
Abstract
Panel based next generation sequencing was performed on a discovery cohort of AML with RUNX1-RUNX1T1. Supervised machine learning identified NRAS mutation and absence of mutations in ASXL2, RAD21, KIT and FLT3 genes as well as a low mutation to be associated with favorable outcome. Based on this data patients were classified into favorable and poor genetic risk classes. Patients classified as poor genetic risk had a significantly lower overall survival (OS) and relapse free survival (RFS). We could validate these findings independently on a validation cohort (n = 61). Patients in the poor genetic risk group were more likely to harbor measurable residual disease. Poor genetic risk emerged as an independent risk factor predictive of inferior outcome. Using an unbiased computational approach based we provide evidence for gene panel-based testing in AML with RUNX1-RUNX1T1 and a framework for integration of genomic markers toward clinical decision making in this heterogeneous disease entity.
Introduction
Acute myeloid leukemia (AML) with t(8;21)(q22;q22) that results in the RUNX1-RUNX1T1 chimeric gene fusion is one of the commonest subtypes of AML. It is characterized by a distinct morphology and a unique immunophenotype and is thus recognized as a specific entity amongst ‘AML with recurrent genetic abnormalities’ [Citation1]. Traditionally, this AML has been recognized as having a favorable outcome as evident by superior survival rates when compared to intermediate and poor cytogenetic risk AMLs [Citation2]. Unfortunately, the treatment outcome in these cases is not homogeneous as evident by relapse in a significant number of patients despite achievement of morphological complete remission (CR) [Citation3,Citation4]. In fact, studies claim that only half the patients of AML with RUNX1-RUNX1T1 get cured [Citation5,Citation6]. This heterogeneous outcome has been explained, in part, by cooperating somatic mutations in genes involved in signaling pathways such as FLT3 and KIT [Citation7].
In the last few years, largely due to next generation sequencing (NGS) technologies, we have identified somatic mutations affecting diverse cellular pathways in AML [Citation8]. Some of these mutations are clinically relevant affecting prognosis or are amenable to targeted therapy. Somatic mutations have been identified in nearly 90% of AML with t(8;21) and commonly include genes encoding for chromatin modifiers (e.g. ASXL1, ASXL2, EZH2, KDM6A), cohesin complex (e.g. RAD21, SMC3, SMC1A) and signaling pathways (e.g. KIT, FLT3, NRAS) [Citation4,Citation9]. Whereas a general consensus exists amongst researchers that the above sets of genes are recurrently mutated in AML with RUNX1-RUNX1T1, an accurate prognostication scheme that guides a treating physician is largely lacking. It is imperative that better approaches be developed so that we can identify patients who are at a high risk of relapse in this rather common subtype of AML.
Machine learning (ML) is a subset of artificial intelligence that holds promise in deciphering complex genomic datasets. ML has been used to develop algorithms for diverse applications such as identification of regulatory regions in the genome to prediction of cancer susceptibility, recurrence and survival [Citation10,Citation11]. ML has also been recently used for prediction of drug response in AML based on gene expression profiles as well as discovery of novel antibiotics [Citation12,Citation13]. We have recently developed a supervised ML based algorithm for prognostication of AML with mutated NPM1 based on the underlying genomic data [Citation14]. To decipher the clinical significance of the large numbers of genetic variables we used an unbiased computational approach and identified that NPM1 mutation type and corrected NPM1 variant allele fraction (VAF), presence of DNMT3A R882 mutation, FLT3 internal tandem duplication VAF and IDH2 mutations were clinically relevant. Based on these ML derived variables we developed a scoring system. The genetic score could classify AML with mutated NPM1 into three classes with vastly different outcomes.
Given the heterogeneity in treatment outcomes that is observed in AML with RUNX1-RUNX1T1 we questioned if such ML based approaches can be applied to AML with RUNX1-RUNX1T1. In this manuscript, we develop a ML based genomics driven prognostication model for AML with RUNX1-RUNX1T1 and demonstrate that this model correlates with measurable residual disease (MRD) and clinical outcome.
