
ABSTRACT
Direct decompositions of nitric oxide (NO) by La0.7Ce0.3SrNiO4, La0.4Ba0.4Ce0.2SrNiO4, and Pr0.4Ba0.4Ce0.2SrNiO4 are experimentally investigated, and the catalysts are tested with different operating parameters to evaluate their activities. Experimental results indicate that the physical and chemical properties of La0.7Ce0.3SrNiO4 are significantly improved by doping with Ba and partial substitution with Pr. NO decomposition efficiencies achieved with La0.4Ba0.4Ce0.2SrNiO4 and Pr0.4Ba0.4Ce0.2SrNiO4 are 32% and 68%, respectively, at 400 °C with He as carrier gas. As the temperature is increased to 600 °C, NO decomposition efficiencies achieved with La0.4Ba0.4Ce0.2SrNiO4 and Pr0.4Ba0.4Ce0.2SrNiO4, respectively, reach 100% with the inlet NO concentration of 1000 ppm while the space velocity is fixed at 8000 hr−1. Effects of O2, H2O(g), and CO2 contents and space velocity on NO decomposition are also explored. The results indicate that NO decomposition efficiencies achieved with La0.4Ba0.4Ce0.2SrNiO4 and Pr0.4Ba0.4Ce0.2SrNiO4, respectively, are slightly reduced as space velocity is increased from 8000 to 20,000 hr−1 at 500 °C. In addition, the activities of both catalysts (La0.4Ba0.4Ce0.2SrNiO4 and Pr0.4Ba0.4Ce0.2SrNiO4) for NO decomposition are slightly reduced in the presence of 5% O2, 5% CO2, or 5% H2O(g). For durability test, with the space velocity of 8000 hr−1 and operating temperature of 600 °C, high N2 yield is maintained throughout the durability test of 60 hr, revealing the long-term stability of Pr0.4Ba0.4Ce0.2SrNiO4 for NO decomposition. Overall, Pr0.4Ba0.4Ce0.2SrNiO4 shows good catalytic activity for NO decomposition.
Implications: Nitrous oxide (NO) not only causes adverse environmental effects such as acid rain, photochemical smog, and deterioration of visibility and water quality, but also harms human lungs and respiratory system. Pervoskite-type catalysts, including La0.7Ce0.3SrNiO4, La0.4Ba0.4Ce0.2SrNiO4, and Pr0.4Ba0.4Ce0.2SrNiO4, are applied for direct NO decomposition. The results show that NO decomposition can be enhanced as La0.7Ce0.3SrNiO4 is substituted with Ba and/or Pr. At 600 °C, NO decomposition efficiencies achieved with La0.4Ba0.4Ce0.2SrNiO4 and Pr0.4Ba0.4Ce0.2SrNiO4 reach 100%, demonstrating high activity and good potential for direct NO decomposition. Effects of O2, H2O(g), and CO2 contents on catalytic activities are also evaluated and discussed.
Introduction
Oxides of nitrogen (NOx) have been regarded as one of the most serious air pollutants, and their emission has received much public concern. They not only cause adverse environmental effects such as acid rain, photochemical smog, and deterioration of visibility and water quality, but also harm human lungs and respiratory system (Bowker, Citation2008). Hence, how to effectively reduce NOx emission at a reasonable cost has become an emerging issue. NOx originate mainly from combustion processes, including mobile and stationary sources. Traditional technologies for effective removal of NOx can be generally divided into precombustion treatment, combustion modification, and postcombustion treatment (Wood, Citation1994). As a postcombustion treatment, selective catalytic reduction (SCR) has the highest nitric oxide (NO) removal efficiency, which is the most effective technique for removing NO from gas streams. However, SCR must rely on the injection of large amount of reducing agent such as ammonia (NH3), which is costly and may cause secondary pollution. Alternatively, direct NOx decomposition with catalysts is of high interest, since no additional reactant is required and the products are exclusively composed of N2 and O2.
Some catalysts, including noble metals (Sugisawa et al., Citation2001; Pietraszek et al., Citation2007; Wu et al., Citation2013), metal oxide catalysts (Teraoka et al., Citation1998; Silveira et al., Citation2008; Hong et al., Citation2011), and zeolite-based catalysts (Iwamoto and Hamada, Citation1991; Iwamoto, Citation1996; Lisi et al., Citation2009), have been investigated for direct NO decomposition. However, several disadvantages associated with these catalysts need to be overcome. For example, most zeolite-based catalysts are poisoned to some extent by water vapor (H2O(g)) or sulfur-containing gases; also, they are of low hydrothermal stability (Van den Brink et al., Citation2001). Therefore, they are difficult to apply in industry, although they may have high activities at low temperatures, whereas NOx decomposition efficiency and yield of N2 achieved with metal oxide catalysts are typically low (Hong et al., Citation2011). Noble metal catalysts, including Pt, Pd, Rh, and Au, are of high activity for NOx decomposition; however, they are expensive (National Association of Securities Dealers Automated Quotations System [NASDAQ], 2014). On the other hand, previous studies indicate that perovskite-type catalysts have the potential to replace noble metal catalysts for NOx removal (Voorhoeve et al., Citation1972; Khan and Frey, Citation1993) due to their high activities in decomposing NOx into N2 and O2 (Ishihara et al., Citation2003; Zhang et al., Citation2006; Russo et al., Citation2007; Zhu et al Citation2009; Zhao et al., Citation2009; Ivanov et al., Citation2011; Wang et al., Citation2012).
