiments (Khelashvili et al., Citation2013Quick et al., 2009, 2012; Shan et al., Citation2011; Shi et al., Citation2008; Zhao et al., Citation2011). It was showed that the S2 site is 10 Å higher than S1 towards the extracellular environment (Shi et al., Citation2008). Although the results were supported by several experimental studies, existence of this S2 binding site is still controversial. For example, in one study, the reason that one could not obtain a structure with the S2 site occupied was attributed to the interference of the detergent β-OG (Quick et al., Citation2009). However, binding experiments performed in detergents and crystals grown in other detergents/micelles (Khelashvili et al., Citation2013; Quick et al., Citation2012; Wang et al., Citation2012) do not solidly support the existence of a S2 site.
The general locations of the substrate binding sites in LeuT (S1 site), ApcT and MhsT seem conserved. The putative substrate binding site in the inward-occluded ApcT overlaps with that of LeuT, mainly formed by residues from non-α-helical regions of TM1 and 6, TM3 and 8, IL1 and EL4 (Shaffer et al., Citation2009). The outward facing DAT is stabilized by a TCA antidepressant nortriptyline, which binds in a pocket that equivalent to the leucine-binding region of LeuT (Penmatsa et al., Citation2013). This pocket is also in close proximity to the densities of sodium and chloride ions. Within the pocket, the substrate nortriptyline is coordinated by residues mainly from TM1, 3, 6 and 8 through hydrophobic interaction (Dibenzocycloheptene ring bound to Val120, Tyr124 and Ala117), hydrogen bond interactions (amine group bound to Phe43, side chain bound to Tyr124), cation–π interaction (amine group to Phe43), as well as the interaction between the tricyclic group to Gly425 (TM8) and Ala479 (TM10). Similar to DAT, a strong density of sodium ion could be observed in SERT coordinated with the inhibitor (S)-citalopram (Coleman et al., Citation2016). However, the ion-coupling and the transport mechanism are still not clear and need further exploration in SERT.
The residues involved in the coordination of sugars are quite different from the above transporters. In vSGLT (Faham et al., Citation2008), although the lactose is bound about halfway across the membrane bilayer similarly to the substrate in the above transporters, the coordination residues are unique. Lactose was found interacting with the Gln428 (TM11), Gln69 (TM2), Lys294 (TM8), Ser79 (TM3), Tyr263 (TM7), Asn260 (TM7) and possibly Tyr87 (TM10) through many hydrogen bonds, which is a common feature in sugar transporters. Interestingly, a similar coordinating role of Lys294 is also observed in the MFS fold LacY and other sugar transporters, indicating a potential conserved substrate binding mechanism within the sugar transporters no matter whether they belong to MFS or LeuT fold.
In the metal transporter ScDMT (Ehrnstorfer et al., Citation2014) the position of the Mn2+ binding site was observed from the strong anomalous difference electron density. The Mn2+ ion is coordinated by the carbonyl oxygen of Ala223 and Thr224 (TM6) and the side chains of Met226 (TM6), Asp49 and Asn52 (TM1). Residues contributing to ion–side-chain interactions are identical in ScaDMT and human DMT1 and are strongly conserved within the family.
Ion binding and coupling
Substrates and ions are in a stoichiometry ranging from 1:1 to 1:3, and ion binding sites of several SLCs superimpose with the Na2 site in LeuT
Two sodium ions are important in stabilizing the LeuT core region (the unwound part of TM1 and 6 together with the bound leucine) (Yamashita et al., Citation2005). Sodium ion binding site 1(Na1) is directly linked to the leucine carboxyl oxygen, and the carbonyl oxygens of Ala22 (TM1) and Thr254 (TM6) (), the hydroxyl oxygen of Thr254, the side-chain carbonyl oxygens of Asn27 (TM1) and Asn286 (TM7). Particularly, Asn27 is fully conserved across the Neurotransmitter Sodium Symporter (NSS) family, whereas Asn286 and Thr254 (TM6) are replaced with aspartate and serine respectively in other NSS transporters. Compared with Na1 site, the Na2 site is less conserved. Na2 is located between the TM8 and TM1 unwound region, a distance of 7.0 Å away from Na1 and 5.9 Å from the substrate α-carbon. It is coordinated by carbonyl oxygens of Gly20 and Val23 (TM1), the side chain hydroxyl oxygens of Ala351 (TM8), Thr354 and Ser355 (, left).
Different from LeuT, which couples the leucine and ions with a stoichiometry of 1:2, vSGLT exhibits a 1:1 stoichiometry with the Na+ binding site corresponding to Na2 site in LeuT (Watanabe et al., Citation2010). Two sodium binding sites were also reported in MhsT (Malinauskaite et al., Citation2014). The Na1 site of MhsT is similar to the LeuT Na1 site, whereas the Na2 site in MhsT differs from the Na2 in LeuT by adding an additional water molecule. The situation in DAT is more complicated since three ion binding sites (two Na+ and one Cl− binding sites) were reported (Penmatsa et al., Citation2013). Na1 site is indirectly bound to Asp46 (TM1) through water. The Na2 site is coordinated between TMs 1 and 8 within a so-called “trigonal bipyramidal fashion” through interaction with main-chain carbonyls of Gly42 (TM1a), Val45 (TM1-hinge), Leu417 (TM8) and the side-chain oxygens of Asp420 and Ser421 (TM8). The Cl3 site is located within a “tetrahedral fashion” formed by residues Ser320, Gln316 (TM6), Ser356 (TM7) and Tyr 69 (TM2). Similarly, the proton binding site in ApcT also superimposed on the Na2 site in LeuT (Shaffer et al., Citation2009), with the protonation site concluded to be at Lys158.
Ion binding/releasing is directly coupled with the substrate binding and release
The Na+ site of vSGLT is located ∼10 Å away from the galactose binding site between TM2 and 9 (Faham et al., Citation2008), which is contributed by Ala62 (TM2, similar to TM1 of LeuT), Ile65 (TM2 discontinuous region), Ala361 (TM9, similar to TM8 of LeuT) and the conserved Ser365 (TM9) (, vSGLT). A similar binding site is also reported in Watanabe et al. (Citation2010) using molecular dynamic simulations. Based on the inward-open substrate-free structure with a resolution of 2.7 Å, the authors indicated that release of sodium would cause a reorientation of TM1 in vSGLT to open the inner gate for substrate’s exit. Compared with LeuT, the space of the additional water molecule in Na2 site of MhsT is provided by the shift of Ser323 side chain. This residue of the second sodium ion connects the Na2 to intracellular environment and was supposed to be ready for next step solvation and Na+ release to the cytoplasmic side.
The substrate binding site in DAT is also in close proximity to the densities of sodium and chloride ions. However, the ion coupling mechanism is unclear based on limited structure information. In the proton transporter ApcT, the positively charged Lys158 was supposed to form a kink in TM1 upon protonation, similarly in LeuT, to recruit the carbonyl oxygen of Ile22 (TM1) and the hydroxyl groups of Ser283 and Ser286 (TM8). A subsequent effect on the movement of TMs (1a, 1b, 6a and 6b) was proposed to trigger the gates’ opening. However, in scaDMT, there is no indication of whether the ion binding sites superimpose the Na sites of LeuT. The two conserved histidines (His228 and His233) close to the substrate-binding site were considered to be correlated with proton coupling (Ehrnstorfer et al., Citation2014), but the coupling seems not strict because uncoupled flows of substrates were observed (Mackenzie et al., Citation2006).
Gate
The LeuT extracellular gate is composed of a conserved charged pair: Arg30 and Asp404 that may form a salt bridge upon rearrangement, and two aromatic residues Tyr108 and Phe253 below (, right) (Yamashita et al., Citation2005). A similar hydrophobic cluster in an extracellular gate was also seen in MhsT (Malinauskaite et al., Citation2014), but the two have differences. LeuT possesses a Leu29, whereas MhsT harbors a Trp33 in the corresponding position, which is conserved across NSS family. The author discussed that the extracellular vestibule holding the bulky Trp33 cannot accommodate an additional ligand. However, for LeuT, a second unidentified substrate site (S2) could accommodate around this Leu29, but the opinions vary in different studies (Khelashvili et al. Citation2013;, Quick et al., Citation2012; Wang et al., Citation2012).
In the inward occluded state MhsT, most of the residues contributing the intracellular gates resembles the corresponding residues in LeuT (Arg9MhsT and Arg5LeuT; Glu10MhsT and Glu6LeuT; Ser244MhsT and Ser267LeuT; Tyr245MhsT and Tyr268LeuT; Glu330MhsT and Gln361LeuT; Asp369LeuT; and Arg344MhsT and Arg375LeuT, as well as the hydrophobic plug Trp12MhsT and Trp8LeuT), except for the unwind region in TM5, which might provide a solvent pathway to the Na2 site in MhsT (Malinauskaite et al., Citation2014).
The extracellular gate of DAT is similar to LeuT, which is composed of Tyr124 and Phe319, and Arg52 and Asp475 (Penmatsa et al., Citation2013). Compared with LeuT, the long distances among these residues suggest that the gate is open to the extracellular side. Residues (Arg27, Trp30 and Asp435) contributing to the intracellular gate of DAT are also strictly conserved in LeuT. Similar mixed polar and non-polar interactions are also reported in the extracellular vestibule of SERT while the intracellular gate was not discussed in detail (Coleman et al., Citation2016).
Different from the NSS family members, the vSGLT exhibits unique features in the sugar porter family (Faham et al., Citation2008). The galactose in vSGLT was found sandwiched between intra- and extracellular hydrophobic residues gates. In the intracellular gate, the Tyr263 (discontinuous helix TM7E) stacks with the pyranose ring which is common in sugar transporters (Ermolova et al., Citation2006; Sujatha & Balaji, Citation2004), while the extracellular gate is defined by hydrophobic residues (Met73, Tyr87 and Phe424). The two gates are also reported conserved in solute-sodium symporter (SSS) family (Faham et al., Citation2008). Based on current studies, most residues contributing the extracellular and intracellular gates are conserved in LeuT, MhsT, DAT and SERT. Besides, the mixture of polar and non-polar interactions on both gates in these LeuT fold proteins seems different from that of MFS fold proteins. However, since the gates of sugar porter vSGLT are quite different from the other four Neurotransmitter/Na+ symporters, then the characteristic of gates seem to more correlate with the function of porters rather than folds.
Conformational changes associated with transport
The mechanism of substrate permeation and conformational changes in LeuT has been discussed in the study by Piscitelli & Gouaux (Citation2012). Structures of the outward facing and occluded states LeuT (Singh et al., Citation2008) suggest that the transition from outward open to the occluded state seems to involve the inward movement of TM1b and 6a towards TM3 and 10, as also supported by EPR measurements (Claxton et al., Citation2010). Subsequently, opening of the intracellular gate from occluded state seems to be contributed by an outward movements of TM1a, as supported by single-molecule FRET assays (Zhao et al., Citation2010, Citation2011) and TM6b, as proved by molecular modeling (Forrest et al., Citation2008; Shaikh & Tajkhorshid, Citation2010), models (Forrest et al., Citation2008; Yamashita et al., Citation2005) and substituted cysteine accessibility studies (Forrest et al., Citation2008; Kniazeff et al., Citation2008). However, before 2012, the transition from occluded state to inward-open state was still unknown due to a lack of inward open structure.
