
ABSTRACT
This article provides a systematic study of the impact of different thermal treatments (62 ± 2°C, without and with relative humidity control, 79%) on soy protein in defatted soy flour and their aqueous dispersions. The effect of dispersing treatments (magnetic stirring, high-speed, and high-pressure homogenization) on dispersions also was assessed. Changes in protein solubility (water and 0.2 g/100 g potassium hydroxide solution), apparent-reactive lysine content, urease and trypsin inhibitor activities, protein denaturation, and Fourier transform infrared spectra were studied. Glycosylation, aggregation, and denaturation of storage and biologically active soy proteins were observed in different degrees, being mainly promoted by the control of relative humidity and the dispersibility of the sample.
Introduction
Soybeans are an abundant and relatively inexpensive source of proteins that are widely recognized for their high nutritional value and excellent functional properties.[Citation1,Citation2] Virtually all soybeans are solvent extracted using hexane to remove the oil. At present, only defatted soybean flakes (DSF) are converted into edible-grade products. Hexane is removed in a vapor desolventizer-deodorizer or flash desolventizer to permit preparation of flakes ranging from raw to fully cooked. Defatted soy flours and grits are made grinding and sieving flakes and they contain ca 56 g/100 g protein and 35 g/100 g total carbohydrates (mainly oligosaccharides and polysaccharides) on a moisture-free basis.[Citation3,Citation4] Defatted soy flakes flour,without moist thermal treatment, has a protein-dispersibility index of ca 90, without inactivation of main anti-nutritional factors, the Kunitz trypsin inhibitor (KTI), and lectin. Moreover, storage soy proteins (glycinin, 11S and β-conglycinin, 7S) also have a low degree of denaturation.[Citation3]
On the other hand, glycosylation has been proposed as an interesting way for improving functional properties of food proteins. Due to proceeding under mild conditions and requiring no strange chemical, this reaction is more desirable than other types of chemical modification for food proteins, and possess a promising application in the food industry.[Citation5] Glycosylation extent is commonly affected by several factors such as temperature, pH, heating time, relative humidity (RH), intrinsic properties of protein and sugar, and amino group-reducing sugar ratio.[Citation6] The conjugation of glycan chains to proteins can affect their thermal aggregation properties.[Citation7] Glycosylation modifies the surface hydrophobicity, net charge, and heat stability, all of which can affect the aggregation process.[Citation5,Citation8] The studies concerned with glycosylation of soy proteins focused on the study of the structure of glycoconjugates and their improved functional properties, including gelation and emulsification. Soy protein isolates and carbohydrates, such as glucose, fructose, and some polysaccharides, such as dextran, were used as starting materials.[Citation8–Citation11]
This article provides a systematic study concerned with the influence of different heating (with and without RH control) and dispersing treatments (magnetic stirring [MS], high-speed homogenization [HSH], and high-pressure homogenization [HPH]) on glycosylation, denaturation, and aggregation of soy proteins in defatted soy flakes flour (DSFF) aqueous dispersions. For comparative purposes, thermal treatments were also carried out on DSFF solid samples. Unlike previous articles, the reaction between soy proteins and carbohydrates naturally present in soy was promoted. The impact of mentioned treatments on the protein solubility in water, the protein solubility in 0.2g/100 g potassium hydroxide solution, and the apparent-reactive lysine (RL) content was assessed. Protein denaturation degree was evaluated by modulated differential scanning calorimetry (MDSC) and structural changes were studied by Fourier transform infrared spectroscopy (FTIR). It is worth noting that the relationship between structure and functional properties of modified proteins, which would require the isolation and characterization of glycoconjugates, was not assessed in this work. Therefore, this article should be considered as basis for further researches.
Materials and methods
Materials
Active DSF, without thermal inactivation of anti-nutritional factors, were provided by Bunge Argentina S.A. (Puerto General San Martín, Santa Fe, Argentina). According to the provider, the solvent of these samples (n-hexane) was evaporated at a low temperature. O-phthaldialdehyde (OPA), dimethyl sulfoxide (DMSO), L-lysine hydrochloride, β-mercaptoethanol (β-ME), sodium dodecyl sulfate (SDS), benzoyl-DL-arginine-p-nitroanilide (BAPA) and crystalline porcine trypsin (Type IX) were purchased from Sigma Co. (MO, USA). Other laboratory grade reactants were purchased from Anedra (Research AG; Buenos Aires, Argentina). The DSF sample was ground in an all-purpose high-speed smashing machine (Chincan FW Model; China). The resultant DSFF was sieved at 500 μm (ASTM E-11-81, Zonytest; Buenos Aires, Argentina) and further stored in sealed flasks at room temperature.
Preparation of aqueous dispersions of DSFF
DSFF was dispersed in distilled water (10.0 g total solids/100 g) by MS during 3 h to ensure a complete dispersion and hydration. Part of the initial dispersion was homogenized by HSH using a rotor/stator dispositive (Ultraturrax T-25, S25N-18G dispersing tool, IKA-Labortechnik; Staufen, Germany) at 20,000 rpm for 1 min. The other part, besides HSH treatment, was treated by HPH (Panda 2K high-pressure valve homogenizer, GEA Niro Soavi; Parma, Italy). Aqueous dispersions were passed four times through homogenization device at 1000 bar. During the HPH treatment the container was cooled using a water–ice bath to avoid the excessive temperature increase.
Thermal treatments
In this work, different thermal treatments with and without RH control and different times and temperatures, were carried out on DSFF aqueous dispersions and DSFF solid samples. For all heating assays, a forced draft oven (Memmert; Schwabach, Germany) at 62 ± 2°C was used. To perform the assays at controlled RH, the corresponding samples were putted in desiccators containing a saturated KBr solution (RH = 79%).[Citation8] Sample acronyms are given according to dispersing and heating conditions. All samples are summarized in . When it was necessary, the samples obtained after heating and/or dispersing treatments were grinded and sieved before characterization assays. Moreover, additional tests were carried out to determine the loss or gain of moisture (g/100 g) during heating of the samples, by means of the determination of sample weight as a function of heating time.