Generally, the mechanism of NO decomposition over perovskite-type catalysts can be described by eqs 1–3 (Teraoka et al., Citation1998). First, NO molecules are adsorbed on oxygen vacancies (active sites of catalyst) at the catalyst surface, and two adjacent N–O bonds can be dissociated to produce a N2, which is subsequently released into the gas stream from the catalyst surface (Teraoka et al., Citation1998). Previous study indicates that adsorption of the second NO molecule on the pair site (eq 2) is the rate-determining step (Imanaka and Masui, Citation2012); hence, NO decomposition would depend on NO adsorption potential and the number of vacancies available.
where O is an oxygen atom and Vo is a surface oxygen vacant site.
Perovskite-type catalyst is of the general form ABO3 or A2BO4, where A can be either an alkaline metal or rare earth element, typically La is applied. On the other hand, 3d transition metal with a relatively small ion such as Cu, Fe, Co, Ni, or Mn (Pérez-Ramírez et al., Citation2003) is generally applied at the B site. The number of vacancies available depends on the B site, and it directly influences catalyst activity. Also, catalytic performance can be enhanced by partial substitution of the A site, because it may lead to formation of lattice vacancy. Further, lattice vacancy increases mobility of oxygen. Therefore, physical and chemical properties of perovskite-type catalysts may vary by substituting different elements at the A and/or B sites.
ABO3-type and A2BO4-type catalysts such as LaMO3 (M = Fe, Mn, and Ni) (Wu et al., Citation2013; Zhang et al., Citation2006; Wang et al., Citation2012) and La2MO4 (M = Ni, Cu) (Zhao et al., Citation1996; Gao et al., Citation2008) have been studied for NO decomposition. Catalysts with rather complex composition are synthesized by partial substitution at A-site and/or B-site to improve the activity. For example, La1-xAxM1-yByO3 (A = Ba, Th; M = Cu, Cr, Fe, Mn; and B = Ga, In) (Ishihara et al., Citation2003; Zhu et al., Citation2006) and La1−xSrxNiO4 (Zhao et al., Citation1996) have been evaluated for the effectiveness in direct NO decomposition. In another study, La1−xCexSrNiO4 is supported on MgO to enhance NO decomposition (Zhu et al., Citation2006).
As high as 100% NO decomposition efficiency could be achieved with perovskite-type catalysts; however, they need to be operated at high temperatures (>700 °C) for a high NO decomposition efficiency. Additionally, NO decomposition is strongly inhibited by the presence of oxygen (O2) and carbon dioxide (CO2). For example, the N2 yield achieved with La0.2Ba0.8Mn0.8Mg0.2O3 reaches 85% in the absence of CO2 at 850 °C, but it decreases dramatically to below 20% in the presence of 5% CO2 (Iwakuni, Citation2008). Furthermore, activity of perovskite-type catalysts may be influenced by the presence of H2O(g) (Tofan et al., 2002). Since CO2, O2, and H2O(g) commonly exist in industrial flue gases, tolerances of CO2, O2, and H2O(g) should be considered for field application.
With respect to the enhancement of perovskite-type catalyst activity, several methods have been proposed. For instance, La at the A site was partially substituted by Sr to improve the activity because the oxide vacancies can be produced by charge compensation as a low-valence cation (Sr2+) occupies the frame at the La3+ site (Zhao et al., Citation1996; Zhu et al., Citation2005). Previous study suggests that nonstoichiometric perovskites (La1−xSrxM1−yMyO3−δ) own several advantages, such as fast electron transfer, high oxygen mobility, and easy oxygen desorption (Tabata and Misono, Citation1990). Also, the activity of perovskite-type catalysts can be enhanced when A is partially substituted with Ce. Ce possesses good properties such as oxygen storing and scavenging, resulting in easy regeneration of active sites. Kirchnerova et al. (Citation2002) show that the activity of LaCoO3 is significantly improved if doped with CeO2 (Kirchnerova et al., Citation2002). Belessi et al. (Citation2001) show that the NO decomposition rate achieved with La1−x−ySrxCeyFeO3 can be improved by increasing CeO2 content (Belessi et al., Citation2001). Nitadori and Misono (Citation1985) applied Sr, Ce, and Th to substitute LaMO3 (M = Mn, Fe, Co) and show that Ce-substituted catalysts increase the oxygen mobility (Nitadori and Misono, Citation1985). Iwakuni et al. (Citation2007) showed that the lattice of A2BO4-type catalyst could accommodate more cerium ions due to larger cavity space; therefore, redox effect of A2BO4-type perovskite catalysts is more prominent than that of ABO3-type (Iwakuni et al., Citation2007).
Ba possesses a high basicity, and it has been applied to partially substitute the A site of perovskite-type catalyst for enhancing NO decomposition (Iwakuni, Citation2008; Iwakuni et al., Citation2007). LaMnO3 doped with Ba also exhibits a higher activity for direct NO decomposition at over 800 °C (Khan and Frey, Citation1993). Praseodymium (Pr) is one of important rare earth elements for preparing perovskite-type catalyst, with its electron distribution and structural characteristics being similar to light rare earth elements such as lanthanum (La) and cerium (Ce). Recently, Pr-based perovskites have been prepared and investigated for their catalytic properties. Ran et al. (Citation2005) applied Pr0.8Ce0.2MnO3 to decompose NO, and the results indicate that NO decomposition efficiency achieved with Pr0.8Ce0.2MnO3 reaches 90% at 650 °C (Ran et al., Citation2005). However, application of Pr-based perovskites-type catalyst is limited, especially for NO decomposition.