In 2012, Krishnamurthy & Gouaux (Citation2012) presented the first inward-open LeuT structure with conformation-specific antibody fragments. This structure suggested that the transition to inward state did not occur strictly along the pseudo-two-fold symmetric axis, but were realized by individual adjustment of TM helices, including bending of TM 2 and 7 at the conserved glycines, the independent movements of TM1a and 6b, as well as the large movement of extracellular loop 4 (EL4) compared with the relative static intracellular loop 1 (IL1). Notably, the TM1a was seen tilting away from scaffold domain and TM 6b, and this was not considered a crystallization artifact since TM1a is neither involved in crystal contacts nor interacts with the Fab. Interestingly, the movement of TM1a was also reported in single-molecule FRET studies (Zhao et al., Citation2010, Citation2011). A hindering to the movement of TM1a by cholesterol was also observed in the structure of neurotransmitter DAT (Penmatsa et al., Citation2013). This suggests that cholesterol could play a structural and perhaps a regulatory role. Furthermore, the LeuT-fold vSGLT captured in an inward-open and substrate-free conformation indicated that the intracellular half of TM1 flexes 13° away compared with the TM1 when in the inward-occluded state (Watanabe et al., Citation2010). The above phenomenon suggests that the movement of TM1a is probably the major event during LeuT’s transition to an inward-open conformation. Clearly, the conformational changes associated with substrate translocation involve more than rigid body movements of protein sub-domains.
How does TM1 initiate the cascade of movement to open the intracellular door? Since TM1 hinges at Leu25 – a location close to substrate and ion binding site – the hinge movement of TM1 is suggested to directly coupled to the substrate and Na+ site. A model was proposed by which changes in ion and substrate binding cause TM1a to tilt away, which further facilitates the closure of extracellular gate by TM5 “pushing” TM7 and 1b. The intracellular gate cannot open without the closure of the extracellular gate. In 2016, an “outward-oriented, Na+-free” conformation of LeuT was captured (Malinauskaite et al., Citation2016). The structure is considered to be in an inward-to-outward return transition. Combined with molecular dynamics simulation results, the authors further proposed that residue Leu25 enables the inward-to-outward transition by compensating for Na+ in the substrate-free state and acts as a gatekeeper for Na+ binding which prevents ion leak during inward–outward return transition.
For other LeuT fold structures, the outward-facing DAT with a bound TCA inhibitor, two sodium ions and one chloride are shown located in the vicinity of the ligand site, indicating that the binding of ions and inhibitor are directly coupled (Penmatsa et al., Citation2013). MhsT differs from LeuT not only in the absence of residue Trp33 in the extracellular gate but also in an additional unwound region in TM5 which was proposed to provide a solvation pathway for the Na2 site from the intracellular side (Malinauskaite et al., Citation2014). With the flexibility of this TM5, TM1a was proposed to be liberated after the release of Na2. A subsequent reunion of the intracellular part of TM5 (TM5I) and extracellular part of TM5 (TM5E) in turn enable the release of Na1.
Disease relationship
Although most crystal structures are from prokaryotic species, they never-the-less provide us with structural information essential to our understanding of the mechanism of diseases caused by mutations in mammalian SLC homologs ().
LeuT fold represents the second largest fold clusters within SLCs proteins, and most of them belong to NSS family. These transporters are correlated with severe diseases including orthostatic intolerance (Shannon et al., Citation2000), depression (Masson et al., Citation1999), attention deficit hyperactivity disorder (Waldman et al., Citation1998), Parkinson’s disease (Masson et al., Citation1999) and infantile parkinsonism dystonia (Kurian et al., Citation2009). Although numerous side effects (Anderson Citation2000) were reported, this family of proteins (NSS) has long been the target of antidepressants. For example, the usage of the tricyclic antidepressant (TCA) imipramine as the antidepressant therapy (Axelrod et al., Citation1961).
For the SLC sugar transporters, the substrate binding site within vSGLT is supported by mutational analysis of the corresponding residues (K321A, Q457R and Q457C) in mammalian protein SGLT1 (Meinild et al., Citation2002; Panayotova-Heiermann et al., Citation1998). Mutation of Gln457 to Arg causes severe glucose-galactose malabsorption among newborn infants (Wright et al., Citation2007), suggesting a conserved role of these sugar interacting residues across homologs in different species.
The structure of ScDMT also maps the location of the mutations which cause diseases in human and rodents (Ehrnstorfer et al., Citation2014). Mutation on a conserved Gly within α-helix 4 to arginine leads to anemia in mice and rats. This mutation was also reported to be correlated with the increase of Ca2+ permeability (Xu et al., Citation2004). Four mutants that cause anemia in humans can be found in the TM domains.
Further studies, including homology modeling of mammalian SLC structures in different conformations, could illustrate the effect that the disease-related mutations may have on the folding and transport activity of these transporters.
Other transporters with “inverted repeat”
In addition to the LeuT-fold proteins, the so called “inverted repeat” with antiparallel orientation was also seen in NhaA (Appel et al., Citation2009; Hunte et al., Citation2005) (12 TMs), ASBT (Hu et al., Citation2011; Zhou et al., Citation2014) (10 TMs), UraA (Lu et al., Citation2011), SLC26Dg (Geertsma et al., Citation2015) (14 TMs) and Band 3 the human red cell anion exchanger (AE1, SLC4A1) (Arakawa et al., Citation2015; Reithmeier et al., Citation2016). Similar to LeuT, they all share a core domain (transport domain) and a scaffold (panel/gate) domain within each protomer ().
ASBT (5 + 5 fold) is a Na+-dependent bile acid symporter expressed on enterocytes in the terminal ileum of humans (Claro da Silva et al., Citation2013). ASBT is also the target of potential therapeutics for the treatment of hypercholesterolemia and type 2 diabetes (Chen et al., Citation2012). Structures of the homolog ASBT from Neisseria meningitides (Hu et al., Citation2011) and Yersinia frederiksenii (Zhou et al., Citation2014) were solved separately. The structure consists of 10 TMs and can be divided into a panel domain (TM1, 2, 6 and 7) and a core domain (TM3–5 and 8–10). TM 1–5 and TM 6–10 were reported to be structurally homologous of inverted topology with an internal two-fold pseudosymmetry axis. Particularly, TM 4 and 9 are unwound in the middle and they cross with each other (Zhou et al., Citation2014). The overall structure of Na+/H+ antiporter NhaA and its rigid body movement between the core and panel domain during the transition between inward and outward is similar to those of ASBT (Hunte et al., Citation2005).
The overall folds of UraA (Uracil Transporter, E. coli) (Lu et al., Citation2011), SLC26Dg (SLC26, Deinococcus geothermalis) (Geertsma et al., Citation2015) and the Band 3 (SLC4A1) (Arakawa et al., Citation2015) structures are similar in containing “7 + 7” inverted TMs repeats which constitute the “core” and “gate” domain. Specifically, all these structures share a unique “beta strand followed by short alpha helix” (Reithmeier & Moraes, Citation2015) in the crossed TM 3 and its symmetry-related TM 10. The N-termini of these two short helices faces each other in the center of the protein providing a binding site for anionic substrates between the positive helix dipoles (Reithmeier et al., Citation2016). A proton-binding site in Band 3 (SLC4A1) that is required for sulfate uptake is identified at Glu681, while the equivalent Glu241 in SLC26Dg is proposed to be the proton-binding site (Reithmeier & Moraes, Citation2015) but this needs verification (Geertsma et al., Citation2015). However, the corresponding residues are not conserved in human SLC26s since SLC26s in human operate as exchangers, rather than as proton (or sodium)-driven co-transporters. Reithmeier & Moraes (Citation2015) further discussed that in sodium-driven bicarbonate cotransporters (NBCs), that Glu681 has been replaced by Asp compared with proton-driven transporters, due to a changing in the driving force. In addition, the smaller Asp can likely accommodate the larger sodium ion than the Glu required for proton binding.
Other transporters with different folds
Structures with “HP domains”
Glutamate transporters catalyze the uptake of nutrients in prokaryotic cells, while in mammals, they mediate the reuptake of glutamate into neuron cells to maintain the low concentration of neurotransmitter outside, which is necessary for the sensitive signal transmission and preventing the detrimental effect of high concentration of neurotransmitter outside (Danbolt, Citation2001; Grewer & Rauen, Citation2005). As shown before (Cesar-Razquin et al., Citation2015), 17 PDB entries of glutamate transporters have been reported since 2004, with 16 of them from Pyrococcus horikoshii (Gltph) and 1 from Thermococcus kodakarensis KOD1 (GltTK). Gltph selectively transports aspartate rather than glutamate by coupling three sodiums (Boudker et al., Citation2007; Groeneveld & Slotboom, Citation2010; Reyes et al., Citation2009; Ryan et al., Citation2009; Verdon & Boudker, Citation2012; Yernool et al., Citation2004). An additional efflux of potassium ion is needed in mammals to drive uptake, but not in prokaryotes (Ryan et al., Citation2009).
The GltPh was shown to be a homotrimer with a three-fold non-crystallographic symmetry (NCS) axis perpendicular to the membrane in different crystal structures (Boudker et al., Citation2007; Jensen et al., Citation2013; Reyes et al., Citation2009, Citation2013; Verdon & Boudker, Citation2012; Verdon et al., Citation2014; Yernool et al., Citation2004) (). The three protomers form a deep solvent-filled bowl exposed to the extracellular side. Each protomer is composed of 8 α-helical TMs and 2 hairpins (HP domain). The structure can be divided into a scaffolding trimerization domain that houses a transport domain (core region) holding the substrate and sodium binding sites. Reyes et al. (Citation2009) reported that a fully bound of Na+ and L-asp facilitate the Gltph transport domain movement of about 15 Å across the membrane during the transition from an outward- to an inward-facing state.
Nucleoside transporters (NTs) transport nucleosides in different cell types; compounds that are important in DNA and RNA synthesis, adenosine-related signaling processes (Johnson et al., Citation2012) and nucleoside-derived drugs uptake (Cano-Soldado et al., Citation2008; Damaraju et al., Citation2003). The structure of vcCNT (concentrative nucleoside transporter from Vibrio cholerae) shares similar local elements to Glts (in 2 HP domains) with some own features.
