
ABSTRACT
Density functional methods were used to predict the antioxidative efficiency of thirteen 4-benzylidenamino-4, 5-dihydro-1H-1,2,4-triazol-5-one derivatives in the gas phase and in the solution phase (water and benzene). Optimized geometries of molecules and reaction thermodynamic energies (enthalpies and reaction-free energies) of three main antioxidant mechanisms (hydrogen atom transfer, single electron transfer-proton transfer, and sequential proton loss electron transfer) were studied at B3LYP/6-31G (d,p) level. Solvent contributions to thermodynamic energies were computed employing integral equation formalism integral equation formalism polarized continuum model method. Obtained results revealed that the three main working mechanisms were endothermic, but not spontaneous especially in the gas phase. We found that the single electron transfer process from the anionic form was more preferable than that from the neutral form in the gas phase. The comparison of the ionization potentials of 4-benzylidenamino-4, 5-dihydro-1H-1,2,4-triazol-5-one derivatives to those of classical antioxidants (gallic acid, caffeic acid, ferulic acid, and ascorbic acid) indicated that the electron transfer mechanism was more predominant in the thirteen 4-benzylidenamino-4, 5-dihydro-1H-1,2,4-triazol-5-one derivative compounds. Thermodynamically, single electron transfer process from the anionic form was the most preferable mechanism in the gas phase. Solvent effect drastically modified thermodynamic energies of mechanisms. The proton transfer process was the thermodynamically favored mechanism as compared to other mechanisms in both solvents. It is worth mentioning that all the mechanisms were found not to be spontaneous in the solution phase except the proton transfer process.
Introduction
The production of the excess of reactive species (reactive oxygen species [ROS], reactive nitrogen species [RNS], and reactive chlorine species [RCS]) in human organisms which react with the DNA, proteins, and other cellular components pose a health hazard.[Citation1] A significant destabilization of the DNA molecules is then expected at the side of its lesion.[Citation2,Citation3] This destabilization results from a localized perturbation of the tacking forces responsible for adhesion with water molecules (throughout hydrogen bonds) and positive ions like Na+ surrounding the DNA double helix.[Citation3,Citation4] In addition, ROS and RNS can attack various key cellular proteins like those participating in the DNA repair and cell cycle control.[Citation5] This latter phenomenon is usually underestimated in the analysis of chronic exposure to oxidative stress.[Citation1] The oxidative stress to the organism can be intensified beyond the basal level by a variety of endogenous and exogenous factors.[Citation6] Free radicals derived from circulating catecholamines are one possible endogenous source.[Citation7] The antioxidant defense system appears to be able to react adaptively in response to oxidative challenges, subject to regulation by the availability of other antioxidant compounds.[Citation8]
This fact is the justification of the considerable effort to find out of novel antioxidant compounds. The results obtained have shown that the antioxidant therapies have been recognized as strategies for preventing degenerative diseases.[Citation9–Citation11] The applications of antioxidants as preservatives in the food industry[Citation12] and skin-protective ingredients in cosmetics[Citation13] have also been observed. For instance, Citrius flavonoids (dietary polyphenolics) have been detected to be more effective antioxidant in vitro than vitamin E and C, and, therefore, used against cancer cell lines.[Citation14–Citation16] Phenolic compounds and its derivatives widely present in plants (vegetables, fruits, grains, and spices).[Citation17] play an important role in the nutrition and the diet. This is the plausible explanation of many experimental[Citation18,Citation19] and theoretical[Citation20–Citation27] studies on the antioxidant activities of phenolic acids (PhAs) observed. More recently, Yuzhen et al. succeeded, through an experimental and theoretical evaluation, to establish a relationship between three factors of selective PhAs and its derivatives: structure-thermodynamic-antioxidant activity.[Citation28] In the same vein, the 1,2,4-triazole derivatives have also been experimentally found out to be a powerful antioxidant with multiple biological activities (antibacterial, antifungal, antitubercular, anti-inflammatory, analgesic, antitumoral, antiviral, and anticonvulsant).[Citation29–Citation34] However, to the best of our knowledge, no study regarding the comparison of the antioxidant activities of 1,2,4-triazole derivatives to those of PhAs. The survey of the literature showed that few molecules containing simultaneously the 1, 2, 4-triazol ones and 3,4-dihydroxyphenyl moieties have been synthesized.[Citation33,Citation35] These synthesized chemicals were screened for their antioxidant properties. But this experimental estimation did not give any information on comparison of the contribution of each moiety to the antioxidant activity of these molecules. In our previous works on the 1,2,4-triazole derivatives, the emphasis was put on the theoretical assessment of the chelation of divalent cations by some 4-benzylidenamino-4,5-dihydro-1H-1,2,4-triazol-5-one derivatives (BDHTD) in various media.[Citation36] The investigation of the complexation of BDHTD by metal cations using the density functional method B3LYP indicated that that k2-O,N structures are most stable in the gas phase. A significant reduces of bond dissociation enthalpies (BDEs) observed when going from isolated BDHTD to complexes obtained confirms the enhancement of the antioxidant activity by the metal chelation.
It is noteworthy to mention that there are three generally accepted mechanisms of antioxidant action.[Citation20,Citation37] For an antioxidant RX-H, single-step hydrogen atom transfer (HAT) mechanism is described according to Eq. (1; X = O, S, N, and C). A numeral thermodynamic parameter associated with HAT mechanism is bond dissociation enthalpy (BDE).
The second possible mechanism is single electron transfer-proton transfer (SET-PT) mechanism by which an antioxidant RX-H can deactivate a free radical. In the first step (single electron transfer), the electron leaves the antioxidant molecule RX-H (Eq. [2]). The ionization potential (IP) is the enthalpy required to withdraw its electron.