Methods
PATIENT DETAILS:
Patient Accrual: The study was cleared by the institutional ethics board (IEC III Project 163 and IEC III Project 900613). We accrued a total of 131 patients of AML with RUNX1-RUNX1T1. These patients were accrued over a 7-year period from March 2012 to April 2019. Diagnosis, immunophenotyping and cytogenetic analysis were performed as previously described [Citation3].
Patient Treatment and Evaluation of Outcome: Patients were divided into a discovery cohort (n = 70) and an independent validation cohort (n = 61). All patients were treated with conventional induction ‘3 + 7’ chemotherapy consisting of daunorubicin (60 mg/m2 D1-D3) and cytarabine (100 mg/m2/day D1-D7). For the validation cohort, 13 out of 61 patients had baseline fungal pneumonia or multidrug resistant bacterial colonization and were treated with oral metronomic chemotherapy to stabilize the patient prior to intensive ‘3 + 7’ chemotherapy. Complete remission (CR), overall survival (OS) and relapse free survival (RFS) were calculated as previously described [Citation3,Citation14,Citation15]. One out of 13 patients not achieving morphological CR was treated with palliation and the rest were treated with conventional therapy, at discretion of the treating physician.
GENETIC TESTING ON DIAGNOSTIC SAMPLE:
Cytogenetics: Only patients who were confirmed to have RUNX1-RUNX1T1 by conventional karyotyping and/or fluorescence in-situ hybridization (FISH) were included.
Panel Based NGS and Data Analysis: Details of the single molecule molecular inversion probe (smMIP) based myeloid sequencing panel and bioinformatics approaches used to analyze this dataset are as previously described in detail (please see supplementary methods) [Citation14].
Machine learning based genetic score: We developed a supervised ML based approach for identification of prognostic variables most likely to influence outcome in AML with RUNX1-RUNX1T1 as described previously [Citation14]. Additional details pertaining to ML can be seen in the Supplementary Methods that accompanies this manuscript. Based on the results of the ML model we scored each variable as ‘-1’ if the results were predictive of an unfavorable outcome and ‘+1’ if otherwise. A sum of these scores was finally derived to generate a final score. Based on this final score patients were classified into favorable and poor genetic risk (GR).
MEASURABLE RESIDUAL DISEASE ASSESSMENT USING MULTIPARAMETRIC FCM (FCM-MRD):
FCM-MRD was detected using a two tube 10 color assay as described previously by our group [Citation3]. Patients were called as FCM-MRD negative if they were negative on two consecutive MRD time points (post induction and post consolidation). Everyone else was MRD positive.
CORRELATION OF ML DERIVED GR WITH FCM-MRD AND TREATMENT OUTCOME:
Chi squared test was used to correlate FCM-MRD with ML derived GR classes. The impact of GR defined classes was also evaluated against OS and RFS using Kaplan–Meier technique and log-rank test.
Results
is a summary of clinical and laboratory parameters of the entire cohort.
Table 1. Prognostic significance of machine learning derived genetic risk in AML with t(8;21).
PATIENT DETAILS:
The median follow-up for the entire cohort (131 patients) was 27.6 months. The median overall survival (OS) was 30.7 months (95% CI: 23.0–38.4 months) and the median relapse free survival (RFS) was 32.9 months (95% CI: 27.7–38.1 months). Out of 131 patients only 4 underwent allogeneic bone marrow transplantation. Due to small numbers their outcome was not different from the rest with respect to OS (p = 0.25) or RFS (p = 0.9). These patients are therefore not considered separately. The clinical and laboratory parameters of the discovery and validation cohorts can be seen in Supplementary Tables 1 & 2 respectively.
B.GENETIC TESTING ON THE DIAGNOSTIC SAMPLE:
Cytogenetics and gene mutations: Details of conventional karyotyping and FISH can be seen in supplementary data accompanying this manuscript. At least one somatic mutation was detected in 85.5% of all patients (median coverage: 983.5, range: 402–2793X). An overview of these mutations can be seen in . Supplementary Figure 1 highlights the frequencies of commonly occurring mutations seen in our cohort.