Mitochondrial carriers
The ADP/ATP carrier is essential in exchanging the ATP generated inside the mitochondria for the cytosolic ADP. Dysfunction of ADP/ATP carrier can cause severe diseases including mitochondrial myopathies or ophthalmoplegia (Fiore et al., Citation2001; Heddi et al., Citation1993; Kaukonen et al., Citation2000; Napoli et al., Citation2001; Torroni et al., Citation1990). In 2003, the first 3D structure of ADP/ATP carrier (bovine ANT1) was determined bound with an inhibitor carboxyatractyloside (Pebay-Peyroula et al., Citation2003). This structure consists of 6 TMs with three similar fold repeats and three short α-helical stretches connecting the odd- and even-numbered helices (). The proline residue located in the conserved sequence PX(D/E)XX(K/R) rendered the odd-numbered helices with a sharp kink, which is a common feature of mitochondrial carriers. This structure is opened towards the cytoplasmic side with a central funnel-shaped cavity composed of strong positive patches at the bottom and weaker positive ones on the matrix side. These basic residues are highly conserved and are predicted to participate in the binding of negatively charged ADP.
Berardi et al. (Citation2011) reported the structure of the mitochondrial proton transporter UCP2 by solution-NMR. The structure resembles the overall fold of bovine ADP/ATP carrier, but with an orientation open to the matrix side. Although there are minor structural differences in repeat three between UCP2 and ANT1, the high structural similarity proved that the large carrier family shared a conserved overall fold. Ruprecht et al. (Citation2014) showed that the three interdomain salt-bridge interactions including the “glutamate brace” at the matrix side in the conserved motif PX(D/E)XX(K/R) are responsible for the closure of the gate, while the cytoplasmic side is regulated by a second salt-bridge network based on mutation analysis. Recently, the structures of the cytosolic regulatory domains of human mitochondrial aspartate/glutamate carriers and ATP-Mg/Pi carrier were reported separately (Harborne et al., Citation2015; Thangaratnarajah et al., Citation2014), which indicated the interaction between the regulatory domain and the amphipathic α-helix is important in “closing” and “opening” of the transmembrane carrier domain (supposed to have similar structural fold to ADP/ATP carrier). This mechanism then adds new insight into the functioning of mitochondrial carrier family proteins.
Zinc transporters (SLC30 and 39)
Zn works as catalytic or structural cofactors of enzymes such as carbonic anhydrase and plays important role in processes such as growth, development, immunity and metabolism (Beach et al., Citation1980; Danks, Citation1988; Festa & Thiele, Citation2011; Lutsenko et al., Citation2007; Pena et al., Citation1999; Prasad et al., Citation1961; Vallee & Falchuk, Citation1993; Vulpe & Packman, Citation1995 ). Lu & Fu (Citation2007) reported the first zinc transporter homolog structure Yiip from E. coli. Yiip is a “V” shaped homodimer held together by the C-terminal cytosolic domain (CTD) with a two-fold crystallographic axis perpendicular to the membrane plane (). Two compact six-helix bundles of each protomer form the transmembrane domain, while the cytosolic domains juxtapose each other and were supposed to be key for the dimerization contact. Another factor that contributes to the dimer interface is from the intracellular loops at the juncture between the cytosolic domain and transmembrane domain juncture. The dimerization also used to be reported in SLC30A5/SLC30A6 (heterodimer) and in SLC30A7 (Suzuki et al., Citation2005). In 2009, a higher resolution (2.9 Å) structure of YiiP was reported (Lu et al., Citation2009).
CaCA fold transporters (SLC8, SLC24)
Ca2+ signaling is essential in different species involving diverse processes like hyper-osmotic stress, seed germination and oxidative stress in plants, and muscle contraction, the release of neurotransmitters and gene expression in animals (Berridge et al., Citation2003; Sanders et al., Citation1999; White & Broadley, Citation2003). The first crystal structure of Na+/Ca2+ antiporter NCX_Mj was reported from Methanococcus jannaschii (Liao et al., Citation2012), followed by the Ca2+/H+ antiporter structures of YfkE and Vcv1 captured in inward-facing conformations in 2013 (Waight et al., Citation2013; Wu et al., Citation2013). All three structures’ protomers exhibit “5 + 5” TM helical bundles with antiparallel orientation and a pseudo-two-fold symmetry perpendicular to the membrane plane (). In contrast to the 10 TMs of NCX_Mj, the YfKE and Vcx1 hold an additional short TM0 (or MR) at their N-terminus that may be involved in protein targeting and signaling in yeast (Pittman & Hirschi, Citation2001; Pittman et al., Citation2002; Shigaki et al., Citation2006). Despite the poor protein sequence homology among the three transporters, the similar overall 5 + 5 fold demonstrates the structural conservation within the Ca2+/H+ or Ca2+/Na+ efflux superfamily.
Channel like transporters
Urea transporters (UT)
UT members have a wide distribution of various species including bacteria, fungi, insects and vertebrates. There are two urea transporter genes in mammals: UT-A (SLC14A2) and UT-B (SLC14A1). The study of urea transport has a long history (Aukland, Citation1961; Gallucci et al., Citation1971; Macey, Citation1984; Mayrand & Levitt, Citation1983; Sands & Knepper, Citation1987; Schafer et al., Citation1974). However, the atomic resolution structure of UT transporter was not resolved until in 2009. The first structure of UT family from Desulfovibrio vulgaris str. Hildenborough was solved by Levin et al. (Citation2009) at 2.40 Å. The trimeric structure showed that each protomer contains two 6 TMs, named a and b, with a rotational pseudo-two-fold symmetry axis in between. The first helix of each domain (Pa and Pb, P means pore helix) is tilted at around 50° angle to the membrane norm and extends about 10 Å into the membrane. Central TM helices (TM1a-4a and TM1b-TM4b) span the entire membrane. The end helices Tm5a and Tm5b, are perpendicular to the membrane with an unwind region in the middle. The interface between the domains was shown largely hydrophobic. Later on, the first X-ray crystal structure from mammals (Bos Taurus), UT-B, was determined by the same group in 2012 at 2.36 Å (Levin et al., Citation2012). The dvUT and UT-B structures shared a similar overall fold with the trimer interface formed by equivalent helices (Levin et al. Citation2009, Citation2012).
Ammonia channel
SLC42A1, A2 and A3 encodes three Rh glycoproteins, RhAG, RhBG and RhCG, respectively. RhAG is found distributed in red blood cells to form a complex with the nonglycosylated Rh proteins RhD and RhCE in humans or with Rh30 in other mammals (Nakhoul & Lee Hamm, Citation2013). RhAG together with band 3 and ankyrin form a complex that may modulate the transport of HCO3− and possibly NH4+ (Nicolas et al., Citation2006). For RhBG, the role as a NH3/NH4+ transporter has been reported in oocytes and mammalian cells with different explanations on the substrate transporting mechanism, concentration and affinity (Ludewig, Citation2004; Mak et al., Citation2006; Nakhoul et al., Citation2005, Citation2006, Citation2010a,Citationb; Zidi-Yahiaoui et al., Citation2005). Function of RhCG is also controversial in whether it transports NH3 (Mouro-Chanteloup et al., Citation2010), NH4+ (Nakhoul et al., Citation2005) or both (Bakouh et al., Citation2004), or, it is an NH4+/H+ antiporter (Ludewig, Citation2004). The Rh proteins are also indicated as gas channels in transporting CO2 (Kaplan et al., Citation2004; Soupene et al., Citation2004) and NH3 (Ripoche et al., Citation2004). However, the mechanism is unclear. A big step in this field has been the crystal structure of E. coli homolog of Rh-AmtB (Khademi et al., Citation2004).
The structure of AmtB from E. coli was shown a threefold symmetric trimer with each monomer containing 11 TM α-helices (). TM1 to TM10 diverge outward from the central plane in a right-handed helical bundle. TM1–TM5 and TM6–TM10 are structural duplications with opposite polarity with respect to the membrane plane. TM11 is an additional straight helix surrounding the lipid contacting face of each monomer. Two constricted hydrophobic regions were found located at the entry and exit of the channel with two highly conserved histidines in between, the His168 and His318, which play an essential role in coordinating the substrate Am (ammonium) molecules.
Discussion
The SLC superfamily is ubiquitous in living organisms and is the largest family of membrane transport proteins. They play essential physiological roles in transporting a large variety of substrates including ions, nutrients and waste products. Genetic variants or mutations of all SLCs except for several limited members (SLC14, 28, 32, 48, 50, 54 and 63) have been correlated with severe human diseases, such as neurodevelopmental disorders, diabetes, psychiatric diseases and cancer (http://www.ncbi.nlm.nih.gov/omim) (). In addition, SLCs are also important factors involved in drug absorption and delivery. However, compared with hot topics of membrane proteins (e.g. GPCRs), SLCs are still understudied (Cesar-Razquin et al., Citation2015) and only limited members of SLCs (∼12) were found to be utilized as therapeutic drug targets currently (Rask-Andersen et al., Citation2013).
There are a number of limitations and challenges in determining the structures of membrane transport proteins. Expressing and purifying membrane proteins in a native state is a major challenge. Structures are usually obtained in detergent solutions and there is no guarantee that they represent the structure of the membrane. Prokaryotic homologs are often used allowing models of their eukaryotic cousins to be built. Often mutant forms of the transporter, conformational antibodies, or inhibitors are used to facilitate crystallization, which may produce a non-native structure. Eukaryotic transport proteins typically have a more complex structure than their bacterial homologs, including N-glycosylation.
The relatively low resolution (∼3.5 Å) of many structures limits the fine detail required to determine the molecular features of the substrate binding sites. Transporters exist in at least two conformational states; outward and inward-facing and it is the details of the dynamics between these two states that are at the heart of the transport mechanism. Here we summarized all available SLC structures to date with the main focus on MFS and LeuT folds. Compared with primary sequence analysis which showed low similarities among different SLC family members, structural analysis seems more reliable since the available structures could be assigned into several similar folds. Structures will also help us visualize SLC proteins at the atomic level, which will enhance our understanding of their function, especially, substrate binding sites which usually constitute characteristic residues that only 3D structure could fully reveal. Further, many functional assay designs are limited without the information of structures as these structures could make the corresponding biochemical studies (e.g. site-directed mutagenesis) feasible (Bai et al., Citation2016).
When discussing the structures, the resolution is an important factor since it indicates the quality of the details that the structure can reveal. Resolutions of SLC structures are varied with most of them ranging from 1.5 to 3.5 Å (Cesar-Razquin et al., Citation2015). A few were reported with lower resolution, including GltPh at 4.5 Å (PDB 4IZM), 4.66 Å (3V8G), 4 Å (4OYE), 4.08 Å (4P6H), XylE at 4.2 Å (4JA4) and LeuT at 4.5 Å (3USK). Notably, the molecular details of the substrate binding site are highly relevant and depend on the resolution. Therefore, interpretations of these binding sites need to be carefully reviewed, since some structures may not be of high enough resolution to tell the details. Substrate-binding sites from speculations based on analyzing of the “pore” region within the structure (Huang et al., Citation2003) or central cavity regions (Newstead et al., Citation2011) are not direct evidence and may need experimental evidence to verify the assigned sites. There are excellent examples which demonstrate that information of binding sites can be obtained directly from high-resolution structures. For example, in a bacterial homolog of the human drug-peptide transporter PepTSo2, 2Fo − Fc electron density maps at 1σ and anomalous difference density map contoured at 3σ clearly showed the binding peptides and its coordinating amino acids (Guettou et al., Citation2014). In the 2.6 Å XylE structure, the orientation the substrate was verified by the bromide anomalous signal of 6-BrGlc (Sun et al., Citation2012). An omit mFobs-DFcalc density was also shown for phosphate in the inward occluded 2.9 Å PiPT (Pedersen et al., Citation2013). Therefore, a high-resolution structure with a binding substrate will surely provide direct evidence of substrate binding information.