Subsequently, the proton is transferred to RX• formed. The enthalpy change of the reaction (Eq. [3]) is called proton dissociation enthalpy (PDE).
The sequential proton loss electron transfer (SPLET) is another two-step mechanism described by the following reactions.
The reaction enthalpy of the first process is usually denoted proton affinity (PA) of the formed anion. The electron transfer enthalpy (ETE) represents the reaction enthalpy of the second step of the SPLET mechanism.
Here, we clarify the contribution of each moiety of BDHTD using DFT calculations. We have performed a systematic study of reaction thermodynamic energies (enthalpy and free energy) to HAT, SET-PT, and SPLET mechanisms for BDHTD. More precisely, five sets of thermodynamic parameters were estimated. Because of no experimental reaction thermodynamic energies for mechanisms of BDHTD are available; the authors integrated the substitution effect in the analysis. The solvation in two solvents (benzene and water) has been explored. From the thermodynamic point-of-view, the preferred mechanism was identified.
Computational details
All calculations were performed using Gaussian 09 program package.[Citation38] The geometries of each structure (RX-H, RX•, RX−, and RX-H+) was optimized using density functional theory (DFT) with B3LYP[Citation39] functional without any constraints. Calculations were performed using 6-31G (d,p) basis set in the gas phase and solution phase. In the same vein, the triple zeta basis set 6-311++G(d,p) in which diffuse s and d polarization function was added on hydrogen atom[Citation40] was used for solvation enthalpies of hydrogen atom or charges. The optimized structures were confirmed to be real minima by frequency calculations. The enthalpies obtained were zero-point corrected with unscaled frequencies. The description of the thermodynamic characteristic of solvation was performed employing an integral equation formalism polarized continuum model (IEF-PCM) method.[Citation41] The authors have determined the enthalpies of all reactions studies (reactions in Eqs. [Citation1–Citation5]) from the calculated total enthalpies at 298.15 K and 1.0 atmosphere pressure:
The calculated gas-phase enthalpy change of proton is () = 6.197 kJ/mol and the gas-phase enthalpy of an electron (
) is 3.1351 kJ/mol.[Citation42] Hydrogen atom, proton, and electron solvation enthalpies for studied solvents (water and benzene) were taken from previous researches.[Citation23,Citation43] From the calculated free energies, we have also determined the following quantities (bond dissociation free energy [BDFE], ionization potential free energy [IPFE], proton dissociation free energy [PDFE], proton affinity free energy [PAFE], and electron transfer free energy [ETFE]) for the reactions in Eqs. (1–5).
For electron (e−) and proton (H+) free energy, we employed the values of –3.72[Citation44] and –26.28 kJ/mol.[Citation45,Citation46] Hydrogen atom, proton, and electron solvation free energies in studied solvents (water and benzene) were taken from the report by Fifen et al.[Citation47]
Results and discussion
Geometrical details
The investigation of the substituent effect has forced the author to use several derivatives (A–M) with electron donating or withdrawing substituent on the 1, 2, 4-triazol-5-one ring of the 4-benzylidenamino-4, 5-dihydro-1H-1, 2, 4-triazol-5-ones (A) as shown in . The hypothesis of configurations with both free meta O1-H1 and O2-H2 bonds of the catechol moiety is excluded because of the zero probability of presence of similar conformations of 3,4-dihydroxyphenylpyruvic acid.[Citation23] As observed in , the authors only take into account the O2-H2…O1 intramolecular interaction that has showed to be stronger than O1H1….O2 for 3,4-dihydroxyphenylpyruvic acid.[Citation23] The optimized parameters obtained in various media (gas, water, and benzene) labeled according to the convention given in , are listed in . Since no experimental data of the molecules have been reported so far, the authors have compared these geometrical parameters with related crystallographically characterized 1-acetyl-4-(p-chlorobenzylideneamino)-3-methyl-4, 5-dihydro-1H-1, 2, 4-triazol-5-one[Citation48] presented in . One finds that the theoretical bond lengths are in general slightly higher than experimental results. This minor deviation may result from the neglected solvent effect or a slight structure difference between experimental and the author’s data.
Figure 1. Numbering system used to designate specific atom of the 4-benzylidenamino-4, 5-dihydro-1H-1, 2, 4-triazol-5-ones. A and specification of electron donating or withdrawing substituent used.
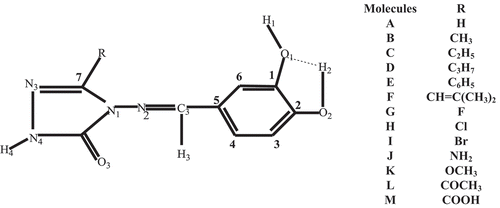
Figure 2. Structure of 1-acetyl-4-(p-chlorobenzylideneamino)-3-methyl-4, 5-dihydro-1H-1, 2, 4-triazol-5-one used for geometrical comparison.