Table 1. Sample nomenclature for heated samples prepared from defatted soy flakes flour (DSFF).
Characterization assays
Determination of chemical composition of DSFF
The proximate composition of DSFF sample was determined. Crude protein was determined by micro-Kjeldahl method (N×6.25);[Citation12] total solids was determined by heating the sample (103 ± 2ºC) for 3 h to constant weight; ash was determined by dry ashing at 550 ± 10ºC; total fat and dietary fiber was measured by using the methods AOAC-2011.25 and AOCS-Am 5-04, respectively.[Citation13,Citation14]
Determination of dispersible material (DM) and particle size
To determine DM, DSFF aqueous dispersions (10.0 g total solids/100 g in distilled water) were centrifuged at 800 × g (Rolco CM 4080 Millenium low-speed centrifuge; Buenos Aires, Argentina) at 20°C for 15 min. Then, the supernatant was separated. The amount of DM, expressed as grams of dispersible solids per 100 g total solids, was calculated as follows:
TSs is the total solid content for the supernatants and TSd is the same parameter determined on non-centrifuged aqueous dispersions. To determine TSs and TSd parameters, the samples were dried to constant weight (103 ± 2ºC, Memmert force draft oven; Schwabach, Germany).
The particle size distribution for DSFF aqueous dispersions was determined as differential volume (range 0.1–1000 μm) by laser diffraction, using a Malvern Mastersizer 2000E analyzer (Malvern Instruments; Worcestershire, UK), associated with a wet dispersion unit (Hydro 2000MU, Malvern Instruments; Worcestershire, UK). Pump speed was set at 2000 rpm. Before measurements, dispersions were carefully mixed by turning the containers upside down to get a droplet size for the whole sample. The optical parameters were the following: relative refractive index, 1.14; adsorption coefficient, 0.1. From particle size distributions, De Brouckere moment mean values (D4,3) were obtained.
Determination of protein solubility in distilled water and KOH solution
Sample aqueous dispersions (0.1 g total solids/100 g) were prepared by dispersing solid samples in distilled water or 0.2 g/100 g (~ 36 mmol/L) potassium hydroxide solution.[Citation15] For all samples, isoionic pH values ranged from 6.57 to 6.95. Later, they were centrifuged at 10,500 × g (20 min, 20°C, Beckman Coulter GS-15R centrifuge, Beckman Coulter Inc., USA) and the supernatants were reserved for soluble protein determination (N×6.25) [Citation12]. Protein solubility, determined in water and potassium hydroxide solution (SW and SKOH, respectively), was expressed as g of soluble protein per 100 g total protein. These parameters were calculated by dividing the protein content of the water or alkali extracted solution by the protein content of the original DSFF sample. Moreover, residual solubility values (r-SW and r-SKOH) were calculated as follows:
SW, C and SKOH, C were the protein solubility values for control DSFF sample, without thermal treatment; SW, S and SKOH, S were the protein solubility values for heated and/or dispersed samples.
Determination of apparent-RL
The amount of apparent-RL was determined by the spectrophotometric assay based on the OPA and β-ME reaction with primary amines in alkaline media.[Citation16,Citation17] Thirty milligrams of the sample was dispersed in 20 g of 1.0 g/100 mL SDS solution; then, the dispersion was heated at 100ºC for 30 min, cooled in a water–ice bath to room temperature and finally centrifuged at 800 × g for 15 min (Rolco CM4080 Milenium centrifuge; Buenos Aires, Argentina). The lipid supernatant (350 μL) of was mixed by inversion with 3 mL OPA reagent (8.0 mmol/L OPA, 0.2 mL/100 mL β-ME, 50.0 mmol/L sodium tetraborate, 1.0 g/100 mL SDS); the absorbance at 340 nm was determined after 2 min incubation at room temperature.[Citation16] The corresponding calibration procedure was performed by using appropriate dilutions of L-lysine hydrochloride (8.0 mmol/L stock solution) in 1.0 g/100 mL SDS solution. The samples’ nitrogen content determination was carried out according to the Micro Kjeldahl method.[Citation12] RL was expressed in g/16 g N. The OPA reaction is specific for free amino groups (ε-amino group of the lysine and α-terminal amino group).[Citation17] The α-terminal amino groups of soy proteins were not blocked before determination. Hence, for all samples, the RL value is an apparent parameter. The residual apparent-RL content, expressed as percentage respect to control (r-RL), was calculated as follows:
RLC and RLS are the apparent-RL contents (g/16 g N) for the control, without thermal treatment, and heated and/or dispersed samples, respectively.
Determination of trypsin inhibitor activity (TIA)
The effect of thermal treatments on TIA was quantified by assaying anti-tryptic activity according to the method of Liu and Markakis with some experimental modifications.[Citation18] The sample (0.5 g) was extracted with 50 mL of distilled water for 30 min under MS. Ten milliliters of the sample dispersion was mixed by adding an equal volume of 50.0 mmol/L Tris/HCl buffer (pH 8.2, 10.0 mmol/L CaCl2). The resultant mixture was filtered through a Whatman No. 2 paper. The filtrate was diluted with water to the point where 1 mL gave 30–70% trypsin inhibition.