In classical in vitro studies, detergents have been widely used for purification and crystallization. However, there has been considerable debate over whether crystals grown in detergent represent the structures in the real membrane. In this respect, many functional assays were performed in environments closely related to membranes to support the structural information. Taking LacY as an example, it was crystallized in the detergent DDM (Kaback et al., Citation2011). To verify whether the conformation of crystal structure is consistent with the protein’s real state in bacterial membranes, site-directed alkylation was carried out in both membrane vesicles and detergent. Based on the observation that tetra-methyl rhodamine maleimide (TMRM) labeling is almost negligible to the periplasmic side of the protein in both vesicles and DDM, the author concluded that LacY in the native membrane environment has the similar conformation to that of the X-ray crystal structures. GkPOT was crystallized in detergent n-dodecyl-β-maltoside. To verify the substrate binding state, functional assays were performed in liposomes, which provide evidence of alafosfalin recognition mechanism. In this case, the crystal structure from detergent reflects its substrate binding state in a liposome (Doki et al., Citation2013). A similar demonstration of the validity of the crystal structure from detergent could also be found in in vivo [3 H]-glycylsarcosine transport assays of PepTSo performed in E. coli cells (Newstead et al., Citation2011), cell-based uptake assay in XylE (Sun et al., Citation2012), nitrate transporter assay of NRT1.1 in oocytes (Sun et al., Citation2014), radioactive substrate transport assay of YbgH in cells (Zhao et al., Citation2014), glucose transport assay of GlcP performed in bacterial strains JM1100 (Iancu et al., Citation2013), in which the assays were designed from structures obtained in detergent, while verification is performed in lipids/liposomes/cells/oocytes. Therefore, structures do reflect the proteins’ real states within the cell in many aspects (e.g. substrate binding). However, the detergent is not identical to lipid bilayer and usually results in the aberrant behavior of the membrane protein. This can be seen from site-directed alkylation experiments on LacY. The changes of signals in TMRM labeling observed in vesicles is 10-fold, while with purified proteins in detergent DDM, the signals are 5- to 6-fold (Kaback et al., Citation2011). Furthermore, protein-protein interactions within the crystals may be due to crystal packings which are non-physiological relevant. These interactions may need to be verified in functional assays. To mimic a more natural lipid environment, novel techniques were developed including nanodisc technology and lipid cubic phase. Structures in these environments may give us information closer to its natural state. In addition, since crystal structures only captured one particular moment of the protein, various biophysical and cellular functional assays are needed to illustrate the dynamic activities of SLCs within cells.
Among all SLC structures, the impact of mammalian ones is significant since the structure will give a direct evidence on the molecular mechanism of mammalian SLCs (especially human SLCs). However, there are only nine mammalian SLCs available in the literature. Several aspects contribute to the fact that very few mammalian SLCs structures are available in the database. First, since SLCs play fundamental roles in transporting various small molecule substrates and waste products in the body, expression of SLC can be difficult, as overexpression may cause toxicity probably due to metabolic disturbance (Cesar-Razquin et al., Citation2015). Second, the stability of membrane proteins may be poor since detergent has been widely used to extract membrane protein from its native membrane, which perturbs the protein’s environment drastically. Finally, for mammalian SLCs, the purification and crystallization difficulty has increased exponentially since mammalian protein need modifications which may not be manufactured properly in non-mammalian expression systems, including bacteria, yeast and insect cells. For different membrane protein, various individualized strategies need to be used to overcome difficulties in getting structures. For example, human band 3 protein was purified straight from human erythrocytes white ghost membranes (Arakawa et al., Citation2015) and it took the authors more than 5 years to select the appropriate antibody used for stabilizing the protein’s conformation for crystallization. Crystallization has been considered full of mystery. Membrane protein crystallography is even more challenging. With the development of technologies including Cryo-EM, serial femtosecond crystallography, lipid cubic phase crystallography, nanodisc crystallography and high throughput detergent screen, truncation design, membrane protein expression system screen, utilization of antibodies and nanobodies, progress in techniques of structure determination and most importantly, the increase in SLC studies, we anticipate a faster speed on the outcome of SLC structures, especially human ones.
| ABBREVIATIONS | ||
| DASS | = | divalent anion/sodium symporter |
| LeuT | = | leucine transporter |
| MFS | = | major facilitator superfamily |
| NRT | = | nitrate transporter |
| NSS | = | Neurotransmitter: Sodium Symporter |
| SLC | = | solute carrier |
| SSS | = | Solute-sodium symporter |
| TM | = | transmembrane |
Disclosure statement
The authors report no declarations of interest.
Additional information
Funding
References
- Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. 2003. Structure and mechanism of the lactose permease of Escherichia coli. Science 301:610–615.
- Alakbarzade V, Hameed A, Quek DQ, Chioza BA, Baple EL, Cazenave-Gassiot A, et al. 2015. A partially inactivating mutation in the sodium-dependent lysophosphatidylcholine transporter MFSD2A causes a non-lethal microcephaly syndrome. Nat Genet 47:814–817.
- Alper SL, Sharma AK. 2013. The SLC26 gene family of anion transporters and channels. Mol Aspects Med 34:494–515.
- Anderson CM, Stahl A. 2013. SLC27 fatty acid transport proteins. Mol Aspects Med 34:516–528.
- Anderson IM. 2000. Selective serotonin reuptake inhibitors versus tricyclic antidepressants: a meta-analysis of efficacy and tolerability. J Affect Disord 58:19–36.
- Ansermet C, Moor MB, Centeno G, Auberson M, Hu DZ, Baron R, et al. 2017. Renal fanconi syndrome and hypophosphatemic rickets in the absence of xenotropic and polytropic retroviral receptor in the nephron. J Am Soc Nephrol 28:1073–1078.
- Appel M, Hizlan D, Vinothkumar KR, Ziegler C, Kuhlbrandt W. 2009. Conformations of NhaA, the Na/H exchanger from Escherichia coli, in the pH-activated and ion-translocating states. J Mol Biol 386:351–365.
- Arakawa T, Kobayashi-Yurugi T, Alguel Y, Iwanari H, Hatae H, Iwata M, et al. 2015. Crystal structure of the anion exchanger domain of human erythrocyte band 3. Science 350:680–684.
- Arroyo JP, Kahle KT, Gamba G. 2013. The SLC12 family of electroneutral cation-coupled chloride cotransporters. Mol Aspects Med 34:288–298.
- Aukland K. 1961. Renal tubular permeability to urea with special reference to accumulation of urea in the renal medulla. Scand J Clin Lab Invest 13:646–660.
- Axelrod J, Whitby LG, Hertting G. 1961. Effect of psychotropic drugs on the uptake of H3-norepinephrine by tissues. Science 133:383–384.
- Bai X, Moraes TF, Reithmeier RA. 2016. Effect of SLC26 anion transporter disease-causing mutations on the stability of the homologous STAS domain of E. coli DauA (YchM). Biochem J 473:615–626.
- Bakouh N, Benjelloun F, Hulin P, Brouillard F, Edelman A, Cherif-Zahar B, Planelles G. 2004. NH3 is involved in the NH4+ transport induced by the functional expression of the human Rh C glycoprotein. J Biol Chem 279:15975–15983.
- Ballatori N, Christian WV, Wheeler SG, Hammond CL. 2013. The heteromeric organic solute transporter, OSTalpha-OSTbeta/SLC51: a transporter for steroid-derived molecules. Mol Aspects Med 34:683–692.
- Beach RS, Gershwin ME, Makishima RK, Hurley LS. 1980. Impaired immunologic ontogeny in postnatal zinc deprivation. J Nutr 110:805–815.
- Ben-Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, Yan H, Gu C. 2014. Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature 509:507–511.
- Berardi MJ, Shih WM, Harrison SC, Chou JJ. 2011. Mitochondrial uncoupling protein 2 structure determined by NMR molecular fragment searching. Nature 476:109–113.
- Bergeron MJ, Clemencon B, Hediger MA, Markovich D. 2013. SLC13 family of Na+-coupled di- and tri-carboxylate/sulfate transporters. Mol Aspects Med 34:299–312.
- Berridge MJ, Bootman MD, Roderick HL. 2003. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol 4:517–529.
- Blauw HM, van Rheenen W, Koppers M, Van Damme P, Waibel S, Lemmens R, et al. 2012. NIPA1 polyalanine repeat expansions are associated with amyotrophic lateral sclerosis. Hum Mol Genet 21:2497–2502.
- Bodoy S, Fotiadis D, Stoeger C, Kanai Y, Palacin M. 2013. The small SLC43 family: facilitator system l amino acid transporters and the orphan EEG1. Mol Aspects Med 34:638–645.
- Boudker O, Ryan RM, Yernool D, Shimamoto K, Gouaux E. 2007. Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature 445:387–393.
- Brandsch M. 2009. Transport of drugs by proton-coupled peptide transporters: pearls and pitfalls. Expert Opin Drug Metab Toxicol 5:887–905.
- Burzle M, Suzuki Y, Ackermann D, Miyazaki H, Maeda N, Clemencon B, et al. 2013. The sodium-dependent ascorbic acid transporter family SLC23. Mol Aspects Med 34:436–454.
- Cano-Soldado P, Molina-Arcas M, Alguero B, Larrayoz I, Lostao MP, Grandas A, et al. 2008. Compensatory effects of the human nucleoside transporters on the response to nucleoside-derived drugs in breast cancer MCF7 cells. Biochem Pharmacol 75:639–648.
- Carstea ED, Polymeropoulos MH, Parker CC, Detera-Wadleigh SD, O'Neill RR, Patterson MC, et al. 1993. Linkage of Niemann-Pick disease type C to human chromosome 18. Proc Natl Acad Sci USA 90:2002–2004.
- Cesar-Razquin A, Snijder B, Frappier-Brinton T, Isserlin R, Gyimesi G, Bai X, et al. 2015. A call for systematic research on solute carriers. Cell 162:478–487.
- Chaptal V, Kwon S, Sawaya MR, Guan L, Kaback HR, Abramson J. 2011. Crystal structure of lactose permease in complex with an affinity inactivator yields unique insight into sugar recognition. Proc Natl Acad Sci USA 108:9361–9366.