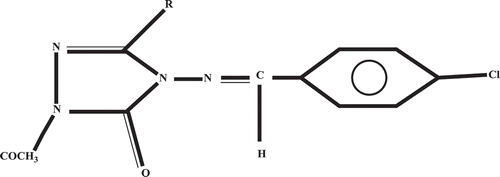
It can be seen from that the substitution effect is minor on the bond distances in various media. But a sensible variation of dihedral angle values can be observed in ESI ‡, –. For instance, the difference of N1-N2=C3-C5 value obtained for A and those of other molecules ranges from 0.6 to 6.4° in gas. This fact exhibits that the rotation about the N2=C3 axis leads to a slight staggering of these bonds. The results obtained (ESI ‡, ) revealed that the planarity of the triazole ring is slightly deformed due to the deviation of N1 ranging 3.6–7.5°. Whereas, the benzene ring in all the optimized structures are planar. This planarity of both rings showed that our optimized geometries is in good agreement with crystallographic data of other triazole derivatives.[Citation48] The N3-C7-N1-N2 values in the gas phase show that the atom N3 is out of the plane defined by C7, N1 and N2 (see , in which N3 is drawn in the plane). Therefore, the authors concluded that the whole molecule (A–N) do not lie in the same plane. It follows from ESI ‡, and that this fact remains the same in the solution phase. In the whole, the optimization processes were characterized by the formation of O2-H2…O1 hydrogen bond. Such an intramolecular hydrogen bond has been observed in many previous investigations dealing with compounds containing the phenolic moieties.[Citation18–Citation27] Recently, Yuzhen et al. demonstrated the important contribution of these intermolecular hydrogen bonds on the stability[Citation28] of the corresponding radicals through a comparison of energy difference of the optimized structures of radicals without hydrogen bond and those with hydrogen bond on the phenolic moiety. ESI ‡, exhibits different X-H bond distances (X = O1, O2, C3, and N3) in various media. Higher values are obtained for C3-H3 bonds because of captodative effect in which, the cause of a synergistic weakening of the C-H bond the carbon center is the simultaneous substitution by an electron-withdrawing substituent and an electron-donating substituent.[Citation49] ESI ‡, shows that the dipole moment of each molecule increases with the increase of the dielectric constant of the solvent. This conclusion corroborates earlier study[Citation50] that has shown the decreasing of the X-H bonds by the solvent’s polarity and the induced enhancement of the antioxidant activity of molecules. Fifen et al.[Citation23] have given the explanation that the electrostatic interactions between charged particles are all the weaker as the dipole moment of the molecular system increases. The plausible argument is the hypothesis of another electronic distribution resulting from the solvation of the molecule. These results are in the line with the observations of Clemens et al.[Citation51] on some organic chromophores. Accordingly, ESI ‡, shows that the vibrational frequencies of the X-H mode decreases with the increasing of dielectric constant of the solvent. For instance, for I (R = Br), v(O2-H2), and v(O1-H1) frequencies are 3768 and 3841 cm−1 in vacuum, respectively, compared to 3759 cm−1 and 3834 cm−1 in benzene. Such a trend shows that the identification in the infrared spectra by a red shift of the X-H stretching mode is probable. It is clear that this could be an identification of a good indication of the easier of the hydrogen bond.
Table 1. Relevant B3LYP/6-31G(d,p) geometrical parameters of BDHTD in various media.
Table 2. Relevant B3LYP/6-31G(d,p) geometrical parameters of BDHTD in various media.
Table 3. B3LYP/6-31G(d,p) reaction thermodynamic energies in kJ/mol in gas-phase: reaction enthalpies and reaction free energies.
Table 4. B3LYP/6-31G(d,p) reaction thermodynamic energies in kJ/mol in benzeneab: reaction enthalpies and reaction free energies.
Gas-phase reaction thermodynamic energies
Calculated B3LYP/6-31G (d,p) reaction thermodynamic energies in the gas phase are compiled in . Noticeably, the BDEs of molecules studied can be arranged in the following order: BDE1 < BDE2 < BDE4. It can be deduce that H1 is easier than the others H atoms. This is in good agreement with prior works on phenolic molecules.[Citation21,Citation23] The authors attributed it to the fact that H1 is a free hydrogen atom bond, whereas H2 is implicated in the O2H2…H1 intermolecular hydrogen. clearly shows that the substituent effect is more pronounced for BDE4 values because of nearness of H4 atom to substitution side. Nevertheless, the BDE values of H1 and H2 are not almost affected by various substitutions on 1,2,4-triazole-5-one moieties. This absence of influence of the activities of O1H1 and O2H2 groups is due to the fact that the catechol moiety is located further away from the substitution side. Therefore, for sake of simplicity, the authors focused the analysis of the influence of the substituent effect only on thermodynamic energies related to H4 atom. Our results are in accordance with the literature[Citation26,Citation52] which revealed that the introduction of electron-donating substituent or electron-withdrawing substituent on the position of hydrogen atom of A (R = H) leads respectively to a decreasing or an increasing of BDE4 values. An increasing of the BDE4 values while the number of carbon atom of the akyl substituent increases is noticeable. The minor augmentation of BDE4 values observed is due to the number of carbon atoms less than five. This fact is consistent with earlier researches[Citation53,Citation54] that demonstrated that the influence of the side-chain on the antioxidant is more pronounced when the number of carbon atoms of the akyl substituent is exceeding five. For that reason, the steric effect may be integrated in the analysis of the scavenging activity of compounds. The higher reduction of BDE4 values observed for E and F compared to those of molecules with alkyl substituent (B, C, and D) is due to the extension of conjugation system with the corresponding substituent (CH=C[CH3]2 or C6H5). These values are also in good accordance with preceding researches that showed that the HAT mechanism is mainly governed by the resonance effect.[Citation55] This effect is highly enlightened by 1,1-diphenyl-2-picryl-hydrazyl (DPPH) scavenging activity values in N, N-dimethylformamide (DMF) of 0.025 and 0.5 mg/mL for B and E, respectively, experimentally obtained by Yüksek et al.[Citation34] The slight BDE4 gap observed between E and F (2 kJ/mol) results from the superposition of resonance effect and the captodative effect[Citation49] related to the presence of C=C group (in the substituent CH=C[CH3]2). This presence of C=C group is shown to favourably stabilize the specific radical species to enhance the radical scavenging activities.[Citation28] The highest decreasing of BDE4 is obtained for J (R = NH2) is attributed to strong electron-donating nature of NH2. also shows that for halogen substituent, the decreasing of BDE3 values is almost constant. Independently of the substituent used, the BDFEi values (i = 1, 2, 3) obtained are positive. This enables the authors to conclude that the HAT mechanism is not spontaneous. revealed that BDFEs are lower than BDEs. BDFE values are lower by 23–38 kJ/mol. The assumption used by other researchers[Citation27,Citation43] that the absolute values of entropy term reach few tens kJ/mol is thus confirmed. Therefore, to analyze the HAT mechanism, free energy or enthalpy can be used as the criterion of the thermodynamically preferred process.