To assay TIA, 2.0 mL of BAPA (0.92 mmol/L in DMSO) was mixed with 1.0 mL of diluted filtrate and 0.5 mL of trypsin solution (50 μg/mL in 1 mmol/l HCl solution containing 2.5 mmol/L CaCl2) at 37ºC. After 20 min of incubation, the reaction was stopped by adding 30 g/100 g acetic acid solution. The absorbance at 410 nm for the sample (As410) was a measure of the trypsin activity in the presence of sample inhibitors. The reaction was also run in the absence of inhibitors by replacing the diluted filtrate with 1 mL of distilled water; the corresponding absorbance was Ar410. Defining a trypsin unit as an A410 increase of 0.01 under the conditions of the assay, the TIA was expressed in trypsin units inhibited per milligram of dry sample (TUI/mg) and calculated as follows:
VF is the volume of diluted filtrate (mL) and CF is its concentration (mg dry sample/mL). Moreover, residual trypsin inhibitor activity (r-TIA), expressed as percentage respect to DSFF control sample, was defined as:
TIAC is the TIA value for control DSFF sample, without any heating, and TIAS is the TIA value for heated and/or dispersed samples.
Determination of urease activity (UA)
UA was determined according to the AOCS-Ba-9-58 official procedure.[Citation14] Each sample (0.2 g of each) was incubated in 10.0 mL of potassium phosphate buffered urea solution (pH 7.0) at 30°C for 30 min. The same procedure was performed in potassium buffered solution (pH 7.0) in the absence of urea. pH values for the sample dispersions without and with substrate were measured after incubation time. UA was defined as the increase in pH units (∆pH) from pH 7.00 under the conditions of the assay. Residual urease activity (r–UA), expressed as percentage respect to control DSFF sample, was calculated as follows:
UAC is the UA value for DSFF control sample, without any heating, and UAS is the UA value for heated and/or dispersed samples.
Determination of relative residual parameters
To evaluate the influence of dispersing methods on protein aggregation, apparent-RL content, TIA and UA of DSFF aqueous dispersions, the relative values of each residual parameter (r-SW, r-SKOH, r-RL, r-TIA, or r-UA) were obtained as:
r-P is the residual parameter for samples dispersed only by MS or by MS + HSH + HPH and r-Pref is the residual parameter obtained from samples dispersed by MS + HSH dispersing treatments.
FTIR spectroscopy
To obtain FTIR spectra, solid samples were previously dispersed in distilled water and (1.0 g total solids/100g) and then homogenized (Ultraturrax T-25, S25N-18G dispersing tool, IKA-Labortechnik; Staufen, Germany) at 20,000 rpm for 1 min. Then, 100 μL of dispersion was putted in the ZnSe ATR device covering the entire crystal and then was gently dried with warm air until it reached total dehydration. FTIR spectra were obtained with a Shimadzu model IR Affinity-1 device (Shimadzu Corporation; Kyoto, Japan). Measurement conditions were: Happ-Genzel Appodization; number of scans: 25, Resolution: 4 cm−1; wavenumber range: 650 to 4000 cm−1.
MDSC
MDSC assays were performed with a Q200 calorimeter (TA Instruments, Waters, L.L.C, New Castle, DE, USA) using concentrated aqueous dispersions (30.0 g/100 g in 1.0 mol/L NaCl).[Citation4,Citation19] Standard hermetic caps with 10–15 mg of each dispersion were heated at 5°C·min−1 from 20 to 120°C with a modulation of ±0.50°C every 50 s. An empty cap was used as a reference. Partial enthalpy of each transition and total enthalpy of each thermogram was calculated. Taking into account the crude protein (N×6.25) content of each sample, denaturation enthalpies (ΔHd) were expressed in J/g of dry protein. Moreover, for each transition, relative denaturation enthalpy values, respect to that of control DSFF sample (rΔHd), were also obtained.
Statistical analysis
All the characterization assays were performed at least in duplicate and the results were expressed as mean ± standard deviation. The statistical analysis was performed by analysis of variance (ANOVA) and test of least significant differences (LSD) using the statistical program Statgraphics Plus V5.1 (Statgraphics Corporation; USA, 2000). Significance was considered at p < 0.05.
Results and discussion
Characterization of DSFF control sample
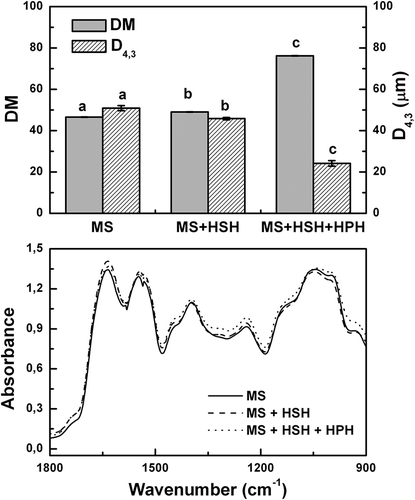
First, the characterization of DSFF sample without any thermal treatment was carried out. The proximate composition (g/100 g in dry basis) of control sample (92.0 ± 0.1 g total solids/100 g) was: crude protein (N × 6.25), 55.1 ± 0.4; ash, 7.7 ± 0.1; lipids 1.9 ± 0.1; insoluble dietary fiber (IDF), 20.6 ± 0.6; soluble dietary fiber precipitated with 78 mL/100 mL ethanol solution (SDF), 3.3 ± 0.2 and soluble dietary fiber not precipitated with 78 mL/100 mL ethanol solution, 10.0 ± 0.2. This latter fraction contains sucrose, oligosaccharides (mainly stachyose and raffinose) and a lesser extent, monosaccharides, in concordance with a previous article.[Citation20] For DSFF control sample, water solubility (SW) at isoionic pH (6.94 ± 0.01) was 49.7 ± 0.6 g/100 g total protein; this value was undoubtedly attributed to protein aggregates generated during oil extraction of flakes with non-polar organic solvent (n-hexane). Nevertheless, these protein aggregates are easily dissociable in alkaline medium according to almost ideal value for protein solubility 0.2g/100 g potassium hydroxide solution (SKOH = 99.8 ± 0.1 g/100 g total protein). This strong alkaline solubilization condition is employed as control of thermal overprocessing in defatted soy flours for animal feed. In this context, the protein aggregation mediated by covalent interactions (Maillard type reactions, thiol-disulfide interchange) induces a loss of SKOH.[Citation16] Then, the total protein solubilization of DSFF sample in this condition reflects that protein aggregates was mainly mediated by non-covalent interactions. The control sample exhibited high values of TIA (TIA = 559.5 ± 12.6 TUI/mg dry sample) and UA (UA = 2.00 ± 0.01 pH units). The apparent-RL content was 6.56 ± 0.11 g/16 g N; this value was similar to typical values for active defatted soy flours.[Citation4,Citation21] Moreover, the effect of various dispersing treatments on dispersing material (DM) and particle size (D4,3 values) is shown in . The additional treatment by HSH and HPH noticeably increased the DM values (p < 0.05). At the same time, D4,3 values were significantly decreased by application of HSH and HPH treatments (p < 0.05). The variation of mentioned parameters was higher when high-energy homogenization devices were used, in agreement with previous works.[Citation22] In addition, FTIR spectra showed the characteristic bands of amide I (AI, C-O stretching, ~1636 cm−1), amide II (AII, N-H deformation, ~1537 cm−1) and amide III (AIII, mainly C-N stretching and N-H deformation, ~1396 cm−1) in the protein region and the bands corresponding to carbohydrate region (900–1200 cm−1).[Citation4] Interestingly, the FTIR spectra for all unheated aqueous dispersions were fairly similar. This fact shows that the intensity of spectral bands was not substantially modified by the variation of DM and D4,3 values as a consequence of the application of various dispersing treatments ().