- Chen L, Yao X, Young A, McNulty J, Anderson D, Liu Y, et al. 2012. Inhibition of apical sodium-dependent bile acid transporter as a novel treatment for diabetes. Am J Physiol Endocrinol Metab 302:E68–E76.
- Chen LY, Pan CJ, Shieh JJ, Chou JY. 2002. Structure-function analysis of the glucose-6-phosphate transporter deficient in glycogen storage disease type Ib. Hum Mol Genet 11:3199–3207.
- Chou JY, Sik Jun H, Mansfield BC. 2013. The SLC37 family of phosphate-linked sugar phosphate antiporters. Mol Aspects Med 34:601–611.
- Claro da Silva T, Polli JE, Swaan PW. 2013. The solute carrier family 10 (SLC10): beyond bile acid transport. Mol Aspects Med 34:252–269.
- Claxton DP, Quick M, Shi L, de Carvalho FD, Weinstein H, Javitch JA, McHaourab HS. 2010. Ion/substrate-dependent conformational dynamics of a bacterial homolog of neurotransmitter:sodium symporters. Nat Struct Mol Biol 17:822–829.
- Coleman JA, Green EM, Gouaux E. 2016. X-ray structures and mechanism of the human serotonin transporter. Nature 532:334–339.
- Dahlqvist J, Klar J, Hausser I, Anton-Lamprecht I, Pigg MH, Gedde-Dahl T Jr, et al. 2007. Congenital ichthyosis: mutations in ichthyin are associated with specific structural abnormalities in the granular layer of epidermis. J Med Genet 44:615–620.
- Damaraju VL, Damaraju S, Young JD, Baldwin SA, Mackey J, Sawyer MB, Cass CE. 2003. Nucleoside anticancer drugs: the role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene 22:7524–7536.
- Danbolt NC. 2001. Glutamate uptake. Prog Neurobiol 65:1–105.
- Dang S, Sun L, Huang Y, Lu F, Liu Y, Gong H, et al. 2010. Structure of a fucose transporter in an outward-open conformation. Nature 467:734–738.
- Danks DM. 1988. Copper deficiency in humans. Annu Rev Nutr 8:235–257.
- Deng D, Sun P, Yan C, Ke M, Jiang X, Xiong L, et al. 2015. Molecular basis of ligand recognition and transport by glucose transporters. Nature 526:391–396.
- Deng D, Xu C, Sun P, Wu J, Yan C, Hu M, Yan N. 2014. Crystal structure of the human glucose transporter GLUT1. Nature 510:121–125.
- Doki S, Kato HE, Solcan N, Iwaki M, Koyama M, Hattori M, et al. 2013. Structural basis for dynamic mechanism of proton-coupled symport by the peptide transporter POT. Proc Natl Acad Sci USA 110:11343–11348.
- Donowitz M, Ming Tse C, Fuster D. 2013. SLC9/NHE gene family, a plasma membrane and organellar family of Na(+)/H(+) exchangers. Mol Aspects Med 34:236–251.
- Ehrnstorfer IA, Geertsma ER, Pardon E, Steyaert J, Dutzler R. 2014. Crystal structure of a SLC11 (NRAMP) transporter reveals the basis for transition-metal ion transport. Nat Struct Mol Biol 21:990–996.
- Endele S, Fuhry M, Pak SJ, Zabel BU, Winterpacht A. 1999. LETM1, a novel gene encoding a putative EF-hand Ca(2+)-binding protein, flanks the Wolf-Hirschhorn syndrome (WHS) critical region and is deleted in most WHS patients. Genomics 60:218–225.
- Ermolova N, Madhvani RV, Kaback HR. 2006. Site-directed alkylation of cysteine replacements in the lactose permease of Escherichia coli: helices I, III, VI, and XI. Biochemistry 45:4182–4189.
- Escary JL, Cecillon M, Maciazek J, Lathrop M, Tournier-Lasserve E, Joutel A. 2000. Mutational analysis of GLUT1 (SLC2A1) in glut-1 deficiency syndrome; dong wang; pamela kranz-eble; darryl C. De vivo; (Article was originally published in human mutation 16:224-231, 2000.). Hum Mutat 16:527.
- Faham S, Watanabe A, Besserer GM, Cascio D, Specht A, Hirayama BA, et al. 2008. The crystal structure of a sodium galactose transporter reveals mechanistic insights into Na+/sugar symport. Science 321:810–814.
- Faria TN, Timoszyk JK, Stouch TR, Vig BS, Landowski CP, Amidon GL, et al. 2004. A novel high-throughput pepT1 transporter assay differentiates between substrates and antagonists. Mol Pharm 1:67–76.
- Festa RA, Thiele DJ. 2011. Copper: an essential metal in biology. Curr Biol 21:R877–R883.
- Fiore C, Arlot-Guilligay D, Trezeguet V, Lauquin GJ, Brandolin G. 2001. Fluorometric detection of ADP/ATP carrier deficiency in human muscle. Clin Chim Acta Int J Clin Chem 311:125–135.
- Fleming MD, Campagna DR, Haslett JN, Trenor CC, III, Andrews NC. 2001. A mutation in a mitochondrial transmembrane protein is responsible for the pleiotropic hematological and skeletal phenotype of flexed-tail (f/f) mice. Genes Dev 15:652–657.
- Fluman N, Ryan CM, Whitelegge JP, Bibi E. 2012. Dissection of mechanistic principles of a secondary multidrug efflux protein. Mol Cell 47:777–787.
- Forrest LR, Kramer R, Ziegler C. 2011. The structural basis of secondary active transport mechanisms. Biochim Biophys Acta 1807:167–188.
- Forrest LR, Zhang YW, Jacobs MT, Gesmonde J, Xie L, Honig BH, Rudnick G. 2008. Mechanism for alternating access in neurotransmitter transporters. Proc Natl Acad Sci USA 105:10338–10343.
- Forster IC, Hernando N, Biber J, Murer H. 2013. Phosphate transporters of the SLC20 and SLC34 families. Mol Aspects Med 34:386–395.
- Fotiadis D, Kanai Y, Palacin M. 2013. The SLC3 and SLC7 families of amino acid transporters. Mol Aspects Med 34:139–158.
- Foulquier F, Amyere M, Jaeken J, Zeevaert R, Schollen E, Race V, et al. 2012. TMEM165 deficiency causes a congenital disorder of glycosylation. Am J Hum Genet 91:15–26.
- Fowler PW, Orwick-Rydmark M, Radestock S, Solcan N, Dijkman PM, Lyons JA, et al. 2015. Gating topology of the proton-coupled oligopeptide symporters. Structure 23:290–301.
- Fredriksson R, Nordstrom KJ, Stephansson O, Hagglund MG, Schioth HB. 2008. The solute carrier (SLC) complement of the human genome: phylogenetic classification reveals four major families. FEBS Lett 582:3811–3816.
- Gallucci E, Micelli S, Lippe C. 1971. Non-electrolyte permeability across thin lipid membranes. Arch Int Physiol Biochim 79:881–887.
- Ganapathy ME, Huang W, Wang H, Ganapathy V, Leibach FH. 1998. Valacyclovir: a substrate for the intestinal and renal peptide transporters PEPT1 and PEPT2. Biochem Biophys Res Commun 246:470–475.
- Geertsma ER, Chang YN, Shaik FR, Neldner Y, Pardon E, Steyaert J, Dutzler R. 2015. Structure of a prokaryotic fumarate transporter reveals the architecture of the SLC26 family. Nat Struct Mol Biol 22:803–808.
- Grewer C, Rauen T. 2005. Electrogenic glutamate transporters in the CNS: molecular mechanism, pre-steady-state kinetics, and their impact on synaptic signaling. J Membr Biol 203:1–20.
- Groeneveld M, Slotboom DJ. 2010. Na(+):aspartate coupling stoichiometry in the glutamate transporter homologue Glt(Ph). Biochemistry 49:3511–3513.
- Gruswitz F, Chaudhary S, Ho JD, Schlessinger A, Pezeshki B, Ho CM, et al. 2010. Function of human Rh based on structure of RhCG at 2.1 A. Proc Natl Acad Sci USA 107:9638–9643.
- Guan L, Kaback HR. 2006. Lessons from lactose permease. Annu Rev Biophys Biomol Struct 35:67–91.
- Guan L, Mirza O, Verner G, Iwata S, Kaback HR. 2007. Structural determination of wild-type lactose permease. Proc Natl Acad Sci USA 104:15294–15298.
- Guettou F, Quistgaard EM, Raba M, Moberg P, Low C, Nordlund P. 2014. Selectivity mechanism of a bacterial homolog of the human drug-peptide transporters PepT1 and PepT2. Nat Struct Mol Biol 21:728–731.
- Guettou F, Quistgaard EM, Tresaugues L, Moberg P, Jegerschold C, Zhu L, et al. 2013. Structural insights into substrate recognition in proton-dependent oligopeptide transporters. EMBO Rep 14:804–810.
- Hagenbuch B, Stieger B. 2013. The SLCO (former SLC21) superfamily of transporters. Mol Aspects Med 34:396–412.
- Halestrap AP. 2013. The SLC16 gene family – structure, role and regulation in health and disease. Mol Aspects Med 34:337–349.
- Harborne SP, Ruprecht JJ, Kunji ER. 2015. Calcium-induced conformational changes in the regulatory domain of the human mitochondrial ATP-Mg/Pi carrier. Biochim Biophys Acta 1847:1245–1253.
- Heddi A, Lestienne P, Wallace DC, Stepien G. 1993. Mitochondrial DNA expression in mitochondrial myopathies and coordinated expression of nuclear genes involved in ATP production. J Biol Chem 268:12156–12163.
- Hediger MA, Romero MF, Peng JB, Rolfs A, Takanaga H, Bruford EA. 2004. The ABCs of solute carriers: physiological, pathological and therapeutic implications of human membrane transport proteinsIntroduction. Pflugers Arch Eur J Physiol 447:465–468.
- Henderson PJ, Maiden MC. 1990. Homologous sugar transport proteins in Escherichia coli and their relatives in both prokaryotes and eukaryotes. Phil Trans R Soc Lond Ser B Biol Sci 326:391–410.
- Hirabayashi Y, Nomura KH, Nomura K. 2013. The acetyl-CoA transporter family SLC33. Mol Aspects Med 34:586–589.
- Holton KL, Loder MK, Melikian HE. 2005. Nonclassical, distinct endocytic signals dictate constitutive and PKC-regulated neurotransmitter transporter internalization. Nat Neurosci 8:881–888.
- Hu NJ, Iwata S, Cameron AD, Drew D. 2011. Crystal structure of a bacterial homologue of the bile acid sodium symporter ASBT. Nature 478:408–411.
- Huang L, Tepaamorndech S. 2013. The SLC30 family of zinc transporters – a review of current understanding of their biological and pathophysiological roles. Mol Aspects Med 34:548–560.