also shows that IP values are significantly greater than the corresponding BDE values. The HAT mechanism is thermodynamically preferred in gas than SET mechanism. To date, neither experimental nor theoretical study of IP values of BDHTDs have been published. The comparison of the IPs of BDHTD to those of phenolics[Citation21] (792.9, 762.9, and 742. 8 kJ/mol obtained, respectively, for gallic acid, caffeic acid, and ferulic acid at the B3LYP/6-311++G[d,p] level) indicates that the ET mechanism is more predominant in BDHTD compounds (investigated in this work). The authors also compared the IP values of BDHTD to that of the ascorbic acid[Citation28] (803.8 kJ/mol obtained B3LYP/6-311++G[2d,2p] /B3LYP/6-31G[d,p]) and concluded that despite the basis set effect, the ET mechanism is also dominant in BDHTD than in ascorbic acid. Our B3LYP results revealed that for halogen substituent (F, Cl and Br), the IP values are higher in comparison to A(R = H). Similar results have observed for electron withdrawing group (COCH3 and COOH), whereas IP values are lower for electron donating group (akyl group, C6H5, NH2, OCH3 and CH=C[CH3]2) in comparison to A(R = H). This trend is good agreement with the findings of Mezsam Najafi[Citation26] obtained during the theoretical analysis of the antioxidant activity of ortho- and meta-substituted daidzein derivatives in the gas phase. Our results (see ) reveal that the IP values are almost close to IPFE values because of the tendency of the entropic term TΔS to disappear. This fact is in better accordance with the results of Holtomo et al.[Citation27] on antioxidative potency of caffeic acid phenethyl ester-Fe2+ complexes. also shows that despite the fact that the IP values calculated from the Koopman theorem (IP = –EHOMO) are lower compared to those calculated using Eq. (7), the prediction of the ordering of the antioxidant activity for BDHTD using the HOMO eigenvalues is similar to that obtained using IP values calculated from the latter procedure. In the same vein, it can be observed from ESI ‡, (a) that J (R = NH2) provided the highest HOMO energy (–541 kJ/mol), followed by F (R = CH=C[CH3]2; –551 kJ/mol; ESI ‡, ). This clearly shows that J has the strongest electron-donating capacity among the molecules studied. Such an ordering of the antioxidant activity is reliable with that obtained from BDE4 values. This parallel has been evoked in prior researches that clearly illustrated the structure-activity relationships of antioxidants.[Citation23,Citation28,Citation56] The exploration of shows that IP values are significantly lower than PDE values in the gas phase. The authors then conclude that the SET mechanism is more dominant than the PT mechanism in this phase. In comparison to A (R = H), it is noticeable that a reduction of PDE values is obtained for halogen substituent and electron withdrawing group (COCH3 and COOH). But, an augmentation of PDEs is observed for electron donating group (akyl group, C6H5, NH2, and CH=C[CH3]2). This trend is contrary to that observed for IPs. ESI ‡, reveals that the differences between PDE values and those of PDFE are almost similar to those obtained in HAT mechanism. The IPFE and PDFE values obtained are positive pointing out that SET-PT mechanism is not spontaneous in the gas phase.
Figure 3. Thermodynamic energies of BDHTD (in kJ/mol) in gas phase for J molecule (R = NH2) in water and benzene: BDE1, IP, PDE1, PA1, and ETE1.
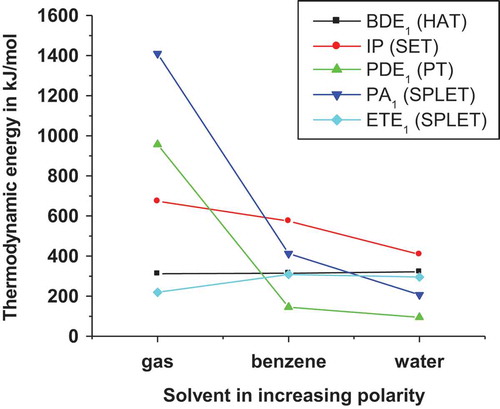
The halogen substituent causes decrease in PA values in comparison to A (R = H). Differences are in 0–28 kJ/mol. In the same vein, PA values are lower than values of A by 1–48 kJ/mol for L(COCH3) and M(COOH). Our B3LYP results also confirm that electron donating substituents increase PA values. It is worth noting that a charged molecule is more sensitive to the substituent effect than its neutral counterpart. In general, electron donating substituents stabilize the anion formed (RX−) but destabilize the parent structures, whereas electron donating groups have an opposite effect.[Citation26] demonstrates that the calculated IPs and PAs of the BDHTD molecules are significantly higher than BDEs illustrating that HAT mechanism represents the most probable process in the gas phase. Such a finding agrees with that reported for polyphenolic deoxybenzoins[Citation21] and natural PhA derivatives.[Citation28] In comparison to PA, ETEs of BDHTD molecules are significantly lower than PAs. The presence of electron withdrawing group in A (R = H) lead to higher ETEs (), while that of the electron donating substituents increases ETE values. This is in line in with the results obtained from previous study on p-phenylenediamine[Citation43] and substituted daidzein derivatives.[Citation26] also illustrated higher ETE4 values obtained in the presence of the electron donating substituents. The authors then concluded that the electron transfer process is more favorable on the 1, 2, 4-triazol-5-one ring in the presence of the electron donating substituents. But, reverse results are obtained in the presence of halogen and electron withdrawing substituents. In both cases, ESI ‡, Fig. 4 lucidly exhibits the ETE4 and ETFE4 are close due to the tendency of the entropic term TΔS to disappear as observed for IPFEs. From the positive free energy values obtained, one can conclude that the SPLET mechanism is not spontaneous. The comparison of IP and ETE for each substituent ( and ESI ‡, Fig. 4) demonstrates that the latter is greatly lower than the former. Consequently, the SET process from the anionic form is more preferable than that from neutral form in the gas phase.