Figure 1. a) Dispersible material (DM, expressed as g of dispersible solids per 100 g total solids), De Brouckere mean diameter (D4,3) values and b) FTIR spectra for or unheated DSFF aqueous dispersions prepared using various dispersing treatments. MS: magnetic stirring, HSH: high-speed homogenization and HPH: high-pressure homogenization. Mean values with different lowercase letters indicate significant differences between aqueous dispersions treated with different dispersing treatments (p<0.05).
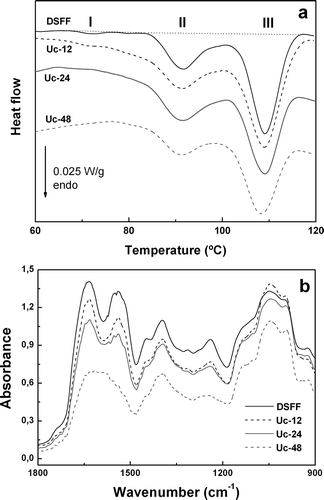
On the other hand, DSFF sample evidenced a typical DSC thermogram for active defatted soy flours (). In the presence of 1.0 mol/l NaCl, three endothermic transitions at Tp = 71–72ºC (peak I), 91–92ºC (peak II), and 108–109ºC (peak III) with denaturation enthalpy values (ΔHd) of 0.11, 2.54, and 4.77 J/g, respectively, were evidenced; total ΔHd for control sample was 7.31 J/g. According to Sorgentini and Wagner,[Citation19] the thermal stability of whey soy proteins (KTI, and lectin) was not significantly affected by ionic strength. Conversely, at 1.0 mol/L NaCl, the endothermic transitions of β-conglycinin (7S fraction) and glycinin (11S fraction) were shifted toward higher temperatures (~20°C). Thus, the peaks I, II, and III would correspond to KTI, 7S/lectin, and 11S, respectively. Moreover, the thermal denaturation of urease and other minor whey soy proteins would contribute to peak I endotherm.[Citation4] The presence of endothermic transition at 71–72ºC was consistent with the high TIA and UA values, as was reported previously.
Figure 2. a) DSC thermograms (30.0 g sample/100 g in 1.0 mol/L NaCl) for defatted soy flakes flour (DSFF control sample, without any previous heating) and heated Uc-12, Uc-24 and Uc-48 samples (62 ± 2 °C, HR = 79% for 12, 24 and 48 h, respectively); b) FTIR spectra for DSFF, Uc-12, Uc-24 and Uc-48 samples. Sample nomenclature was defined in . : a) DSC thermograms (30.0 g sample/100 g in 1.0 mol/L NaCl) for defatted soy flakes flour (DSFF control sample, without any previous heating) and heated Uc-12, Uc-24 and Uc-48 samples (62 ± 2 °C, HR = 79% for 12, 24 and 48 h, respectively); b) FTIR spectra for DSFF, Uc-12, Uc-24 and Uc-48 samples. Sample nomenclature was defined in .