In this study, A2BO4-type perovskite catalyst (La0.7Ce0.3SrNiO4) is prepared and evaluated for the effectiveness in direct NO decomposition. Nickel (Ni) is frequently applied due to a variety of oxygen-involving reactions (Kirchnerova and Klvana, Citation1994; Klvana et al., Citation1994). The A site of La0.7Ce0.3SrNiO4 is partially substituted by doping with Ba to prepare La0.4Ba0.4Ce0.2SrNiO4 catalyst, and then the La site is further substituted by Pr to prepare Pr0.4Ba0.4Ce0.2SrNiO4. It is expected that the properties and activities of the catalysts can be improved to further reduce the reaction temperature needed for NO decomposition. Influences of space velocity, O2, H2O(g), and CO2 on NO decomposition are also evaluated with a laboratory-scale experimental system.
Experimental
Catalyst preparation
A2BO4-type perovskite catalysts, including La0.7Ce0.3SrNiO4, La0.4Ba0.4Ce0.2SrNiO4, and Pr0.4Ba0.4Ce0.2SrNiO4, were prepared by the Pechini method (Pechini, Citation1967). The corresponding metal nitrates were used as starting material for preparing aqueous solution with appropriate stoichiometry. Citric acid was added into the mixed aqueous solution of metal nitrates in a designated proportion so that the molar ratio of citric acid to the total metal cations was 4:1. Ethylene glycol was then added into the mixed aqueous solution to control the molar ratio of citric acid to the ethylene glycol as 1:1. The mixed aqueous solution was then heated and stirred to evaporate the water. Thereafter, the residual solid was dried at 140 °C for 24 hr, followed by heat treatment at 600 °C for 1 hr in air, and finally calcination in air at 950 °C for 6 hr, and then manually milled to powders by using an agate mortar and pestle. Subsequently, the synthesized particles were pulverized to the final 100 mesh size and used as such.
Characterization of the catalysts prepared
Brunauer-Emmett-Teller (BET) surface areas of the catalysts prepared were measured by using N2 adsorption at −196 °C with a Micromeritics ASAP 2010 analyzer (Micromeritics, Norcross, GA, USA). Scanning electron microscopy (SEM) was utilized to identify the morphology and structure of catalysts, and powder X-ray diffraction (XRD) data were obtained using an X-ray diffractometer (model D8AXRD; Bruker, Germany) that was operated at 40 kV and 10 mA by using Cu-Kα radiation combined with a nickel filter. Diffraction patterns were obtained within a 2θ rang of 10°–80° at a scanning rate of 6° min−1. Additionally, oxygen mobility of the catalysts prepared was evaluated via temperature-programmed desorption of oxygen (O2-TPD), which was carried out by using thermal conductivity detector (TCD), in which the sample (0.2 g) was treated at 1000 °C in the presence of O2 for 1 hr. Subsequently, it was cooled down to the room temperature and swept with helium (He) at a rate of 11.8 mL/min. Finally, the sample was heated with a rate of 20 °C/min in He and the data profile was recorded.
Catalytic activity measurement
Direct NO decomposition test was performed with a fixed-bed quartz reactor (inner diameter [i.d.] 20 mm) containing 4 g of catalyst. The gas stream containing 1000 ppm NO with He as carrier was introduced into the reactor continuously. The total gas flow rate was fixed at 628 mL/min, corresponding to a gas hourly space velocity (GHSV) of 8000 hr−1. Also, two space velocities (15,000 and 20,000 hr−1) were tested to evaluate the space velocity effect. As mentioned previously, O2, H2O(g), and CO2 commonly exist in real flue gas; therefore, effects of O2, H2O(g), and CO2 on the activities of catalysts for direct NO decomposition are also investigated. Contents of O2, H2O(g), and CO2 of the feeding gas stream were controlled within the ranges of 1–5%, 5–10%, and 5%, respectively. Sources of NO, O2, and CO2 were from gas cylinders, and designated NO, O2, and CO2 concentrations could be obtained by adjusting the flow rate of individual gas stream. In addition, a peristaltic pump was used to inject H2O(l) into the gas stream to control the H2O(g) content. Mass-flow controllers (MFCs) were utilized to adjust and control the gas flow rates, and all gases were well mixed before reaching the reactor. The operating temperature was varied from 300 to 900 °C to evaluate the temperature effect. NO concentrations were analyzed before and after the reaction by using Fourier transform infrared spectrophotometer (Nicolet 6700; Thermo Scientific, USA) and NOx detector (Testo 350; Testo, Germany), whereas analysis of N2 was conducted using gas chromatography (model 6890N; Agilent Technologies, USA) with a TCD and a column packed with 5A molecular sieve (capillary: 30.0 m × 530 μm × 25 μm nominal). All experimental data were taken after ca. 3–4 hr on stream at each temperature, while the catalytic reaction practically reached a steady state. Decomposition efficiency of NO and yield of N2 were calculated by eqs 4 and 5, respectively:
where and
are NO concentrations measured before and after the reactor, respectively, and
is the N2 concentration measured at reactor outlet.