Reaction thermodynamic energies in water and benzene
Reaction thermodynamic energies obtained in water and benzene are presented in and , respectively. On the whole, BDE4 values in the solution phase (benzene and water) are lower than that in the gas phase. This decrease is more pronounced in water by 2–13 kJ/mol. The plausible explanation of this observation is the formation of strong hydrogen bond OH2…O1 which is weakened more and more when the solvent polarity increases. In reality, the higher the solvent polarity, the easier the separation of charges.[Citation21,Citation23] Although deviations in individual values can be found, the trend found for both solvents are analogous. In contrast to BDE1-2 values are higher in water by 8–17 kJ/mol in comparison to their gas-phase homologs, exception made for M (R = COOH). BDFEs displayed in and which are lower than the BDEs, as observed in the gas phase. On average, reaction free energies are higher by 33.1 and 36.6 kJ/mol than corresponding BDEs of BDHTD molecules for water and benzene, respectively. The BDFEs are positive denoting that the HAT mechanism is not spontaneous in the solution phase.
Our calculations showed that solvent effects stronger influence upon IPs than BDEs. The IPs in the solution phase, are lower than those obtained in the gas phase. The drop in IP varies from 84 to 103 kJ/mol in benzene. In water, the IPs are dramatically lower by 240–269 kJ/mol in comparison to those obtained in the gas phase. The average difference in IP is 257.5 kJ/mol in water and 94.8 kJ/mol in benzene. The authors attributed this relative order of IP (water < benzene < gas) to the great sensitivity of cation radicals to the polarity of solvent used. shows that PDEs are significantly decreasing when moving from the gas phase to solution phase for J (R = NH2). This trend is identical for others molecules. The average differences between the solution phase and gas phase reach 857 and 817 kJ/mol for water and benzene, respectively. The significant drop related to the increase of solvent polarity is attributed to the high solvation enthalpies of proton.[Citation23,Citation43] This fact is in conformity with that reported polyphenolic deoxybenzoins by Yunsheng et al.[Citation21] Compared to BDEs, the corresponding PDE values turn out to be drastically low in the solution phase, whatever the molecule analyzed. – and displayed IP values that are significantly higher than BDEs. The authors therefore concluded that PT is thermodynamically favored mechanism compared to HAT whereas SET is least preferred mechanism in the solution phase. Overall, the IPFE values are positive illustrating that the SET mechanism is not also spontaneous in the solution phase. But, the PDFE values are negative indicating that the PT mechanism is spontaneous in both solvents, with the exception of PDFE4 values in water.
Proton affinities in the solution phase are significantly lower than corresponding gas-phase PAs. Differences are ranging: 1179–1225 kJ/mol (in water) and 988–1009 kJ/mol (benzene). The average shift is 1205 and 996 kJ/mol for water and benzene, respectively. These noteworthy drops are also related to the high solvation enthalpies of proton.[Citation43] – and showed that PAn values are higher than PDEn values (i = 1, 2, 4) in both solvents. This is in accordance with the fact that cations easily liberate proton than neutral systems. This results are consistent with previous studies on caffeic acid phenethyl ester,[Citation50] 3,4-dihydroxyphenylpyruvic acid,[Citation23] and other molecular system.[Citation56]
In the solution phase, electron transfer enthalpies are higher than corresponding gas-phase values. The differences between ETE in the gas phase and solution phase lie in the 60–157 and 5–112 kJ/mol range, respectively, for benzene and water. This shows that both solvents do not facilitate electron transfer from anionic compounds. Similarly to observation made in the gas phase, a comparison with the solution-phase IP values (– and ) revealed that the corresponding ETE values were greatly lower. Hence, the SET from the neutral form is less favored than that of the anionic form. Such a conclusion is in agreement with results obtained from prior studies.[Citation21,Citation57] In benzene, the PAFE values are significantly higher than the corresponding PA values by 167–183 kJ/mol, the average shift is approximately 180 kJ/mol. The differences between PA and PAFE values lie in 77–86 kJ/mol intervals in water. On the whole, the solution-phase PAFE and ETE values obtained are positive illustrating that the SPLET mechanism is not spontaneous in both solvents.
Conclusion
In this article, the authors have presented the quantum chemical calculations of the thermodynamic energies (reaction enthalpies and reaction free energies) of antioxidant mechanisms of 13 BDHTD at B3LYP/6-31G (d,p) level in the gas phase and solution phase. The optimization processes were characterized by the formation of an intramolecular hydrogen bond in the phenolic moieties that affects the stability of the corresponding radicals. The variation of BDEs depends on three factors: the nature of the substituent group, the influence of the side chain for alkyl substituents, and the extension of conjugation system with the corresponding substituent (CH=C[CH3]2 or C6H5). The HAT mechanism was thermodynamically preferred in gas than SET mechanism. The comparison of the IPs of BDHTD to those of classical antioxidants (gallic acid, caffeic acid, ferulic acid, and ascorbic acid) indicates that the ET mechanism is the most predominant in the 13 BDHTD compounds studied, despite the basis set effect. Thermodynamically, the SET mechanism is more dominant than the PT mechanism in the gas phase. Moreover, the comparison of IP and ETE for each substituent demonstrates that the latter is greatly lower than the former indicating that the SET process from the anionic form is more preferable than that from neutral form in the gas phase. The positive reaction free energies obtained for all the three mechanisms in gas showed that these mechanisms are not spontaneous in gas. Solvent effects stronger influence upon IPs than BDEs. The IPs in the solution phase, are lower than those obtained in the gas phase. The order of IP (water < benzene < gas) is related to the great sensitivity of cation radicals to the polarity of solvent used. Our results illustrated that PT is thermodynamically favored mechanism compared to HAT, whereas SET is least preferred mechanism in the solution phase. Globally, the solution PAFE and ETFE values obtained are positive illustrating that the SPLET mechanism is not spontaneous in both solvents.