Thermal and dispersing treatments on DSFF aqueous dispersions
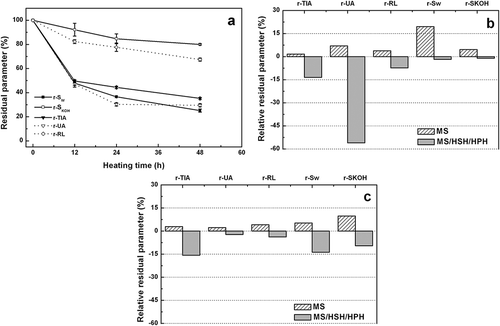
The combined effect of thermal and dispersing treatments on glycosylation, aggregation, and denaturation degree of soy proteins in DSFF aqueous dispersions (10.0 g total solids/100 g) was assessed. The influence of heating time (12–48 h, 62 ± 2ºC, RH = 79%) on different residual parameters for aqueous dispersions (Uc-12, Uc-24, and Uc-48 samples) is shown in . During thermal treatment, an important loss of SW was observed. After 12 h of heating, the r-SW value was higher than 50%, which was further decreased at longer times. Conversely, a gradual and slight loss of SKOH was evidenced throughout the entire heating time period. This fact would be consistent with protein aggregation processes mediated by covalent interactions.[Citation4] The RL content was gradually decreased with increasing heating time; after 48 h of thermal treatment, residual apparent-reactive lysine (r-RL) value was <70%. Although thermal treatments caused a loss of SKOH and SW, the decrease of r-RL values was not necessarily attributed to a lesser amount of soluble protein, due to each sample being totally solubilized by boiling in 1.0 g/100 mL SDS solution during RL determination. Hence, the decrease of r-RL values would be associated to the blockage of ε-amino groups of lysine and, a lesser extent, to the blockage of α-terminal amino groups. A high reaction temperature and prolonged heating times greatly contribute to extent to glycosylation. In fact, glycosylation takes place in both wet and dry conditions.[Citation5] At the beginning of thermal treatment, the protein and carbohydrate concentration is relatively low but both concentrations increased with increasing heating time, as a consequence of gradual water evaporation; the moisture contents after heating for 12, 24, and 48 h were 86.0, 67.3, and 24.5 g/100 g, respectively. In aqueous solutions, the glycosylation proceeds slowly and the reaction rate should be increased during the thermal treatment. However, the decrease of r-RL values takes place at similar rate within the entire range of heating time. Hence, the glycosylation would be limited by the relatively low carbohydrate–protein ratio in DSFF sample. The chemical modification of proteins in DSFF aqueous dispersions induced a significant loss of UA and TIA after 12 h of heating (r-UA and r-TIA ~ 50%), which was gradually increased at longer heating times. This chemical modification also was associated to protein denaturation of KTI and urease (): The denaturation enthalpy values of peak I, relative to that of DSFF control sample (rΔHd), was decreased with increasing heating time and an almost total denaturation was reached after 48 h of thermal treatment (). Interestingly, although the heating temperature was relatively low (62 ± 2°C), rΔHd values for peaks II and III also were decreased without significant changes of peak temperatures (Tp). Therefore, the partial protein unfolding induced by glycosylation, would contribute to decrease of r-SW and r-SKOH values, as a consequence of a hydrophobic interactions and covalent interactions between proteins and carbohydrates. It should be noted that the glycinin and β-conglycinin, initially located in the protein bodies, are released and hydrated during the preparation of aqueous dispersions. Hence, during thermal treatments, both storage soy proteins and whey soy proteins would be available to undergo glycosylation, aggregation, and denaturation, in agreement with the obtained data (, ).
Table 2. DSC and FTIR parameters for heated DSFF aqueous dispersions (62 ± 2°C, without and with relative humidity control, RH = 79%).
Figure 3. a) Effect of heating time on residual parameters (%) for heated DSFF aqueous dispersions treated by MS/HSH dispersing treatments (10.0 g total solids/100 g, 62 ± 2 °C, RH = 79%) (Uc samples). Residual parameters were: trypsin inhibitor activity (r-TIA), urease activity (r-UA), reactive lysine (r-RL), protein solubility in water (r-SW) and 0.2 g/100 g potassium hydroxide solution (r-SKOH); b) Relative residual parameters (%) for DSFF aqueous dispersions treated by MS and MS/HSH/HPH, heated for 24 h (10.0 g total solids/100 g, 62 ± 2 °C, RH = 79%), corresponding to Mc-24 and Vc-24 samples, respectively; c) Idem b) for 48 h, corresponding to Mc-48 and Vc-48 samples, respectively.
In regard to FTIR spectra, the intensity of all bands for Uc samples was significantly reduced with increasing heating time both in the protein and in the carbohydrate region (). The maximum wavenumber for AI and AII bands also was shifted toward lower and higher wavenumbers, respectively. Besides, for Uc-48 sample, AI and AII bands were overlapped: a low-intensity broad peak (1620.2 cm−1) was effectively observed (). According to previous works, a decrease of absorbance of AI and AII bands of whey protein isolate/glucose and soy protein isolate/carboxymethyl cellulose glycoconjugates was observed due to Maillard reaction.[Citation23,Citation24] Although in this work a good correlation (R2 > 0.8) was evidenced between r-RL and the intensity of AI and AII bands, a similar correlation (R2 > 0.7) was also seen with other parameters, such as r-SKOH and r-SW. Hence, the decrease of band intensities would also be associated to protein aggregation. This fact would be reinforced by the relative low protein/carbohydrate ratio of raw material. Indeed, the importance of protein aggregation in the decrease of AI and AII band intensities was previously reported by Sobral et al.[Citation4] for defatted soy flakes subjected to hydrothermal treatments for inactivation of anti-nutritional factors.
Uc-12, Uc-24, and Uc-48 samples were obtained from aqueous dispersions prepared by MS followed by HSH, using a rotor/stator device (). Now, the influence of dispersing method on structural properties of DSFF aqueous dispersions treated at 62 ± 2ºC (RH = 79%) for 24 and 48 h will be examined. The relative values of each residual parameter for samples dispersed only by MS (Mc-24 and Mc-48) or with an additional HPH treatment (Vc-24 and Vc-48) with respect to those of Uc-24 and Uc-48, respectively, are shown in and . Broadly, it can be observed that the dispersing treatments had an important impact on residual values on all assayed parameters. All relative residual parameters were positive for Mc samples and negative for Vc samples, regardless the heating time (24 or 48 h). This behavior would be consistent with a higher DM values and a concomitant decrease of particle size, which increased the reactivity of protein and carbohydrates (). After HPH treatment, residual r-UA and r-RL values showed a higher decrease after heating for 24 h; in contrast, the protein solubility values were more affected after 48 h. In addition, the combination of HPH and heating time also promoted the protein denaturation. For Vc-48 sample, ΔHd value for peak I was <4% and an important degree of denaturation also was reached for lectin/7S and 11 fractions (peaks II-III). The overlapping of AI and AII bands was consistent with protein aggregation processes, as was previously mentioned ().