Results and discussion
Characterization of the catalysts
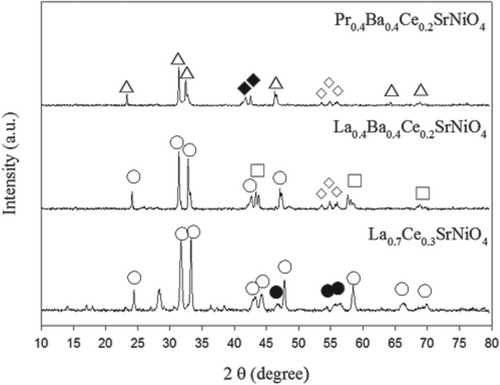
The XRD patterns of the three catalysts prepared are presented in , indicating that all catalysts prepared are of perovskite-like structure (A2BO4-type). As shown in , La0.7Ce0.3SrNiO4 diffraction peaks are attributed to crystalline of LaSrNiO4, but there exists one abrupt peak at 2θ ~ 28°, which could be attributed to CeO2 according to XRD database. However, the characteristic peaks of CeO2 should include 2θ = 28.5°, 33.0°, 47.6°, 56.4°, 58°, and 69.5°, respectively. In this study, characteristic peaks of CeO2 barely appear at 2θ = 28°, 47°, 56°, 58°, whereas other diffraction peaks are mainly assigned to LaSrNiO4. Previous study indicates that diffraction peak at 2θ ~ 28° might imply La1−xCexSrNiO4 (Zhu et al., Citation2005). This result demonstrates that Ce occupies the La site without destroying the matrix structure. In addition, La0.4Ba0.4Ce0.2SrNiO4 shows strong main diffraction peaks that are attributed to those from LaSrNiO4. Some peaks are also observed in the XRD pattern of La0.4Ba0.4Ce0.2SrNiO4, and they belong to La2NiO4 and BaO, respectively, Crystalline of BaO formed due to excess addition of Ba element, although the lattice of A2BO4-type catalyst has larger cavity space to accommodate other ions. On the other hand, characteristic peaks of Pr0.4Ba0.4Ce0.2SrNiO4 are mainly assigned to Pr2NiO4. Clearly, crystalline of Pr1-xMxNiO4-like is not detected. Possibly, Sr is incorporated into the lattice of Pr-based catalyst to form a good solid solution, and additional peaks of Pr2O3 and BaO are observed in the XRD pattern of the Pr-based catalyst. In addition, characteristic peaks of CeO2 are not observed for La0.4Ba0.4Ce0.2SrNiO4 and Pr0.4Ba0.4Ce0.2SrNiO4 due to addition of low content of Ce.
Figure 1. X-ray diffraction patterns of the catalysts (Δ:Pr2NiO4, La2NiO4, ○: LaSrNiO4, ●:CeO2, ♦:Pr2O3, ◊:BaO).
The BET surface areas of La0.7Ce0.3SrNiO4, La0.4Ba0.4Ce0.2SrNiO4, and Pr0.4Ba0.4Ce0.2SrNiO4 are 5.2, 25.5, and 29.9 m2/g (see ), respectively, indicating that La0.7Ce0.3SrNiO4 has the smallest BET surface area. Generally, BET surface areas of perovskite-type catalysts are in the range of 5–50 m2/g. However, the BET surface area is significantly increased by the addition of Ba and substitution of Pr. displays the SEM patterns of all three catalysts, including three catalysts present as pellet with slight agglomeration. Obviously, the morphology of Pr-based catalysts is more compact and uniform when compared with La-based catalysts.
Table 1. BET surface areas of the catalysts prepared.
Figure 2. SEM patterns of the catalysts (a) La0.7Ce0.3SrNiO4, (b) La0.4Ba0.4Ce0.2SrNiO4, and (c) Pr0.4Ba0.4Ce0.2SrNiO4.

O2 temperature-programmed desorption (O2-TPD)
The role of perovskite-type oxides for NO decomposition can be explored by O2-TPD analysis because it provides the information on oxygen nonstoichiometry of substituted perovskite (Kumar et al., Citation2012). Typically, two kinds of oxygen desorption peaks are observed in the O2-TPD process and can be denoted as α oxygen and β oxygen, respectively. The peak of α oxygen appears at a relatively lower temperature and generally it is weaker when compared with the peak of β oxygen.
Peaks of α oxygen and β oxygen vary with different perovskite-type catalysts, implying that their peaks may appear at lower or higher temperatures, sometimes peaks of α oxygen are not even observed in the O2-TPD profile. As previously reported, α oxygen is chemically desorbed from the surface whereas β oxygen is desorbed from the oxygen of vacancy and lattice. Hence, the intensity of β oxygen can be an indicator of catalytic performance (Ferri and Forni, Citation1998; Belessi et al., Citation2000). The O2 desorption profiles of the three catalysts prepared are shown in . For La0.7Ce0.3SrNiO4, there exists one abrupt desorption peak of oxygen, which is β oxygen corresponding to the temperature of 590–810 °C, whereas β oxygen of La0.4Ba0.4Ce0.2SrNiO4 is located in the region of 550–820 °C. For Pr-based catalyst, the desorption peak of β oxygen is found at 450–780 °C. As presented in , α oxygen does not appear as significant desorption peaks for the three catalysts prepared.
Figure 3. O2-TPD profiles for La0.7Ce0.3SrNiO4, La0.4Ba0.4Ce0.2SrNiO4, and Pr0.4Ba0.4Cec0.2SrNiO4, respectively.