Nomenclature
| BDE | = | bond dissociation energy |
| BDFE | = | bond dissociation free energy |
| BDHTD | = | benzylidenamino-4, 5-dihydro-1H-1,2,4-triazol-5-one derivatives |
| ETE | = | electron transfer enthalpy |
| ETFE | = | electron transfer free energy |
| HAT | = | hydrogen atom transfer |
| IEF-PCM | = | integral equation formalism polarized continuum model |
| IP | = | ionization potential |
| IP | = | ionization potential |
| PA | = | proton affinity |
| PAFE | = | proton affinity free energy |
| PDE | = | proton dissociation enthalpy |
| PDFE | = | proton dissociation free energy |
| PhA | = | phenolic acids |
| RCS | = | reactive chlorine species |
| RNS | = | reactive nitrogen species |
| ROS | = | reactive oxygen species |
| SET | = | single electron transfer |
| SET-PT | = | single electron transfer-proton transfer |
| SPLET | = | sequential proton loss electron transfer |
Electronic_supplementary_information.docx
Download MS Word (163.2 KB)Acknowledgments
The authors are grateful to Bénoît Champagne (Université Catholique de Louvain [UCL]; Belgium) and Olivier Holtomo (University of Bamenda; Cameroon).
Funding
This work was supported by the Ministry of Higher Education of Cameroon.
Additional information
Funding
Related Research Data
References
- Krystona, T.B.; Georgieva, A.B.; Pissis, P.; Georgakilasa, A.G. Role of Oxidative Stress and DNA Damage in Human Carcinogenesis. Mutation Research/Fundamental and Molecular Mechanism of Mutagenesis 2011, 711, 193–201.
- Jiranusornkul, S.; Laughton, C.A. Destabilization of DNA Duplexes by Oxidative Damage at Guanine: Implications for Lesion Recognition and Repair. Journal of the Royal Society Interface 2008, 5, 191–198.
- Konsta, A.A.; Visvardis, E.E.; Haveles, K.S.; Georgakilas, A.G.; Sideris. E.G. Detecting Radiation-Induced DNA Damage: From Changes in Dielectric Properties to Programmed Cell Death. Journal of Non-Crystalline Solids 2003, 305, 295–302.
- Spassky, A.; Angelov, D. Influence of the Local Helical Conformation on the Guanine Modifications Generated from One-Electron DNA Oxidation. Biochemistry 1997, 36, 6571–6576.
- Gillard, N.; Begusova, M.; Castaing, B.; Potheim-Maurizot, M. Radiation Effects Binding of Fgp Repair Protein to An Abasic Site Containing DNA. Radiation Research 2004, 162, 566–571.
- Boveris, A. Superoxide Radical and Hydrogen Peroxide in Mitochondria. Free Radicals in Biology; Pryor, W.A.; Ed.; Academic Press: New York, NY, 1982; 65–89.
- Misra, H.P.; Fridovich, I. The Role of Superoxide Anion in the Autooxidation of Epinephrine and a Simple Assay for Superoxide Dismutase. Journal of Biological Chemistry 1972, 247, 3170–3175.
- Levine, S.A.; Kidd, P.M. Antioxidant Adaptation: A Unified Disease Theory. Journal of Orthomolecular Psychiatry 1983, 14, 19–38.
- Cuzzocrea, S.; Riley, D.P.; Caputi, A.P.; Salvemini, D. Antioxidant Therapy: A New Pharmacological Approach in Shock, Inflammation, and Ischemia/Reperfusion Injury. The American Society for Pharmacology and Experimental 2001, 53, 135–159.
- Gilgun-Sherki, Y.; Rosenbaum, Z.; Melamed, E.; Offen, D. Antioxidant Therapy in Acute Central Nervous System Injury: Current State. Pharmacological Reviews 2002, 54, 271–284.
- Kirkham, P.; Rahman, I. Oxidative Stress in Asthma and COPD: Antioxidants As a Therapeutic Strategy. Pharmacology and Therapeutics 2006, 111, 476–494.
- Winkler, C.; Frick, B.; Schroecksnadelm, K.; Schennach, H.; Fuchs, D. Food Preservatives Sodium Sulfite and Sorbic Acid Suppress Mitogen-Stimulated Peripheral Blood Mononuclear Cells. Journal of Food and Chemical Toxicology 2006, 44, 2003–2007.
- Lupo, M.P. Antioxidants and Vitamins in Cosmetics. Journal of Clinical Dermatatology 2001, 19, 467–473.
- Merken, H.M.; Beecher, G.R. Measurement of Food Flavonoids by High-Performance Liquid Chromatography. Journal of Agricultural and Food Chemistry 2000, 48, 578–599.
- Kawaii, S.; Tomono, Y.; Katase, E.; Ogawa, K.; Yano, M. Antiproliferative Effects of Readily Extractable Fraction Prepared from Citrius Juices on Several Cancer Cell Lines. Journal of Agricultural and Food Chemistry 1999, 47, 2509–2512.