When heating was performed at 62 ± 2ºC without RH control, a fast sample dehydration was reached after ca 4 h of thermal treatment; the moisture content was lower than 3.0 g/100 g after heating for 48 h. The influence of heating time on various parameters of aqueous dispersions (U-12, U-24, and U-48) is shown in . During dehydration, no significant changes in r-SKOH were evidenced; only a slight decrease of r-SW was effectively observed at longer heating times (24 and 48 h). r-TIA and r-UA decreased reaching a minimum value after 24 h of heating. In this period, r-RL gradually decreased, but at longer heating times no additional glycosylation was evidenced due to almost total sample dehydration. This result was supported by Liu et al.,[Citation5] which reported that the optimum RH for the Maillard reaction ranges from 50 to 80%. For all samples, denaturation enthalpy values for peaks I, II, and III were only slightly lower than that of control, which reflects that proteins were slightly denatured. Moreover, no substantial changes of intensity of AI, AII, and AIII bands were observed respect to control sample (). Undoubtedly, the glycosylation, denaturation, and aggregation processes were inhibited at longer heating times, due to sample dehydration. In this condition, the molecular mobility is noticeably constrained.
Figure 4. a) Effect of heating time on residual parameters (%) for heated DSFF aqueous dispersions treated by MS/HSH dispersing treatments (10.0 g total solids/100 g, 62 ± 2 °C without RH control) (U samples). Residual parameters were: trypsin inhibitor activity (r-TIA), urease activity (r-UA), reactive lysine (r-RL), protein solubility in water (r-SW) and 0.2 g/100 g potassium hydroxide solution (r-SKOH); b) Relative residual parameters (%) for DSFF aqueous dispersions treated by MS and MS/HSH/HPH, heated for 12 h (10.0 g total solids/100 g, 62 ± 2 °C without RH control) corresponding to M-12 and V-12 samples, respectively.
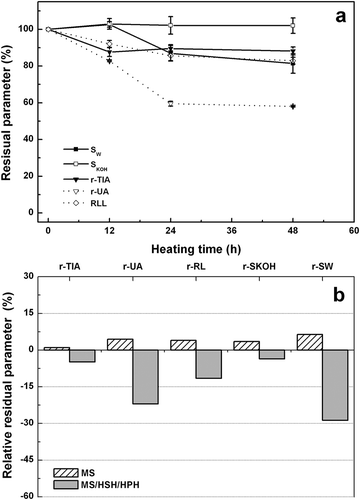
For DSFF aqueous dispersions heated without RH control, the impact of dispersing method on structural properties of DSFF aqueous dispersions was also investigated. In this condition, the most important changes in all parameters were observed at short heating times (). Hence, to analyze the influence of dispersing method, a heating time of 12 h was selected. The relative values of each parameter for samples dispersed only by MS (M-12) or additional HPH (V-12) respect to those of U-12 are shown in . The HPH treatment promoted a higher degree of glycosylation and protein aggregation after heating; in the latter case, this phenomenon was evidenced through a noticeable loss of r-SW. At the same time, r-TIA and r-UA values were additionally reduced after HPH treatment. The tendency was opposite when samples were only dispersed by MS (). This behavior was similar to those observed for dispersions heated with RH control ( and ). As previously mentioned, the size reduction of insoluble particles and the concomitant increase of their exposed area as a consequence of additional homogenization steps (HSH/HPH) would increase the reactivity of protein and carbohydrates. No important changes were detected by MDSC and FTIR as a consequence of dispersing method (data not shown).
Thermal treatments on DSFF sample
In this section, the impact of thermal treatments on glycosylation, aggregation, and denaturation degree of soy proteins in DSFF solid samples also was assessed. As mentioned previously, the initial moisture content of control DSFF sample was 8.0 g/100 g (92.0 ± 0.1 g total solids/100 g). The effect of heating time (12–48 h) at 62 ± 2ºC for samples treated both at RH = 79% (Fc-12, Fc-24, and Fc-48 samples) and without RH control (F-12, F-24, and F-48 samples) is shown in . It worth noting that, during the heating at RH = 79%, the moisture content increased from 13.7 g/100 g at 2 h to 20.0 g/100 g at the end of thermal treatment. In contrast, when heating was performed without RH control, the sample was practically dehydrated after treatment for 12 h. This moisture content (2.0 g/100 g) remains constant at longer heating times. In addition, for DSFF solid samples, storage soy globulins (glycinin and β-conglycinin), the main proteins of soybean seed, are found in protein bodies.[Citation3] Thus, soy globulins were not extracted from their bodies during thermal treatment whatever the RH control.
Figure 5. a) Effect of heating time on residual parameters (%) for heated DSFF solids samples: a) 62 ± 2 °C, RH= 79% and b) 62 ± 2 °C, without RH control. Residual parameters were: trypsin inhibitor activity (r-TIA), urease activity (r-UA), reactive lysine (r-RL), protein solubility in water (r-SW) and 0.2 g/100 g potassium hydroxide solution (r-SKOH).