- Huang Y, Lemieux MJ, Song J, Auer M, Wang DN. 2003. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science 301:616–620.
- Hunte C, Screpanti E, Venturi M, Rimon A, Padan E, Michel H. 2005. Structure of a Na+/H + antiporter and insights into mechanism of action and regulation by pH. Nature 435:1197–1202.
- Iancu CV, Zamoon J, Woo SB, Aleshin A, Choe JY. 2013. Crystal structure of a glucose/H + symporter and its mechanism of action. Proc Natl Acad Sci USA 110:17862–17867.
- Jardetzky O. 1966. Simple allosteric model for membrane pumps. Nature 211:969–970.
- Jensen S, Guskov A, Rempel S, Hanelt I, Slotboom DJ. 2013. Crystal structure of a substrate-free aspartate transporter. Nat Struct Mol Biol 20:1224–1226.
- Jeong J, Eide DJ. 2013. The SLC39 family of zinc transporters. Mol Aspects Med 34:612–619.
- Jiang D, Zhao Y, Wang X, Fan J, Heng J, Liu X, et al. 2013. Structure of the YajR transporter suggests a transport mechanism based on the conserved motif A. Proc Natl Acad Sci USA 110:14664–14669.
- Jiang X, Smirnova I, Kasho V, Wu J, Hirata K, Ke M, et al. 2016. Crystal structure of a LacY-nanobody complex in a periplasmic-open conformation. Proc Natl Acad Sci USA 113:12420–12425.
- Johnson ZL, Cheong CG, Lee SY. 2012. Crystal structure of a concentrative nucleoside transporter from Vibrio cholerae at 2.4 Å. Nature 483:489–493.
- Kaback HR, Sahin-Toth M, Weinglass AB. 2001. The kamikaze approach to membrane transport. Nat Rev Mol Cell Biol 2:610–620.
- Kaback HR, Smirnova I, Kasho V, Nie Y, Zhou Y. 2011. The alternating access transport mechanism in LacY. J Membr Biol 239:85–93.
- Kahn BB. 1992. Facilitative glucose transporters: regulatory mechanisms and dysregulation in diabetes. J Clin Invest 89:1367–1374.
- Kanai Y, Clemencon B, Simonin A, Leuenberger M, Lochner M, Weisstanner M, Hediger MA. 2013. The SLC1 high-affinity glutamate and neutral amino acid transporter family. Mol Aspects Med 34:108–120.
- Kanda M, Shimizu D, Tanaka H, Shibata M, Iwata N, Hayashi M, et al. 2016. Metastatic pathway-specific transcriptome analysis identifies MFSD4 as a putative tumor suppressor and biomarker for hepatic metastasis in patients with gastric cancer. Oncotarget 7:13667–13679.
- Kantcheva AK, Quick M, Shi L, Winther AM, Stolzenberg S, Weinstein H, et al. 2013. Chloride binding site of neurotransmitter sodium symporters. Proc Natl Acad Sci USA 110:8489–8494.
- Kaplan A, Lieman-Hurwitz J, Tchernov D. 2004. Resolving the biological role of the Rhesus (Rh) proteins of red blood cells with the aid of a green alga. Proc Natl Acad Sci USA 101:7497–7498.
- Kaukonen J, Juselius JK, Tiranti V, Kyttala A, Zeviani M, Comi GP, et al. 2000. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science 289:782–785.
- Khademi S, O'Connell J, III, Remis J, Robles-Colmenares Y, Miercke LJ, Stroud RM. 2004. Mechanism of ammonia transport by Amt/MEP/Rh: structure of AmtB at 1.35 A. Science 305:1587–1594.
- Khan AA, Quigley JG. 2013. Heme and FLVCR-related transporter families SLC48 and SLC49. Mol Aspects Med 34:669–682.
- Khananshvili D. 2013. The SLC8 gene family of sodium-calcium exchangers (NCX) - structure, function, and regulation in health and disease. Mol Aspects Med 34:220–235.
- Khelashvili G, LeVine MV, Shi L, Quick M, Javitch JA, Weinstein H. 2013. The membrane protein LeuT in micellar systems: aggregation dynamics and detergent binding to the S2 site. J Am Chem Soc 135:14266–14275.
- Kim H, Wu X, Lee J. 2013. SLC31 (CTR) family of copper transporters in health and disease. Mol Aspects Med 34:561–570.
- Kniazeff J, Shi L, Loland CJ, Javitch JA, Weinstein H, Gether U. 2008. An intracellular interaction network regulates conformational transitions in the dopamine transporter. J Biol Chem 283:17691–17701.
- Ko J, Lee YH, Hwang SY, Lee YS, Shin SM, Hwang JH, et al. 2003. Identification and differential expression of novel human cervical cancer oncogene HCCR-2 in human cancers and its involvement in p53 stabilization. Oncogene 22:4679–4689.
- Koepsell H. 2013. The SLC22 family with transporters of organic cations, anions and zwitterions. Mol Aspects Med 34:413–435.
- Krishnamurthy H, Gouaux E. 2012. X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature 481:469–474.
- Kristensen AS, Andersen J, Jorgensen TN, Sorensen L, Eriksen J, Loland CJ, et al. 2011. SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol Rev 63:585–640.
- Kroncke BM, Horanyi PS, Columbus L. 2010. Structural origins of nitroxide side chain dynamics on membrane protein α-helical sites. Biochemistry 49:10045–10060.
- Kumar H, Kasho V, Smirnova I, Finer-Moore JS, Kaback HR, Stroud RM. 2014. Structure of sugar-bound LacY. Proc Natl Acad Sci USA 111:1784–1788.
- Kurian MA, Zhen J, Cheng SY, Li Y, Mordekar SR, Jardine P, et al. 2009. Homozygous loss-of-function mutations in the gene encoding the dopamine transporter are associated with infantile parkinsonism-dystonia. J Clin Invest 119:1595–1603.
- Law CJ, Maloney PC, Wang DN. 2008. Ins and outs of major facilitator superfamily antiporters. Annu Rev Microbiol 62:289–305.
- Lefevre C, Bouadjar B, Karaduman A, Jobard F, Saker S, Ozguc M, et al. 2004. Mutations in ichthyin a new gene on chromosome 5q33 in a new form of autosomal recessive congenital ichthyosis. Hum Mol Genet 13:2473–2482.
- Legati A, Giovannini D, Nicolas G, Lopez-Sanchez U, Quintans B, Oliveira JR, et al. 2015. Mutations in XPR1 cause primary familial brain calcification associated with altered phosphate export. Nat Genet 47:579–581.
- Levin EJ, Cao Y, Enkavi G, Quick M, Pan Y, Tajkhorshid E, Zhou M. 2012. Structure and permeation mechanism of a mammalian urea transporter. Proc Natl Acad Sci USA 109:11194–11199.
- Levin EJ, Quick M, Zhou M. 2009. Crystal structure of a bacterial homologue of the kidney urea transporter. Nature 462:757–761.
- Li FY, Chaigne-Delalande B, Kanellopoulou C, Davis JC, Matthews HF, Douek DC, et al. 2011. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature 475:471–476.
- Liao J, Li H, Zeng W, Sauer DB, Belmares R, Jiang Y. 2012. Structural insight into the ion-exchange mechanism of the sodium/calcium exchanger. Science 335:686–690.
- Liu KH, Tsay YF. 2003. Switching between the two action modes of the dual-affinity nitrate transporter CHL1 by phosphorylation. EMBO J 22:1005–1013.
- Lu F, Li S, Jiang Y, Jiang J, Fan H, Lu G, et al. 2011. Structure and mechanism of the uracil transporter UraA. Nature 472:243–246.
- Lu M, Chai J, Fu D. 2009. Structural basis for autoregulation of the zinc transporter YiiP. Nat Struct Mol Biol 16:1063–1067.
- Lu M, Fu D. 2007. Structure of the zinc transporter YiiP. Science 317:1746–1748.
- Ludewig U. 2004. Electroneutral ammonium transport by basolateral rhesus B glycoprotein. J Physiol 559:751–759.
- Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY. 2007. Function and regulation of human copper-transporting ATPases. Physiol Rev 87:1011–1046.
- Lyons JA, Parker JL, Solcan N, Brinth A, Li D, Shah ST, et al. 2014. Structural basis for polyspecificity in the POT family of proton-coupled oligopeptide transporters. EMBO Rep 15:886–893.
- Macey RI. 1984. Transport of water and urea in red blood cells. Am J Physiol 246:C195–C203.
- Mackenzie B, Ujwal ML, Chang MH, Romero MF, Hediger MA. 2006. Divalent metal-ion transporter DMT1 mediates both H+-coupled Fe2+ transport and uncoupled fluxes. Pflugers Arch Eur J Physiol 451:544–558.
- Maiden MC, Davis EO, Baldwin SA, Moore DC, Henderson PJ. 1987. Mammalian and bacterial sugar transport proteins are homologous. Nature 325:641–643.
- Mak DO, Dang B, Weiner ID, Foskett JK, Westhoff CM. 2006. Characterization of ammonia transport by the kidney Rh glycoproteins RhBG and RhCG. Am J Physiol Renal Physiol 290:F297–F305.
- Malinauskaite L, Quick M, Reinhard L, Lyons JA, Yano H, Javitch JA, Nissen P. 2014. A mechanism for intracellular release of Na + by neurotransmitter/sodium symporters. Nat Struct Mol Biol 21:1006–1012.
- Malinauskaite L, Said S, Sahin C, Grouleff J, Shahsavar A, Bjerregaard H, et al. 2016. A conserved leucine occupies the empty substrate site of LeuT in the Na(+)-free return state. Nature Commun 7:11673.
- Masson J, Sagne C, Hamon M, El Mestikawy S. 1999. Neurotransmitter transporters in the central nervous system. Pharmacol Rev 51:439–464.
- Mayrand RR, Levitt DG. 1983. Urea and ethylene glycol-facilitated transport systems in the human red cell membrane. Saturation, competition, and asymmetry. J Gen Physiol 81:221–237.
- Meinild AK, Hirayama BA, Wright EM, Loo DD. 2002. Fluorescence studies of ligand-induced conformational changes of the Na(+)/glucose cotransporter. Biochemistry 41:1250–1258.
- Mirza O, Guan L, Verner G, Iwata S, Kaback HR. 2006. Structural evidence for induced fit and a mechanism for sugar/H + symport in LacY. EMBO J 25:1177–1183.
- Montalbetti N, Simonin A, Kovacs G, Hediger MA. 2013. Mammalian iron transporters: families SLC11 and SLC40. Mol Aspects Med 34:270–287.
- Motohashi H, Inui K. 2013. Multidrug and toxin extrusion family SLC47: physiological, pharmacokinetic and toxicokinetic importance of MATE1 and MATE2-K. Mol Aspects Med 34:661–668.