- Manthey, J.A.; Guthrie, N. Antiproliferative Activities of Citrius Flavonoids Against Six Cancer Cell Lines. Journal of Agricultural and Food Chemistry 2002, 50, 5837–5843.
- Kuhnau, J. The Flavonoids. A Class of Semi-Essential Food Components: Their Role in Human Nutrition. World Review of Nutrition and Dietetics 1976, 24, 117–191.
- Manach, C.; Scalbert, A.; Morand, C.; Remesy, C.; Jimenez, L. Polyphenols: Food Sources and Bioavailability. The American Journal of Clinical Nutrition 2004, 79, 727–747.
- Manach, C.; Williamson, G.; Morand, C.; Scalbert, A.; Remesy, C. Bioavailability and Bioefficacy of Polyphenols in Humans. American Journal of Clinical Nutrition 2005, 81, S230–S242.
- Wright, J.S.; Johnson, E.R.; DiLabio, G.A. Predicting the Antioxidant Activity of Phenolic Antioxidant: Theoretical Method, Analysis of Substituent Effects, and Application to Major Families of Antioxidants. Journal of the American Chemical Society 2001, 123, 1173–1183.
- Yunsheng, X.; Youguang, Z.; Lin, A.; Yunyan, D.;Yi, L. Density Functional Theory of the Structure-Antioxidant Activity of Polyphenolic Deoxybenzoins. Journal of Food Chemistry 2014, 151, 198–206.
- Javan, A.J.; Javan, M.J.; Tehrani, A.A. Theoretical Investigation on Antioxidant Activity of Bromophenol from the Marine Red Alga Rhodomela Confervoides: H-Atom Versus Electron Tranfer Mechanism. Journal of Agricultural and Food Chemistry 2013, 61, 1534–1541.
- Fifen, J.J.; Nsangou, M.; Dhaouadi, N.; Motapon, O.; Jaidane, N. Solvent Effects on the Antioxidant Activity of 3,4-Dihydroxyphenylpyruvic Acid: DFT and TD-DFT Studies. Journal of Computional Chemistry 2011, 966, 232–243.
- Leopoldini, M.; Russo, N.; Toscano, M. Iron Chelation by the Powerful Antioxidant Flavonoid Quercetin. Journal of Agricultural and Food Chemistry 2006, 54, 3078–3085.
- Leopoldini, M.; Russo, N.; Toscano, M. The Molecular Basis of Working Mechanism of Natural Polyphenolic Antioxidants. Food Chemistry 2011, 125, 288–306.
- Mezsam, N. On the Antioxidant Activity of the Ortho and Meta Substituted Daidzein Derivatives in the Gas Phase and Solvent Environment. Journal of the Mexican Chemical Society 2014, 58, 36–45.
- Holtomo, O.; Nsangou, M.; Fifen, J.J.; Motapon, O. Antioxidative Potency and UV-Vis Spectra of the Compounds Resulting from the Chelation Fe2+ by Caffeic Phenethyl Ester and Two of Its Derivatives. Computional and Theoretical Chemistry 2015, 1067, 135–147.
- Yuzhen, C.; Huizhu, X.; Jie, Z.; Guizhao, L. Structure-Thermodynamics-Antioxidant Activity Relationship of Selected Natural Phenolic Acids and Derivatives: An Experimental and Theoretical Evaluation. Plos One 2015, 10, 012276–012295.
- Demirbaş, N.; Uğurluoğlu, R. Synthesis and Antitumor Activities of Some 4-(1-Naphthylidenamino)-and 4-(1-Naphthylmethylamino)-1,2,4-Triazol-5-One Derivatives. Turkish Journal of Chemistry 2004, 28, 679–690.
- Ikizler, A.A.; Uçar, F.; Yüksek, H.; Aytin, A.; Yasa, I.; Gezer, T. Synthesis and Antifungal Activity of Some New Arylidenamino Compounds. Acta Poloniae Pharmaceutica Drug Research 1997, 54, 135–140.
- Bhat, A.R.; Bhat, G.V.; Shenoy, G.G. Synthesis and in-Vitro Antimicrobial Activity of New 1,2,4-Triazoles. Journal of Pharmacy and Pharmacology 2001, 53, 267–272.
- Yüksek, H.; Küçük, M.; Alkan, M.; Bahçeci, Ş.; Kolaylı, S.; Ocak, Z.; Ocak, U.; Şahinbaş, E.; Ocak, M. Synthesis and Antioxidant Activities of Some New 4-(4-Hydroxybenzylidenamino)-4,5- Dihydro-1H-1,2,4-Triazol-5-One Derivatives with Their Acidic Properties. Asian Journal of Chemistry 2006, 18, 539–550.
- Yüksek, H.; Kolaylı, S.; Küçük, M.; Yüksek, M.O.; Ocak, U.; Şahinbaş, E.; Sivrikaya, E.; Ocak, M. Synthesis and Antioxidant Activities of Some 4-Benzylidenamino-4,5-Dihydro-1H-1,2,4-Triazol-5-One Derivatives. Indian Journal of Chemistry 2006, 45B, 715–718.
- Yüksek, H.; Demibaş, A.; Ikizler, A.; Johansson, C.B.; Çelik, C.; Ikizler, A.A. Synthesis and Antibacterial Activities of Some 4,5-Dihydro-1H-1,2,4-Triazol-5-Ones. Arzneimittel Forschung–Drug Research 1997, 47, 405–409.
- Ünver, Y.; Meydanal, S.; Sancak, K.; Ünlüer, D.; Ustabas, R.; Dügdü, E. Synthesis, Cristal Structure and Antioxidant Properties of Novel 1,2,4-Triazol-5-Ones Containing 3,4-Dimethoxyphenyl and 3,4 Dihydroxyphenyl Moiety. Turkish Journal of Chemistry 2011, 35, 265–277.