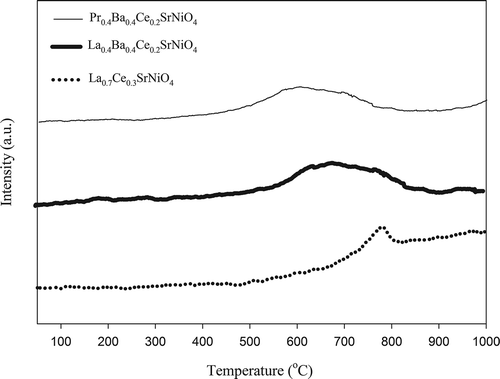
The O2-TPD results indicate that La0.7Ce0.3SrNiO4 has a smaller area of β oxygen desorption when compared with the modified catalysts. Previous study indicates that area of β oxygen desorption is related to the number of oxygen vacancies. In other words, a perovskite-type catalyst would have excellent performance for NO decomposition if it has a wide area of β oxygen desorption (Zhu et al., Citation2005). As shown in , the highest peak for La0.7Ce0.3SrNiO4 is located at 790 °C, whereas those of La0.4Ba0.4Ce0.2SrNiO4 and Pr0.4Ba0.4Ce0.2SrNiO4 are located at 670 and 590 °C, respectively. The O2-TPD results show that β oxygen peaks are shifted to lower temperature with partial substitution at the A site. Presumably, physical and chemical properties of La0.7Ce0.3SrNiO4 are improved due to Ba substitution. In addition, the highest β oxygen peak of La0.4Ba0.4Ce0.2SrNiO4 is shifted from 670 to 590 °C when Pr is substituted for the A site of La0.4Ba0.4Ce0.2SrNiO4. The order of the mobility of lattice oxygen in the three perovskite-type catalysts tested is Pr0.4Ba0.4Ce0.2SrNiO4 > La0.4Ba0.4Ce0.2SrNiO4 > La0.7Ce0.3SrNiO4.
Activities of catalysts for NO decomposition
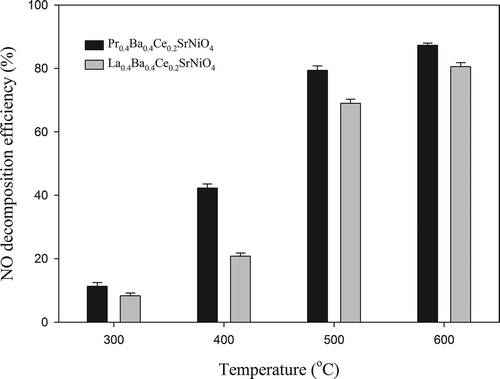
The catalytic activities of the three perovskite-type catalysts prepared were tested at various temperatures (300–900 °C) to evaluate the effect on NO decomposition. NO decomposition efficiencies achieved with La0.7Ce0.3SrNiO4, La0.4Ba0.4Ce0.2SrNiO4, and Pr0.4Ba0.4Ce0.2SrNiO4, respectively, are presented in . The results indicate that the activities of the three catalysts for NO decomposition all increase with increasing temperature. At initial temperature of 300 °C, NO decomposition efficiencies achieved with Pr0.4Ba0.4Ce0.2SrNiO4, La0.4Ba0.4Ce0.2SrNiO4, and La0.7Ce0.3SrNiO4 are 24.5%, 17.2%, and 2.0%, respectively. The activity of Pr0.4Ba0.4Ce0.2SrNiO4 is significantly higher than that of La0.7Ce0.3SrNiO4 and La0.4Ba0.4Ce0.2SrNiO4. NO decomposition efficiencies achieved with the three catalysts at 400 °C are 68.3%, 32.6%, and 10.2%, respectively. As the temperature is further increased to 600 °C, the NO decomposition efficiencies achieved with La0.4Ba0.4Ce0.2SrNiO4 and Pr0.4Ba0.4Ce0.2SrNiO4, respectively, reach 100%. On the other hand, La0.7Ce0.3SrNiO4 has to be operated at 700 °C to achieve 100% NO decomposition efficiency. The reaction rates per surface area for the three catalysts operated at 600 °C are presented in . The result indicates that Pr0.4Ba0.4Ce0.2SrNiO4 has the highest value (6.58 × 10−5 mg/L·min·m2) when compared with La0.4Ba0.4Ce0.2SrNiO4 and La0.7Ce0.3SrNiO4 (5.33 × 10−5 and 2.02 × 10−5 mg/L·min·m2).
Table 2. Catalytic activities (the reaction rate) per surface area for the three catalysts (T = 500 °C).
Figure 4. Activities of La0.7Ce0.3SrNiO4, La0.4Ba0.4Ce0.2SrNiO4, and Pr0.4Ba0.4Ce0.2SrNiO4 for NO decomposition at different temperatures with He as the carrier gas (CNO = 0.1%, GHSV = 8000 hr−1).
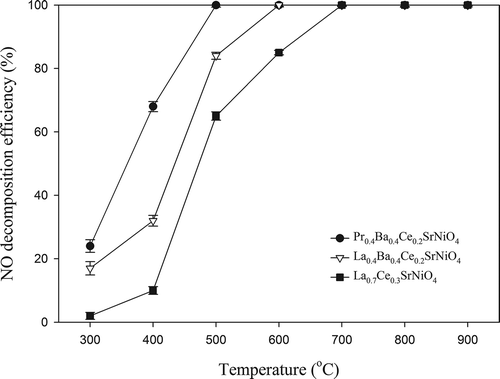
In addition, the efficiency curves are frequently characterized by two parameters, i.e., T50 and T90. T50 is defined as the temperature required to convert 50% of the pollutant, whereas T90 is the temperature needed to achieve 90% removal. As shown in , T50 for NO decomposition with La0.7Ce0.3SrNiO4 is 480 °C, whereas T50 values of La0.4Ba0.4Ce0.2SrNiO4 and Pr0.4Ba0.4Ce0.2SrNiO4 are 450 and 350 °C, respectively. The results indicate that Pr0.4Ba0.4Ce0.2SrNiO4 has the lowest T50. Likewise, T90 with Pr0.4Ba0.4Ce0.2SrNiO4 is also the lowest when compared with the other two catalysts, indicating that the performance of the three perovskite-type catalysts for NO decomposition can be ranked in the order of Pr0.4Ba0.4Ce0.2SrNiO4 > La0.4Ba0.4Ce0.2SrNiO4 > La0.7Ce0.3SrNiO4 at same operating conditions. Accordingly, the modified perovskite-type catalysts have more active sites for NO decomposition.