Figure 1. The above circos plot (A) highlights the spectrum of mutations and their interaction in AML with RUNX1-RUNX1T1. Commonly occurring gene mutations are colored. The machine learning derived scoring system is described in (B). The Kaplan–Meier plot in the top right section (C) shows the clinical impact on overall survival (OS) and for relapse free survival (RFS, D), lower right).
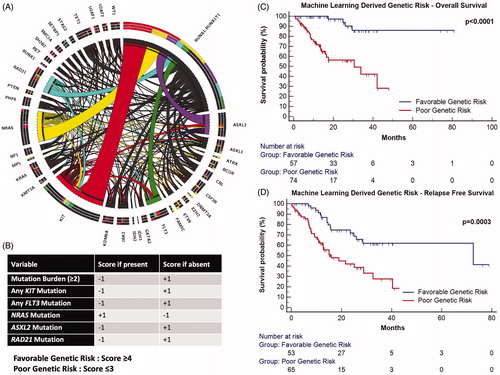
Machine Learning Based Modeling: Performance characteristics of ML model as well as results of feature selection can be seen in supplementary methods (Supplementary Tables 3–5 and Supplementary Figure 2) accompanying this manuscript. Based on the ML modeling on the discovery cohort, mutations in FLT3, NRAS, ASXL2, RAD21, KIT genes as well as mutation burden (≥2 as high mutation burden) were determined as important variables likely to predict outcome.
Machine Learning Derived Genetic Score: High mutation burden, mutations in FLT3, RAD21, ASXL2 and KIT genes were associated with an inferior prognosis and assigned a negative score whereas mutations in NRAS were associated with a favorable outcome and a positive score if present. These features were used to generate a scoring system as is seen in . Based on the prognostic impact of these variables a score was allotted to each feature [Citation14] the sum of which resulted in classification of patients into favorable (score ≥4) and poor genetic risk (score ≤3). In the discovery cohort, patients classified as poor genetic risk were associated with inferior OS and RFS (Supplementary Table 1 and Supplementary Figure 3). We reconfirmed the clinical relevance of these findings by an independent validation on a cohort of pediatric RUNX1-RUNX1T1 rearranged AML (Supplementary Table 2 and Supplementary Figure 4).
C.MEASURABLE RESIDUAL DISEASE ASSESSMENT USING MULTIPARAMETRIC FCM (FCM-MRD):
The presence of FCM-MRD was significantly associated with inferior OS and RFS (Supplementary Figure 5).
D.CORRELATION OF ML DERIVED GR WITH FCM-MRD AND TREATMENT OUTCOME:
We observed a strong correlation of ML derived genetic risk classes with FCM-MRD where cases which were classified as poor genetic risk were more likely to be MRD positive (Supplementary Figure 6). Lastly ML derived GR based risk classes were highly predictive of outcome as seen in and . The clinical relevance of individual components of the ML derived scoring system can be visualized in Supplementary Figure 7.
Discussion
In the last decade several studies have analyzed the prognostic relevance of signaling pathway mutations in core binding factor AML as reviewed by Boissel et al. [Citation16]. However, there is little consensus amongst investigators with respect to clinical relevance of these mutations. This could be attributed to various reasons, such as technical issues associated with low sensitivity assays like sanger sequencing. Some investigators have indicated that allelic abundances of mutations are important [Citation9,Citation17]. Others have indicated that even within a single gene, mutational hotspots such as KIT D816 [Citation18] or FLT3-TKD [Citation19] may have different prognostic connotation in core binding factor AML. Krauth and colleagues indicated mutation burden may be an additional determinant of outcome [Citation20]. Studies employing high throughput sequencing technologies have indicated that beyond signaling pathways the mutational landscape of AML with t(8;21) may be unique characterized by high frequencies of mutations in genes encoding for cohesin complex and chromatin modeling pathways [Citation9,Citation21]. As a result of lack of consensus, current guidelines group AML with RUNX1-RUNX1T1 as a single disease entity [Citation22,Citation23].