- Moura DA, Oliveira JR. 2015. XPR1: a gene linked to primary familial brain calcification might help explain a spectrum of neuropsychiatric disorders. J Mol Neurosci 57:519–521.
- Mouro-Chanteloup I, Cochet S, Chami M, Genetet S, Zidi-Yahiaoui N, Engel A, et al. 2010. Functional reconstitution into liposomes of purified human RhCG ammonia channel. PLoS One 5:e8921.
- Mueckler M, Thorens B. 2013. The SLC2 (GLUT) family of membrane transporters. Mol Aspects Med 34:121–138.
- Nakhoul NL, Abdulnour-Nakhoul SM, Boulpaep EL, Rabon E, Schmidt E, Hamm LL. 2010a. Substrate specificity of Rhbg: ammonium and methyl ammonium transport. Am J Physiol Cell Physiol 299:C695–C705.
- Nakhoul NL, Abdulnour-Nakhoul SM, Schmidt E, Doetjes R, Rabon E, Hamm LL. 2010b. pH sensitivity of ammonium transport by Rhbg. Am J Physiol Cell Physiol 299:C1386–C1397.
- Nakhoul NL, Dejong H, Abdulnour-Nakhoul SM, Boulpaep EL, Hering-Smith K, Hamm LL. 2005. Characteristics of renal Rhbg as an NH4(+) transporter. Am J Physiol Renal Physiol 288:F170–F181.
- Nakhoul NL, Lee Hamm L. 2013. Characteristics of mammalian Rh glycoproteins (SLC42 transporters) and their role in acid-base transport. Mol Aspects Med 34:629–637.
- Nakhoul NL, Schmidt E, Abdulnour-Nakhoul SM, Hamm LL. 2006. Electrogenic ammonium transport by renal Rhbg. Transfus Clin Biol 13:147–153.
- Napoli L, Bordoni A, Zeviani M, Hadjigeorgiou GM, Sciacco M, Tiranti V, et al. 2001. A novel missense adenine nucleotide translocator-1 gene mutation in a Greek adPEO family. Neurology 57:2295–2298.
- Newstead S, Drew D, Cameron AD, Postis VL, Xia X, Fowler PW, et al. 2011. Crystal structure of a prokaryotic homologue of the mammalian oligopeptide-proton symporters, PepT1 and PepT2. EMBO J 30:417–426.
- Nicolas V, Mouro-Chanteloup I, Lopez C, Gane P, Gimm A, Mohandas N, et al. 2006. Functional interaction between Rh proteins and the spectrin-based skeleton in erythroid and epithelial cells. Transfus Clin Biol 13:23–28.
- Nomura N, Verdon G, Kang HJ, Shimamura T, Nomura Y, Sonoda Y, et al. 2015. Structure and mechanism of the mammalian fructose transporter GLUT5. Nature 526:397–401.
- Nurnberg P, Thiele H, Chandler D, Hohne W, Cunningham ML, Ritter H, et al. 2001. Heterozygous mutations in ANKH, the human ortholog of the mouse progressive ankylosis gene, result in craniometaphyseal dysplasia. Nat Genet 28:37–41.
- Nury H, Dahout-Gonzalez C, Trezeguet V, Lauquin G, Brandolin G, Pebay-Peyroula E. 2005. Structural basis for lipid-mediated interactions between mitochondrial ADP/ATP carrier monomers. FEBS Lett 579:6031–6036.
- Oka Y, Asano T, Shibasaki Y, Lin JL, Tsukuda K, Katagiri H, et al. 1990. C-terminal truncated glucose transporter is locked into an inward-facing form without transport activity. Nature 345:550–553.
- Palmieri F. 2013. The mitochondrial transporter family SLC25: identification, properties and physiopathology. Mol Aspects Med 34:465–484.
- Panayotova-Heiermann M, Loo DD, Lam JT, Wright EM. 1998. Neutralization of conservative charged transmembrane residues in the Na+/glucose cotransporter SGLT1. Biochemistry 37:10522–10528.
- Pao SS, Paulsen IT, Saier MH, Jr. 1998. Major facilitator superfamily. Microbiol Mol Biol Rev MMBR 62:1–34.
- Parker JL, Newstead S. 2014. Molecular basis of nitrate uptake by the plant nitrate transporter NRT1.1. Nature 507:68–72.
- Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trezeguet V, Lauquin GJ, Brandolin G. 2003. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 426:39–44.
- Pedersen BP, Kumar H, Waight AB, Risenmay AJ, Roe-Zurz Z, Chau BH, et al. 2013. Crystal structure of a eukaryotic phosphate transporter. Nature 496:533–536.
- Pena MM, Lee J, Thiele DJ. 1999. A delicate balance: homeostatic control of copper uptake and distribution. J Nutr 129:1251–1260.
- Penmatsa A, Wang KH, Gouaux E. 2013. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 503:85–90.
- Piscitelli CL, Gouaux E. 2012. Insights into transport mechanism from LeuT engineered to transport tryptophan. EMBO J 31:228–235.
- Pittman JK, Hirschi KD. 2001. Regulation of CAX1, an arabidopsis Ca(2+)/H + antiporter. Identification of an N-terminal autoinhibitory domain. Plant Physiol 127:1020–1029.
- Pittman JK, Sreevidya CS, Shigaki T, Ueoka-Nakanishi H, Hirschi KD. 2002. Distinct N-terminal regulatory domains of Ca(2+)/H(+) antiporters. Plant Physiol 130:1054–1062.
- Pramod AB, Foster J, Carvelli L, Henry LK. 2013. SLC6 transporters: structure, function, regulation, disease association and therapeutics. Mol Aspects Med 34:197–219.
- Prasad AS, Halsted JA, Nadimi M. 1961. Syndrome of iron deficiency anemia, hepatosplenomegaly, hypogonadism, dwarfism and geophagia. Am J Med 31:532–546.
- Quick M, Shi L, Zehnpfennig B, Weinstein H, Javitch JA. 2012. Experimental conditions can obscure the second high-affinity site in LeuT. Nat Struct Mol Biol 19:207–211.
- Quick M, Winther AM, Shi L, Nissen P, Weinstein H, Javitch JA. 2009. Binding of an octylglucoside detergent molecule in the second substrate (S2) site of LeuT establishes an inhibitor-bound conformation. Proc Natl Acad Sci USA 106:5563–5568.
- Quistgaard EM, Low C, Moberg P, Tresaugues L, Nordlund P. 2013. Structural basis for substrate transport in the GLUT-homology family of monosaccharide transporters. Nat Struct Mol Biol 20:766–768.
- Rainier S, Chai JH, Tokarz D, Nicholls RD, Fink JK. 2003. NIPA1 gene mutations cause autosomal dominant hereditary spastic paraplegia (SPG6). Am J Hum Genet 73:967–971.
- Rask-Andersen M, Masuram S, Fredriksson R, Schioth HB. 2013. Solute carriers as drug targets: current use, clinical trials and prospective. Mol Aspects Med 34:702–710.
- Reichenberger E, Tiziani V, Watanabe S, Park L, Ueki Y, Santanna C, et al. 2001. Autosomal dominant craniometaphyseal dysplasia is caused by mutations in the transmembrane protein ANK. Am J Hum Genet 68:1321–1326.
- Reimer RJ. 2013. SLC17: a functionally diverse family of organic anion transporters. Mol Aspects Med 34:350–359.
- Reithmeier RA, Casey JR, Kalli AC, Sansom MS, Alguel Y, Iwata S. 2016. Band 3, the human red cell chloride/bicarbonate anion exchanger (AE1, SLC4A1), in a structural context. Biochim Biophys Acta 1858:1507–1532.
- Reithmeier RA, Moraes TF. 2015. Solute carriers keep on rockin'. Nat Struct Mol Biol 22:752–754.
- Reyes N, Ginter C, Boudker O. 2009. Transport mechanism of a bacterial homologue of glutamate transporters. Nature 462:880–885.
- Reyes N, Oh S, Boudker O. 2013. Binding thermodynamics of a glutamate transporter homolog. Nat Struct Mol Biol 20:634–640.
- Ripoche P, Bertrand O, Gane P, Birkenmeier C, Colin Y, Cartron JP. 2004. Human Rhesus-associated glycoprotein mediates facilitated transport of NH(3) into red blood cells. Proc Natl Acad Sci USA 101:17222–17227.
- Romero MF, Chen AP, Parker MD, Boron WF. 2013. The SLC4 family of bicarbonate (HCO3-) transporters. Mol Aspects Med 34:159–182.
- Ruprecht JJ, Hellawell AM, Harding M, Crichton PG, McCoy AJ, Kunji ER. 2014. Structures of yeast mitochondrial ADP/ATP carriers support a domain-based alternating-access transport mechanism. Proc Natl Acad Sci USA 111:E426–E434.
- Ryan RM, Compton EL, Mindell JA. 2009. Functional characterization of a Na+-dependent aspartate transporter from Pyrococcus horikoshii. J Biol Chem 284:17540–17548.
- Sahni J, Scharenberg AM. 2013. The SLC41 family of MgtE-like magnesium transporters. Mol Aspects Med 34:620–628.
- Sanders D, Brownlee C, Harper JF. 1999. Communicating with calcium. Plant Cell 11:691–706.
- Sands JM, Knepper MA. 1987. Urea permeability of mammalian inner medullary collecting duct system and papillary surface epithelium. J Clin Invest 79:138–147.
- Schafer JA, Troutman SL, Andreoli TE. 1974. Osmosis in cortical collecting tubules. ADH-independent osmotic flow rectification. J Gen Physiol 64:228–240.
- Schioth HB, Roshanbin S, Hagglund MG, Fredriksson R. 2013. Evolutionary origin of amino acid transporter families SLC32, SLC36 and SLC38 and physiological, pathological and therapeutic aspects. Mol Aspects Med 34:571–585.
- Schnetkamp PP. 2013. The SLC24 gene family of Na(+)/Ca(2)(+)-K(+) exchangers: from sight and smell to memory consolidation and skin pigmentation. Mol Aspects Med 34:455–464.
- Shaffer PL, Goehring A, Shankaranarayanan A, Gouaux E. 2009. Structure and mechanism of a Na+-independent amino acid transporter. Science 325:1010–1014.
- Shaikh SA, Tajkhorshid E. 2010. Modeling and dynamics of the inward-facing state of a Na+/Cl- dependent neurotransmitter transporter homologue. PLoS Comput Biol 6:e1000905.
- Shan J, Javitch JA, Shi L, Weinstein H. 2011. The substrate-driven transition to an inward-facing conformation in the functional mechanism of the dopamine transporter. PLoS One 6:e16350.
- Shannon JR, Flattem NL, Jordan J, Jacob G, Black BK, Biaggioni I, et al. 2000. Orthostatic intolerance and tachycardia associated with norepinephrine-transporter deficiency. N Engl J Med 342:541–549.