Table 3. Temperatures for 50% and 90% NO decomposition.
Previous study indicates that activity of La0.7Ce0.3SrNiO4 is mainly from a complex active site described as Ce3+-(O)-(Ni2+-[]-Ni3+), in which valence of Ce is alterable as Ce4+ ↔ Ce3+ during reaction (Zhu et al., Citation2005). This redox reaction promotes regeneration of active sites. Additionally, basicity of catalyst surface is an important factor affecting the chemical reactivity of NO, and the catalyst with a high basicity generally has excellent performance for NO decomposition (Zhang et al., Citation2006). Due to its high basicity, Ba is frequently applied to prepare NO reduction catalysts, such as TWCs (three-way catalysts) and NSR (NOx storage and reduction) catalysts. Since NO is acidic, adsorption of NO can be significantly enhanced by increasing the basicity of catalyst. Indeed, activity of La0.7Ce0.3SrNiO4 is improved as the A site is partly substituted with Ba. In addition, addition of Ba may also produce oxygen vacancies by charge compensation (generally, Ba is bivalent). Ce promotes the generation of active sites, without destroying the structure of the lattice, and forms a complementary system. Previous study indicates that the activity of Pr-based catalyst can be improved by partial substituting with Sr and Ce (Ran et al., Citation2005) to induce structural default, further increasing the number of oxygen vacancies. Indeed, Pr0.4Ba0.4Ce0.2SrNiO4 possesses a broad β oxygen desorption peak, as shown in .
Overall, the modified catalysts (La0.4Ba0.4Ce0.2SrNiO4 and Pr0.4Ba0.4Ce0.2SrNiO4) show better performance in decomposing NO at a temperature range of 500–900 °C compared with that reported in literature (Russo et al., Citation2007; Pérez-Ramírez et al., Citation2003; Gao, et al., Citation2008). Experimental results indicate that properties of La0.7Ce0.3SrNiO4 are significantly improved by doping with Ba and partial substitution with Pr at the La site; hence, the modified catalysts (La0.4Ba0.4Ce0.2SrNiO4 and Pr0.4Ba0.4Ce0.2SrNiO4) are mainly applied for the follow-up experimental tests.
Influences of space velocity on NO decomposition
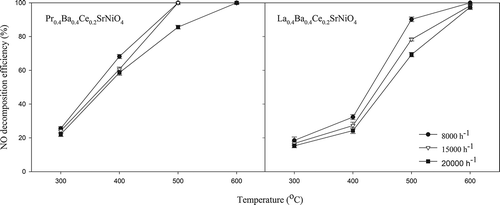
shows the effects of space velocity on NO decomposition achieved with La0.4Ba0.4Ce0.2SrNiO4 and Pr0.4Ba0.4Ce0.2SrNiO4, respectively. The results indicate that NO decomposition efficiency decreases with increasing space velocity at a temperature range of 300–600 °C for both catalysts. Obviously, NO decomposition efficiency might suffer from a larger space velocity due to diffusion limitation. However, NO decomposition efficiency achieved with Pr0.4Ba0.4Ce0.2SrNiO4 reaches 100% at 600 °C, implying that activity of Pr-based catalyst is relatively high at T ≥ 600 °C. Higher activity observed at a higher operating temperature may be attributed to higher mobility of lattice oxygen. O2-TPD results indicate that La0.4Ba0.4Ce0.2SrNiO4, and Pr0.4Ba0.4Ce0.2SrNiO4 have significant desorption peak of lattice oxygen at about 600 °C; it is believed that this is one of the reasons for high activity. Overall, NO decomposition efficiency achieved with La0.4Ba0.4Ce0.2SrNiO4 is affected more significantly by decreasing residence time at a temperature lower than 600 °C compared with Pr0.4Ba0.4Ce0.2SrNiO4. However, influence of space velocity can be improved by loading the active phase on the support with a large surface area.
Figure 5. Influences of space velocity on activities of La0.4Ba0.4Ce0.2SrNiO4, and Pr0.4Ba0.4Ce0.2SrNiO4, respectively, for NO decomposition (CNO = 0.1%; He as carrier gas).