Instead of selecting individual genes, we analyzed all commonly (>5%) occurring mutations in AML with RUNX1-RUNX1T1 in an unbiased manner using a supervised machine learning algorithm. The threshold of including mutations occurring at a frequency of >5% (from a 50 gene panel) is chosen empirically. This is keeping in mind a balance between applicability of the model to AML with RUNX1-RUNX1T1 and inclusion of nongeneralizable data (typically seen with rare mutations). This will presumably prevent ‘overfitting’ into the dataset [Citation24]. ML approaches allow us to identify interactions between data that are not readily visible using legacy approaches. Our approach enabled us to develop a scoring system based which we could classify AML with RUNX1-RUNX1T1 into two prognostic subgroups with different outcomes. In a multivariate analysis this was found to be an independently important predictor of outcome.
Based on these data, we propose a risk stratification of AML with RUNX1-RUNX1T1 that incorporates somatic mutations in FLT3, NRAS, ASXL2, RAD21, KIT genes as well as mutation burden. From data that has been published previously we expected that KIT [Citation18] and FLT3 (exon 20) [Citation19] gene mutations would be prognostically relevant. In addition, based on scant published data we also suspected that mutation burden would influence outcome [Citation20]. However, using AI we could additionally infer the prognostic impact of NRAS, RAD21, FLT3-ITD, ASXL2 mutations. The latter were not expected from legacy data. Micol et al and other studies, have previously demonstrated a high frequency of ASXL2 mutations in this subset of AML and possible inferior outcome [Citation20,Citation25]. Recently Ishikawa et al indicated that only exon 17 mutations were prognostically relevant in AML with RUNX1-RUNX1T1. In comparison, we determined that all KIT mutations may be relevant as determined by ML modeling. Cohesin gene mutations have been associated with inferior outcome in myeloid malignancies [Citation26]. In our study, we identify RAD21 mutations as a mutation associated with possible inferior outcome, especially in the context of ML derived scoring system. NRAS mutations have been described in AML with RUNX1-RUNX1T1, however, have failed to demonstrate a clear survival advantage [Citation27,Citation28]. We think, a more global approach which takes into account the complex interaction of these mutations rather than a simplistic evaluation as evident by our scoring system is warranted for prognostication of this seemingly homogeneous AML.
A disadvantage of our study could possibly be not including recently described newer gene mutations including ZBTB7A [Citation29]. Nonetheless, our approach provides additional evidence for gene panel-based testing in AML with RUNX1-RUNX1T1 and a general framework for the integration of genomic markers toward clinical decision making. The potential limitations of this study include a retrospective analysis and a limited number of patients. This machine learning derived genomics score for AML with RUNX1-RUNX1T1 should be validated prospectively by other investigators.
GLAL-2020-0767-File004.docx
Download MS Word (23.7 KB)GLAL-2020-0767-File002.docx
Download MS Word (2.2 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405.
- Grimwade D, Walker H, Oliver F, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children's Leukaemia Working Parties. Blood. 1998;92(7):2322–2333.
- Patkar N, Kakirde C, Bhanshe P, et al. Utility of immunophenotypic measurable residual disease in adult acute myeloid leukemia-real-world context. Front Oncol. 2019;9:450.
- Christen F, Hoyer K, Yoshida K, et al. Genomic landscape and clonal evolution of acute myeloid leukemia with t(8;21): an international study on 331 patients. Blood. 2019;133(10):1140–1151.
- Marcucci G, Mrozek K, Ruppert AS, et al. Prognostic factors and outcome of core binding factor acute myeloid leukemia patients with t(8;21) differ from those of patients with inv(16): a Cancer and Leukemia Group B study. J Clin Oncol. 2005;23(24):5705–5717.
- Schlenk RF, Benner A, Krauter J, et al. Individual patient data-based meta-analysis of patients aged 16 to 60 years with core binding factor acute myeloid leukemia: a survey of the German Acute Myeloid Leukemia Intergroup. J Clin Oncol. 2004;22(18):3741–3750.
- Care RS, Valk PJ, Goodeve AC, et al. Incidence and prognosis of c-KIT and FLT3 mutations in core binding factor (CBF) acute myeloid leukaemias. Br J Haematol. 2003;121(5):775–777.
- Ley TJ, Miller C, Ding L, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074.