- Shi L, Quick M, Zhao Y, Weinstein H, Javitch JA. 2008. The mechanism of a neurotransmitter:sodium symporter–inward release of Na + and substrate is triggered by substrate in a second binding site. Mol Cell 30:667–677.
- Shigaki T, Rees I, Nakhleh L, Hirschi KD. 2006. Identification of three distinct phylogenetic groups of CAX cation/proton antiporters. J Mol Evol 63:815–825.
- Singh SK, Piscitelli CL, Yamashita A, Gouaux E. 2008. A competitive inhibitor traps LeuT in an open-to-out conformation. Science 322:1655–1661.
- Singh SK, Yamashita A, Gouaux E. 2007. Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature 448:952–956.
- Smirnova I, Kasho V, Kaback HR. 2011. Lactose permease and the alternating access mechanism. Biochemistry 50:9684–9693.
- Smith DE, Clemencon B, Hediger MA. 2013. Proton-coupled oligopeptide transporter family SLC15: physiological, pharmacological and pathological implications. Mol Aspects Med 34:323–336.
- Solcan N, Kwok J, Fowler PW, Cameron AD, Drew D, Iwata S, Newstead S. 2012. Alternating access mechanism in the POT family of oligopeptide transporters. EMBO J 31:3411–3421.
- Song Z. 2013. Roles of the nucleotide sugar transporters (SLC35 family) in health and disease. Mol Aspects Med 34:590–600.
- Soupene E, Inwood W, Kustu S. 2004. Lack of the Rhesus protein Rh1 impairs growth of the green alga Chlamydomonas reinhardtii at high CO2. Proc Natl Acad Sci USA 101:7787–7792.
- Sujatha MS, Balaji PV. 2004. Identification of common structural features of binding sites in galactose-specific proteins. Proteins 55:44–65.
- Sun J, Bankston JR, Payandeh J, Hinds TR, Zagotta WN, Zheng N. 2014. Crystal structure of the plant dual-affinity nitrate transporter NRT1.1. Nature 507:73–77.
- Sun L, Zeng X, Yan C, Sun X, Gong X, Rao Y, Yan N. 2012. Crystal structure of a bacterial homologue of glucose transporters GLUT1-4. Nature 490:361–366.
- Suzuki T, Ishihara K, Migaki H, Ishihara K, Nagao M, Yamaguchi-Iwai Y, Kambe T. 2005. Two different zinc transport complexes of cation diffusion facilitator proteins localized in the secretory pathway operate to activate alkaline phosphatases in vertebrate cells. J Biol Chem 280:30956–30962.
- Tamai I, Nakanishi T, Hayashi K, Terao T, Sai Y, Shiraga T, et al. 1997. The predominant contribution of oligopeptide transporter PepT1 to intestinal absorption of beta-lactam antibiotics in the rat small intestine. J Pharm Pharmacol 49:796–801.
- Thangaratnarajah C, Ruprecht JJ, Kunji ER. 2014. Calcium-induced conformational changes of the regulatory domain of human mitochondrial aspartate/glutamate carriers. Nat Commun 5:5491.
- Torroni A, Stepien G, Hodge JA, Wallace DC. 1990. Neoplastic transformation is associated with coordinate induction of nuclear and cytoplasmic oxidative phosphorylation genes. J Biol Chem 265:20589–20593.
- Toufaily C, Vargas A, Lemire M, Lafond J, Rassart E, Barbeau B. 2013. MFSD2a, the Syncytin-2 receptor, is important for trophoblast fusion. Placenta 34:85–88.
- Traiffort E, O'Regan S, Ruat M. 2013. The choline transporter-like family SLC44: properties and roles in human diseases. Mol Aspects Med 34:646–654.
- Tsui HW, Inman RD, Paterson AD, Reveille JD, Tsui FW. 2005. ANKH variants associated with ankylosing spondylitis: gender differences. Arthritis Res Ther 7:R513–R525.
- Vallee BL, Falchuk KH. 1993. The biochemical basis of zinc physiology. Physiol Rev 73:79–118.
- Verdon G, Boudker O. 2012. Crystal structure of an asymmetric trimer of a bacterial glutamate transporter homolog. Nat Struct Mol Biol 19:355–357.
- Verdon G, Oh S, Serio RN, Boudker O. 2014. Coupled ion binding and structural transitions along the transport cycle of glutamate transporters. eLife 3:e02283.
- Vidaver GA. 1966. Inhibition of parallel flux and augmentation of counter flux shown by transport models not involving a mobile carrier. J Theoret Biol 10:301–306.
- Vitavska O, Wieczorek H. 2013. The SLC45 gene family of putative sugar transporters. Mol. Aspects Med 34:655–660.
- Vulpe CD, Packman S. 1995. Cellular copper transport. Annu Rev Nutr 15:293–322.
- Waight AB, Pedersen BP, Schlessinger A, Bonomi M, Chau BH, Roe-Zurz Z, et al. 2013. Structural basis for alternating access of a eukaryotic calcium/proton exchanger. Nature 499:107–110.
- Waldman ID, Rowe DC, Abramowitz A, Kozel ST, Mohr JH, Sherman SL, et al. 1998. Association and linkage of the dopamine transporter gene and attention-deficit hyperactivity disorder in children: heterogeneity owing to diagnostic subtype and severity. Am J Hum Genet 63:1767–1776.
- Wang H, Elferich J, Gouaux E. 2012. Structures of LeuT in bicelles define conformation and substrate binding in a membrane-like context. Nat Struct Mol Biol 19:212–219.
- Wang H, Goehring A, Wang KH, Penmatsa A, Ressler R, Gouaux E. 2013. Structural basis for action by diverse antidepressants on biogenic amine transporters. Nature 503:141–145.
- Wang H, Gouaux E. 2012. Substrate binds in the S1 site of the F253A mutant of LeuT, a neurotransmitter sodium symporter homologue. EMBO Rep 13:861–866.
- Wang JZ, Xiao N, Zhang YZ, Zhao CX, Guo XH, Lu LM. 2016. Mfsd2a-based pharmacological strategies for drug delivery across the blood-brain barrier. Pharmacol Res 104:124–131.
- Watanabe A, Choe S, Chaptal V, Rosenberg JM, Wright EM, Grabe M, Abramson J. 2010. The mechanism of sodium and substrate release from the binding pocket of vSGLT. Nature 468:988–991.
- Wenzel U, Thwaites DT, Daniel H. 1995. Stereoselective uptake of beta-lactam antibiotics by the intestinal peptide transporter. Br J Pharmacol 116:3021–3027.
- White PJ, Broadley MR. 2003. Calcium in plants. Ann Bot 92:487–511.
- Widdas WF. 1952. Inability of diffusion to account for placental glucose transfer in the sheep and consideration of the kinetics of a possible carrier transfer. J Physiol 118:23–39.
- Williams CJ, Zhang Y, Timms A, Bonavita G, Caeiro F, Broxholme J, et al. 2002. Autosomal dominant familial calcium pyrophosphate dihydrate deposition disease is caused by mutation in the transmembrane protein ANKH. Am J Hum Genet 71:985–991.
- Wisedchaisri G, Park MS, Iadanza MG, Zheng H, Gonen T. 2014. Proton-coupled sugar transport in the prototypical major facilitator superfamily protein XylE. Nat Commun 5:4521
- Wright EM. 2013. Glucose transport families SLC5 and SLC50. Mol Aspects Med 34:183–196.
- Wright EM, Hirayama BA, Loo DF. 2007. Active sugar transport in health and disease. J Intern Med 261:32–43.
- Wu M, Tong S, Waltersperger S, Diederichs K, Wang M, Zheng L. 2013. Crystal structure of Ca2+/H + antiporter protein YfkE reveals the mechanisms of Ca2+ efflux and its pH regulation. Proc Natl Acad Sci USA 110:11367–11372.
- Xu H, Jin J, DeFelice LJ, Andrews NC, Clapham DE. 2004. A spontaneous, recurrent mutation in divalent metal transporter-1 exposes a calcium entry pathway. PLoS Biol 2:E50.
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. 2005. Crystal structure of a bacterial homologue of Na+/Cl–dependent neurotransmitter transporters. Nature 437:215–223.
- Yan N. 2015. Structural biology of the major facilitator superfamily transporters. Annu Rev Biophys 44:257–283.
- Yernool D, Boudker O, Jin Y, Gouaux E. 2004. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature 431:811–818.
- Yin Y, He X, Szewczyk P, Nguyen T, Chang G. 2006. Structure of the multidrug transporter EmrD from Escherichia coli. Science 312:741–744.
- Yonezawa A, Inui K. 2013. Novel riboflavin transporter family RFVT/SLC52: identification, nomenclature, functional characterization and genetic diseases of RFVT/SLC52. Mol Aspects Med 34:693–701.
- Young JD, Yao SY, Baldwin JM, Cass CE, Baldwin SA. 2013. The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol Aspects Med 34:529–547.
- Zeevaert R, de Zegher F, Sturiale L, Garozzo D, Smet M, Moens M, et al. 2013. Bone dysplasia as a key feature in three patients with a novel congenital disorder of glycosylation (CDG) type II due to a deep intronic splice mutation in TMEM165. JIMD Rep 8:145–152.
- Zhao R, Goldman ID. 2013. Folate and thiamine transporters mediated by facilitative carriers (SLC19A1-3 and SLC46A1) and folate receptors. Mol Aspects Med 34:373–385.
- Zhao Y, Mao G, Liu M, Zhang L, Wang X, Zhang XC. 2014. Crystal structure of the E. coli peptide transporter YbgH. Structure 22:1152–1160.
- Zhao Y, Terry D, Shi L, Weinstein H, Blanchard SC, Javitch JA. 2010. Single-molecule dynamics of gating in a neurotransmitter transporter homologue. Nature 465:188–193.
- Zhao Y, Terry DS, Shi L, Quick M, Weinstein H, Blanchard SC, Javitch JA. 2011. Substrate-modulated gating dynamics in a Na+-coupled neurotransmitter transporter homologue. Nature 474:109–113.
- Zhou X, Levin EJ, Pan Y, McCoy JG, Sharma R, Kloss B, et al. 2014. Structural basis of the alternating-access mechanism in a bile acid transporter. Nature 505:569–573.
- Zhou Z, Zhen J, Karpowich NK, Goetz RM, Law CJ, Reith ME, Wang DN. 2007. LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science 317:1390–1393.
- Zhou Z, Zhen J, Karpowich NK, Law CJ, Reith ME, Wang DN. 2009. Antidepressant specificity of serotonin transporter suggested by three LeuT-SSRI structures. Nat Struct Mol Biol 16:652–657.
- Zidi-Yahiaoui N, Mouro-Chanteloup I, D'Ambrosio AM, Lopez C, Gane P, L, van Kim C, Cartron JP, et al. 2005. Human Rhesus B and Rhesus C glycoproteins: properties of facilitated ammonium transport in recombinant kidney cells. Biochem J 391:33–40.