Influences of O2 and H2O(g) on NO decomposition
O2 and H2O(g) exist in typical flue gases and they may cause poisoning of catalyst. Therefore, their influences on catalyst active cannot be ignored. The lifetime of catalyst is successfully increased if it can effectively tolerate O2 and H2O(g). The suppression of NO decomposition by oxygen remains one of the important obstacles to overcome in effective NO decomposition. Inhibition of NO decomposition by oxygen can be explained by the reversible dissociative adsorption of oxygen or via molecular adsorption of oxygen, as shown in eq 5:
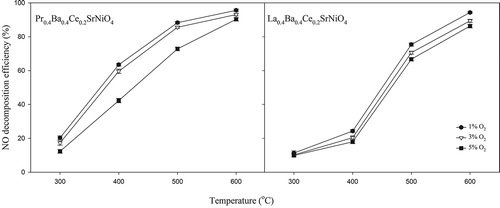
Effects of oxygen content on the NO decomposition over La0.4Ba0.4Ce0.2SrNiO4 and Pr0.4Ba0.4Ce0.2SrNiO4, respectively, are presented in . With 1–3% oxygen in the gas stream, the NO decomposition efficiencies achieved with both catalysts decrease slightly when compared with the case without oxygen at low temperatures (300–400 °C). NO decomposition efficiency achieved with La0.4Ba0.4Ce0.2SrNiO4 decreases from 32.0% to 24.3% at 400 °C as 1% oxygen is introduced into the gas stream, whereas that achieved with Pr0.4Ba0.4Ce0.2SrNiO4 decreases from 68.0% to 63.5%. As oxygen content is further increased to 5%, NO decomposition efficiencies achieved with La0.4Ba0.4Ce0.2SrNiO4 and Pr0.4Ba0.4Ce0.2SrNiO decrease to 20.4% and 42.3%, respectively, at 400 °C. However, influence of O2 content decreases gradually with increasing temperature for both catalysts. The NO decomposition efficiency achieved with Pr0.4Ba0.4Ce0.2SrNiO4 reaches 95.6% at 600 °C with 1% of O2, whereas that achieved with La0.4Ba0.4Ce0.2SrNiO4 is 94.3%. With 5% O2, NO decomposition efficiencies achieved with Pr0.4Ba0.4Ce0.2SrNiO4 and La0.4Ba0.4Ce0.2SrNiO4 decrease to 90.3% and 86.3%, respectively, at 600 °C. The same trend also appears in the space velocity test, and both catalysts show excellent performance for NO decomposition when operated at a temperature higher than 600 °C. However, activities of both catalysts for NO decomposition would be slightly affected at a high oxygen content.
Figure 6. Effects of oxygen contents on NO decomposition at various temperatures (CNO = 0.1%, CO2 = 1–5%, GHSV = 8000 hr−1; He as carrier gas).
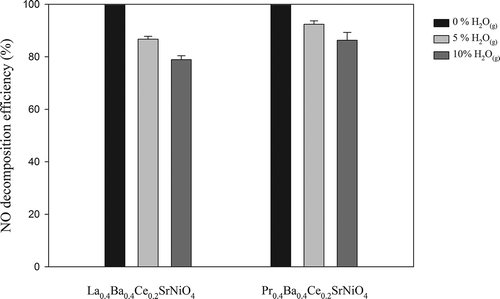
shows the effects of 5% and 10% H2O(g) on NO decomposition achieved with La0.4Ba0.4Ce0.2SrNiO4 and Pr0.4Ba0.4Ce0.2SrNiO4, respectively, at 600 °C. The results indicate that NO decomposition efficiencies achieved with La0.4Ba0.4Ce0.2SrNiO4 and Pr0.4Ba0.4Ce0.2SrNiO4 decrease from 100% to 86.7% and 92.4%, respectively, as 5% H2O(g) is incorporated into the gas stream. With 10% H2O(g), the NO decomposition efficiencies achieved with La0.4Ba0.4Ce0.2SrNiO4 and Pr0.4Ba0.4Ce0.2SrNiO4 further decrease to 78.9% and 86.3%, respectively. Tofan et al. (2002) suggest that influence of H2O(g) is mainly from adsorption effect because H2O(g) is easily adsorbed on the catalyst surface, resulting in the reduction of active sites (Tofan et al., 2002). Nevertheless, the NO decomposition efficiency achieved with Pr0.4Ba0.4Ce0.2SrNiO4 maintains at 86% with 10% H2O(g) in the gas stream. The results indicate that of Pr0.4Ba0.4Ce0.2SrNiO4 has good tolerance for H2O(g) when operated at 600 °C.
Figure 7. Effects of H2O(g) contents on NO decomposition achieved with La0.4Ba0.4Ce0.2SrNiO4 and Pr0.4Ba0.4Ce0.2SrNiO4, respectively, at 600 °C (CNO = 0.1%, = 5–10%, GHSV = 8000 hr−1; He as carrier gas).
Influences of CO2 on NO decomposition
shows the effect of CO2 in the gas stream on NO decomposition achieved with the two catalysts at 600 °C. The results indicate that NO decomposition efficiencies achieved with La0.4Ba0.4Ce0.2SrNiO4 and Pr0.4Ba0.4Ce0.2SrNiO4 decrease from 100% to 80.6% and 87.3%, respectively, as 5% CO2 is introduced into the system. As temperature is controlled at 400 °C, NO decomposition efficiencies achieved with La0.4Ba0.4Ce0.2SrNiO4 and Pr0.4Ba0.4Ce0.2SrNiO4 are greatly reduced to 20.8% and 42.3%, respectively. Obviously, the activities of both catalysts are severely affected by CO2 at low temperatures. Previous study indicates that high basicity of catalyst facilitates adsorption of CO2 in addition to NO (Zhang et al., Citation2006). Therefore, catalyst poisoning by CO2 adsorption can be severe. Pr0.4Ba0.4Ce0.2SrNiO4 shows higher tolerance to O2, CO2, and H2O(g) when compared with La0.4Ba0.4Ce0.2SrNiO4. Characterization of the catalysts indicates that Pr0.4Ba0.4Ce0.2SrNiO4 may have higher oxygen mobility to reduce poison. In addition, previous study also indicates that Pr-based catalyst can induce structural defaults by partial substituting with Sr and Ce, further increasing the number of oxygen vacancies (Ran et al., Citation2005).