- Duployez N, Marceau-Renaut A, Boissel N, et al. Comprehensive mutational profiling of core binding factor acute myeloid leukemia. Blood. 2016;127(20):2451–2459.
- Kourou K, Exarchos TP, Exarchos KP, et al. Machine learning applications in cancer prognosis and prediction. Comput Struct Biotechnol J. 2015;13:8–17.
- Libbrecht MW, Noble WS. Machine learning applications in genetics and genomics. Nat Rev Genet. 2015;16(6):321–332.
- Lee SI, Celik S, Logsdon BA, et al. A machine learning approach to integrate big data for precision medicine in acute myeloid leukemia. Nat Commun. 2018;9(1):42.
- Stokes JM, Yang K, Swanson K, et al. A deep learning approach to antibiotic discovery. Cell. 2020;180(4):688–702.e13.
- Patkar N, Shaikh AF, Kakirde C, et al. A novel machine-learning-derived genetic score correlates with measurable residual disease and is highly predictive of outcome in acute myeloid leukemia with mutated NPM1. Blood Cancer J. 2019;9(10):79.
- Patkar N, Kodgule R, Kakirde C, et al. Clinical impact of measurable residual disease monitoring by ultradeep next generation sequencing in NPM1 mutated acute myeloid leukemia. Oncotarget. 2018;9(93):36613–36624.
- Boissel N, Leroy H, Brethon B, et al. Incidence and prognostic impact of c-Kit, FLT3, and Ras gene mutations in core binding factor acute myeloid leukemia (CBF-AML). Leukemia. 2006;20(6):965–970.
- Allen C, Hills RK, Lamb K, et al. The importance of relative mutant level for evaluating impact on outcome of KIT, FLT3 and CBL mutations in core-binding factor acute myeloid leukemia. Leukemia. 2013;27(9):1891–1901.
- Schnittger S, Kohl TM, Haferlach T, et al. KIT-D816 mutations in AML1-ETO-positive AML are associated with impaired event-free and overall survival. Blood. 2006;107(5):1791–1799.
- Cher CY, Leung GM, Au CH, et al. Next-generation sequencing with a myeloid gene panel in core-binding factor AML showed KIT activation loop and TET2 mutations predictive of outcome. Blood Cancer J. 2016;6(7):e442–e442.
- Krauth MT, Eder C, Alpermann T, et al. High number of additional genetic lesions in acute myeloid leukemia with t(8;21)/RUNX1-RUNX1T1: frequency and impact on clinical outcome. Leukemia. 2014;28(7):1449–1458.
- Faber ZJ, Chen X, Gedman AL, et al. The genomic landscape of core-binding factor acute myeloid leukemias. Nat Genet. 2016;48(12):1551–1556.
- Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–447.
- Tallman MS, Wang ES, Altman JK, OCN, et al. Acute myeloid leukemia, version 3.2019, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2019;17(6):721–749.
- Radakovich N, Nagy M, Nazha A. Machine learning in haematological malignancies. Lancet Haematol. 2020;7(7):e541–e550.
- Micol JB, Duployez N, Boissel N, et al. Frequent ASXL2 mutations in acute myeloid leukemia patients with t(8;21)/RUNX1-RUNX1T1 chromosomal translocations. Blood. 2014;124(9):1445–1449.
- Thota S, Viny AD, Makishima H, et al. Genetic alterations of the cohesin complex genes in myeloid malignancies. Blood. 2014;124(11):1790–1798.
- Bowen DT, Frew ME, Hills R, et al. RAS mutation in acute myeloid leukemia is associated with distinct cytogenetic subgroups but does not influence outcome in patients younger than 60 years. Blood. 2005;106(6):2113–2119.
- Berman JN, Gerbing RB, Alonzo TA, et al. Prevalence and clinical implications of NRAS mutations in childhood AML: a report from the Children's Oncology Group. Leukemia. 2011;25(6):1039–1042.
- Opatz S, Bamopoulos SA, Metzeler KH, et al. The clinical mutatome of core binding factor leukemia. Leukemia. 2020;34(6):1553–1562.