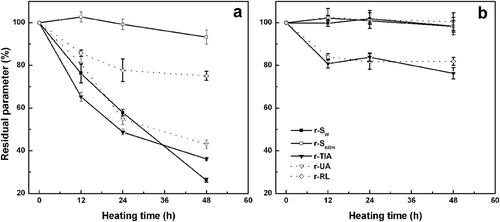
When heating was carried out at RH = 79%, r-SW decreased at the same rate throughout the range of heating time. However, this reduction of protein solubility was not accompanied by a similar decrease of r-SKOH (). Undoubtedly, the protein aggregation during heating at HR = 79% was mainly mediated by non-covalent interactions between polypeptides chains. The RL content gradually decreased due to glycosylation: r-RL was slightly lower than 80% at the end of assay. In this condition, where molecular mobility is not constrained due to sample hydration, r-UA and r-TIA progressively declined at the same rate (~43 and 36%, respectively, at 48 h). As previously mentioned, in solid DSFF samples, glycinin and β-conglycinin reside within the protein bodies. Therefore, it is expected that glycosylation occurs more easily in whey proteins, such as KTI, BBTI, urease, and lectin. Indeed, in model systems KTI/glucose and BBTI/glucose, the TIA was inhibited (40–65%) by heating at 50ºC (RH = 65%) for 48 h. [Citation25] Despite the greater complexity of the composition of DSFF sample, in respect to that of model systems, the degree of inactivation of TIA was similar.
The intensity of AI-AIII bands decreased with increasing heating time, in agreement with the protein aggregation associated to reduction of r-SW values. Although ΔHd parameter significantly decreased for peak I (>50%) after 48 h due to thermal denaturation of KTI and urease, higher ΔHd values were effectively observed for transitions involving 7S/lectin and 11S fractions (). This increase was accompanied by a slight increase of Tp values (data not shown). As the authors mentioned previously, the storage proteins were not dispersed during thermal treatment. Hence, the chemical modifications on protein molecules were made within protein bodies. In this condition, the structural modifications due to aggregation and glycosylation increased the thermal stability of 7S and 11S fractions.
Table 3. DSC and FTIR parameters for DSFF solid samples (62 ± 2°C, without and with relative humidity control, RH = 79%).
On the other hand, the effect of heating time on residual parameters for heated samples without RH control is shown in . It is worth noting that, at the beginning of thermal treatment, DSFF sample exhibited a moisture content of 8.0 g/100 g. On the basis of sorption isotherms of defatted soy flour, this moisture value corresponds to a water activity value of 0.5, which is favorable to glycosylation.[Citation26] Thus, the r-RL value decreased near 16% in this initial heating period, whereas r-TIA evidenced a maximum decrease of 20%. This result suggests that the TIA loss could be attributed to chemical modification in active site of trypsin inhibitors as result of glycosylation reactions, rather than a classic denaturation process. Indeed, when the DSFF moisture content is lower than 20 g/100 g, the denaturation of KTI and storage soy proteins is possible only at temperatures higher than 100ºC.[Citation27] As a consequence of total dehydration after 12 h of heating, the molecular mobility is restricted, and, hence, the glycosylation is almost totally inhibited. All the other parameters (r-SKOH, r-SW, and r-UA) were not significantly affected during heating without RH control.
The absence of changes for r-SW was in agreement with the occurrence of similar FTIR spectra for F-12, F-24, and F-48 samples. This fact reinforces the clear dependence of intensity of AI-AIII bands with the protein aggregation. At the same time, the decrease of ΔHd values for peak I (~20%) would be consistent with a reduction of r-TIA values. In this condition, the active site for urease would be not substantially affected by glycosylation. Moreover, a comparative analysis between the ΔHd values (peaks II and III) for F-24 and F-48 samples, revealed a similar behavior respect to samples heated with RH control (). The increase of ΔHd values for 7S/lectin and 11S fractions would be consistent with structural modifications increasing the thermal stability of these proteins.
Conclusion
In the present work, unlike previous artilces, the glycosylation reactions between proteins with the carbohydrates naturally present in soy were promoted through thermal treatments performed on DSFFs and their aqueous dispersions. The glycosylation, aggregation, and denaturation of soy proteins were more favored when heating was performed at controlled RH, where the molecular mobility is not restricted due to relatively high moisture content at the end of treatment. In contrast, the heating without RH control promoted the almost total sample dehydration both in aqueous dispersions and solid samples. When the heating was carried out on solid samples at a controlled RH, the inactivation of anti-tryptic factors was attributed to a combined effect of glycosylation and denaturation. In contrast, when thermal treatment was performed without control of RH, the lower degree of inactivation of anti-tryptic factors was mainly due to chemical modifications induced by glycosylation at beginning of assay. On the other hand, the increase of DM and a concomitant decrease of particle size favored all mentioned processes regardless the RH control during heating. In spite of the great complexity of the soy flour composition, FTIR was able to detect the protein aggregation through the decrease of intensity of all spectral bands in the protein and carbohydrate regions. The results obtained in this article show that, under mild heating conditions and controlled RH, it is possible reach an advanced degree of inactivation of anti-tryptic factors and glycosylation degree in DSFFs and their aqueous dispersions. The results obtained in this article should be used as basis for the assessment of the further isolation and characterization of corresponding glycoconjugates between soy proteins and carbohydrates.
Funding
The authors wish to acknowledge the financial support of Consejo Nacional de Investigaciones Científicas y Técnicas (PIP 2012-2014 N° 11220110100398) and Universidad Nacional de Quilmes (53/1007 I+D grant).
Additional information
Funding
References
- L’Hocine, L.; Boye, J.I.; Arcand, Y. Composition and Functional Properties of Soy Protein Isolate Prepared Using Alternative Defatting and Extraction Procedures. Journal of Food Science 2006, 71(3), 137–145.
- Perrechil, F.A.; Ramos, V.A.; Cunha, R.L. Synergistic Functionality of Soybean 7S and 11S Fractions in Oil-in-Water Emulsions. Effect of Protein Heat Treatment. International Journal of Food Properties 2015, 18, 2593–2602.
- Balaban, S.M.; Palermo, J.S. Soybeans and Soybean Processing. In Encyclopedia of Food Science and Technology. Vol. 4; Hui, Y.H., Ed.; John Wiley & Sons, Inc.: New York, NY, 1992; 2389–2396 pp.