- Bikélé, M.D.; Zobo, J.M.; Lissouck, D.; Younang, E.; Nono, J.H.; Mbaze, L.M. DFT Investigation of Chelation of Divalent Cations by Some 4-Benzylidenamino-4,5-Dihydro-1h-1,2,4-Triazol-5-One Derivatives. Journal of Chemistry and Chemical Engineering 2015, 9, 1–14.
- Stepanć, V.; Trošelj, K.G.; Lučić, B.; Markoviç, Z.; Amić, D. Bond Dissociation Energy As General for Flavonoid Radical Scavenging Activity. Food Chemistry 2013, 141, 1562–1570.
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; et al. Gaussian 09 2009: Revision A.2. Gaussian, Inc.: Walllingford, CT.
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. Journal of Chemical Physics 1993, 98, 5648–5652.
- Binley, J.S.; Pople, J.A.; Hehre, W.J. Self Consistent Molecular Orbital Method. 21. Small Split-Valence Basis Sets for First-Row Elements. Journal of the American Chemical Society 1980, 102, 939–947.
- Cances, E.; Mennucci, B.; Tomasi, J. A New Integral Equation for Polarizable Model: Theoretical Background and Applications to Isotropic and Anisotropic Dielectrics. Journal of Chemical Physic 1997, 107, 3032–3041.
- Bartmess, J.E. Thermodynamic of the Electron and the Proton. Journal of Physical Chemistry 1994, 98, 6420–6424.
- Rimarčik, J.; Lukeš, V.; Klein, E.; Ilçin, M. Study of the Solvent Effect on the Enthalpies of Hemolytic and Heterolytic N-H Bond Cleavage in P-Phenyllenediamine and Tetracyano-P-Phenylenediamine. Journal of Molecular Structure (Theochem) 2010, 952, 25–30.
- Hwang, S.; Chung, D.S. Calculation of the Solvation Free Energy of the Proton in Methanol. Bulletin of Korean Chemistry Society 2005, 26, 589–593.
- Tissandier, M.D.; Cowen, K.A.; Feng, W.Y.; Gundlach, E.; Cohen, M.H.; Earhart, A.D.; Coe, J.V.; Tuttle, T.R.Jr. The Proton’s Absolute Aqueous Enthalpy and Gibbs Free Energy of Solvation from Cluster-Ion Solvation Data. The Journal of the Physical Chemistry. A 1998, 102(40), 7787–7794.
- Jortener, J.; Noyes, R.M. Some Thermodynamic Properties of the Hydrated Electron. Journal of Physical Chemistry 1966, 70(3), 770–774.
- Fifen, J.J.; Dhaouadi, Z.; Nsangou, M.; Holtomo, O.; Jaidane, N. Proton-Coupled Electron Transfer in the Reaction of 3,4-Dihydroxyphenylpyruvic Acid with Reactive Species in Various Media. International Journal of Chemical Physics 2015 2015, 1–3.
- Çoruh, U.; Kahveci, B.; Şaşmaz, S.; Ağar, E.; Kim, Y. 1-Acettyl-4-(P-Chlorobenzylideneamino)-3-Methyl-4,5-Dihydro-1H-1,2,4 Triazol-5-One. Acta Crystallography Sect E 2003, 59, 0530–0532.
- Wright, J.S.; Shadnia, H.; Chepelev, L.L. Stability of Carbon-Centered Radicals: Effect of Functional Group on the Energetic of Addition of Molecular Oxygen. Journal of Computational Chemistry 2008, 30, 1016–1026.
- Holtomo, O.; Nsangou, M.; Fifen, J.J.; Motapon, O. DFT Study of the Solvent Effects ON The Structure, UV-Vis Spectra and the Antioxidant Activity of Caffeic Acid Phenethyl Ester and Some of Its Derivatives. Journal of Chemistry and Chemical Engineering 2013, 7, 910–923.
- Clemens, O.; Basters, M.; Wild, M.; Wilbrand, S.; Reichert, C.; Bauer, M.; Springborg, M.; Jung, G. Solvent Effects on the Absorption/Emission Spectra of An Organic Chromophore. Journal of Molecular Structure (Theochem) 2008, 866, 15–20.
- Jin, R. A DFT Study of the Radical Scavenging Activity of Juglone and Its Derivatives. Journal of Molecular Structure (Theochem) 2010, 939, 9–13.
- Lu, Z.; Nie, G.; Belton, P.S.; Tang, H.; Zhao, B. Structure-Activity Relationship Analysis of Antioxidant Ability and Neuroprotective Effect of Gallic Acid Derivatives. Neurochemistry International 2006, 48, 263–274.
- Kagan, V.E.; Elena, A.; Serbinova, L.P.; Recycling and Antioxidant Activity of Tocopherol Homologs of Differing Hydrocarbon Chain Lengths in Liver Microsomes. Archives of Biochemistry and Biophysic Biochemistry 1990, 282, 221–225.
- Zang, H.-Y.; Structure-Activity Relationships and Rational Design Strategies for Radical-Scavenging Antioxidants. Current Computer-Aided Drug Design 2005, 1, 257–273.
- Lengyel, J.; Rimarčik, J.; Vagánek, A.; Klein, E. On the Radical Scavenging Activity of Isoflavones: Thermodynamics of O-H Bond Cleavage. Physical Chemistry. Chemical Physic 2013, 15, 10895–10903.
- Xue, Y.S.; Zheng, Y.G.; Zhang, L.; Wu, W.Y.; Yu, D.; Liu, Y. Theoretical Study on the Antioxidant Properties of 2-Hydroxychalcone: H-Atom Vs. Electron Transfer Mechanism. Journal of Molecular Modeling 2013, 19, 3851–3862.