- Sobral, P.A.; Palazolo, G.G.; Wagner, J.R. Evaluación de Cambios Estructurales en las Proteínas de Soja Durante el Proceso de Obtención de Harina Desgrasada de Soja (Evaluation of Structural Changes in Soy Proteins during the Industrial Production of Defatted Soybean Flour). Aceites y Grasas. 2012, 87, 82–90.
- Liu, J.; Ru, Q.; Ding, Y. Glycosylation a Promising Method for Food Protein Modification: Physicochemical Properties and Structure: A Review. Food Research International 2012, 49(1), 170–183.
- Van Boekel, M.A.J.S. Kinetic Aspects of the Maillard Reaction. A Critical Review. Nahrung 2001, 45, 150–159.
- Aoki, T.; Hiidome, Y.; Kitahata, K.; Sugimoto, Y. Improvement of Heat Stability and Emulsifying Activity of Ovoalbumin by Conjugation with Glucuronic Acid Through the Maillard Reaction. Food Research International 1999, 32, 129–133.
- Xu, C.-H.; Yang, X.-Q.; Yu, S.-J.; Qi, J.-R.; Guo, R.; Sun, W.W. The Effect of Glycosylation with Dextran Chains of Differing Lengths on the Thermal Aggregation of β-Conglycinin and Glycinin. Food Research International 2010, 43, 2270–2276.
- Diftis, N.; Kiosseoglou, V. Stability Against Heat Induced Aggregation of Emulsions Prepared with a Dry-Heated Soy Protein-Dextran Mixture. Food Hydrocolloids 2006, 20, 787–792.
- Diftis, N.; Kiosseoglou, V. Physicochemical Properties of Dry-Heated Soy Protein Isolate-Dextran. Food Chemistry 2006, 96, 228–233.
- van de Lagemaat, J.; Silván, J.M.; Moreno, M.F.; Olanoa, A.; del Castillo, M.D. In Vitro Glycosylation and Antigenicity of Soy Proteins. Food Research International 2007, 40, 153–160.
- Nkonge, C.; Murray-Balance, G. A Sensitive Colorimetric Procedure for Nitrogen Determination in Micro-Kjeldahl Digests. Journal of Agricultural and Food Chemistry 1982, 30, 416–420.
- AOAC. AOAC Official Methods of Analysis, 18th Ed; AOAC International: Gaithersburg, MD, 2012.
- AOCS. Official Methods and Recommended Practices of American Oil Chemists’ Society, 5th Ed; AOCS Press: Champaign, Illinois, USA, 2005.
- Araba, M.; Dale, N.M. Evaluation of Protein Solubility as an Indicator of Overprocessing Soybean Meal. Poultry Science 1990, 69, 76–83.
- Church, F.C.; Swaisgood, H.E.; Porter, D.H.; Catignani, G.L. Spectrophotometric Assay Using O-Phthaldialdehyde for Determination of Proteolysis in Milk and Isolated Milk Proteins. Journal of Dairy Science 1983, 66, 1219–1227.
- Medina Hernández, M.J.; García Alvarez-Coque, M.C. Available Lysine in Protein, Assay Using O-Phthalaldehyde/N-Acetyl-L-Cysteine Spectrophotometric Method. Journal of Food Science 1992, 57, 503–505.
- Liu, K.; Markakis, P. An Improved Colorimetric Method for Determining Antitryptic Activity in Soybean Products. Cereal Chemistry 1989, 66, 415–422.
- Sorgentini, D.A.; Wagner, J.R. Comparative Study of Structural Characteristics and Thermal Behavior of Whey and Isolate Soybean Proteins. Journal of Food Biochemistry 1999, 23, 498–507.
- Hayakawa, K.; Mizutani, J.; Wada, K.; Masai, T.; Yoshihara, I.; Mitsuoka, T. Effects of Soybean Oligosaccharides on Human Faecal Flora. Microbial Ecology in Health and Disease 1990, 3, 293–303.
- Chen, X.C. The Use of Soy Protein in Human Nutrition in China. In Soy Protein in Human Nutrition; Innoue, G.; Ed.; The Research Committee of Soy Protein Nutrition: Osaka, Japan, 1987; 79–88 pp.
- Karki, B.; Lamsal, B.P.; Jung, S.; van Leuween, J.; Pometto III, A.L.; Grewell, D.; Khanal, S.K. Enhancing Protein and Sugar Release from Defatted Soy Flakes Using Ultrasound Technology. Journal of Food Engineering 2010, 96, 270–278.
- Su, J.F.; Huang, Z.; Yuan, X.Y.; Wang, X.Y.; Li, M. Structure and Properties of Carboxymethyl Cellulose/Soy Protein Isolate Blend Edible Films Crosslinked by Maillard Reactions. Carbohydrate Polymers 2010, 79, 145–153.
- Liu, Q.; Kong, B.; Han, J.; Sun, C.; Li, P. Structure and Antioxidant Activity of Whey Protein Isolate Conjugated with Glucose C Kunitz Trypsin Inhibitor Via the Maillard Reaction Under Dry-Heating Conditions. Food Structure 2014, 1, 145–154.
- Kato, Y.; Matsuda, T. Glycation of Proteinous Inhibitors: Loss in Trypsin Inhibitory Activity by the Blocking of Arginine and Lysine Residues at Their Reactive Sites. Journal of Agricultural and Food Chemistry 1997, 45, 3826–3831.
- Riganakos, K.A.; Demertzis, P.G.; Kontominas, M.G. Water Sorption by Wheat and Soy Flour: Comparison of Three Methods. Journal of Cereal Science 1994, 20, 101–106.
- Sessa, D.J. Hydration Effects on Thermal Stability of Proteins in Cracked Soybeans and Defatted Soy Flours. LWT–Food Science and Technology 1992, 25, 365–370.