Figure 8. Effects of CO2 on NO decomposition achieved with La0.4Ba0.4Ce0.2SrNiO4 and Pr0.4Ba0.4Ce0.2SrNiO4, respectively, at 600°C (CNO = 0.1%, CO2= 5%, GHSV = 8000 hr−1; He as carrier gas).
Catalytic tests in the presence of O2, CO2, and H2O(g)
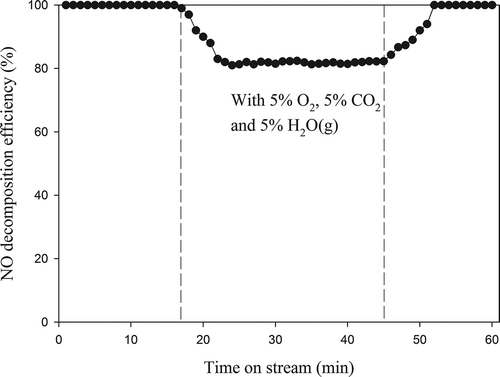
Testing of catalytic activity with real gas is very important in determining the catalysts’ practical usefulness. Therefore, 5% O2, 5% CO2, and 5% H2O(g) are simultaneously introduced into the catalysis system to investigate the effect on activity of Pr0.4Ba0.4Ce0.2SrNiO4. As shown in , NO decomposition efficiency could maintain 100% at initial period of the reaction, i.e., without O2, CO2, and H2O(g) condition. As 5% O2, 5% CO2, and 5% H2O(g) are simultaneously introduced into the gas streams, NO decomposition efficiency gradually decreases from 100% to 82%. Obviously, activity of Pr0.4Ba0.4Ce0.2SrNiO4 can be affected by simultaneous presence of O2, CO2, and H2O(g). However, the NO decomposition efficiency returns to 100% as the supplies of O2, CO2, and H2O(g) are terminated.
Figure 9. Catalytic test of Pr0.4Ba0.4Ce0.2SrNiO4 for NO removal in the absence or presence of O2, CO2, and H2 (CNO = 0.1%, GHSV= 8000 hr−1, T = 600 °C; He as carrier gas).
Product analysis and durability test
summarizes the N2 yields from NO decomposition achieved with the three selected catalysts. The results indicate that N2 yield clearly depends on the temperature. Among the three catalysts, Pr0.4Ba0.4Ce0.2SrNiO4 shows the best NO decomposition efficiency (68%) and the highest N2 yield (54%) at 400 °C. As the temperature is increased to 600 °C, N2 yield achieved with Pr0.4Ba0.4Ce0.2SrNiO4 reached 92%. For La0.4Ba0.4Ce0.2SrNiO4, N2 yield increases from 15% to 88% as the temperature is increased from 400 to 600 °C, and in the meantime, N2 yield increases from 3% to 63% for La0.7Ce0.3SrNiO4. Results indicate that Pr0.4Ba0.4Ce0.2SrNiO4 possesses the best activity in decomposing NO due to its excellent oxygen mobility.
Table 4. Products analysis and efficiencies of NO decomposition achieved with the three catalysts at various temperatures.
Also, effects of 5% O2 and 5% H2O(g) on N2 yield are evaluated, respectively. The results indicated that N2 yields achieved with Pr-based catalyst is 89% at 600 °C in the presence of 5% O2, whereas N2 yields is 90% in the presence of 5% H2O(g). Furthermore, with 5% CO2 in the gas stream, N2 yield is 82% at 600 °C for Pr-based catalyst.
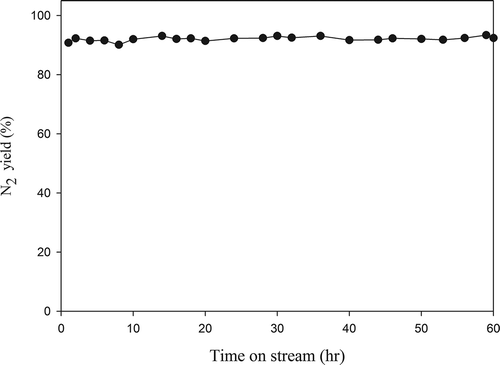
The durability of catalysts is important in determining their practical application, and shows the results of durability test with Pr0.4Ba0.4Ce0.2SrNiO4 as the catalyst. The results indicate that N2 yield maintains 92% at 600 °C and no significant deactivation is observed during the continuous operation of 60 h.
Figure 10. N2 yield during the durability test of Pr0.4Ba0.4Ce0.2SrNiO4 catalyst for NO decomposition (CNO = 0.1%, GHSV = 8000 hr−1, T = 600 °C; He as carrier gas).
Conclusions
Development of NO decomposition technology is essential for reducing the environmental impact associated with NOx. This study discusses the activities of perovskite-type catalysts for NO decomposition and evaluates the influences of O2, H2O(g), and CO2. The results indicate that catalytic activities of La-based catalysts and their tolerance to the complex composition of typical flue gases can be improved by partial substitution with Pr at the La site. Important conclusions can be drawn from this study as follows:
NO decomposition efficiencies achieved with three perovskite-type oxide catalysts prepared rank in the order of Pr0.4Ba0.4Ce0.2SrNiO4 > La0.4Ba0.4Ce0.2SrNiO4 > La0.7Ce0.3SrNiO4 at the same operating condition.
O2-TPD results show that the order of the mobility of lattice oxygen in the three perovskite-type catalysts tested is Pr0.4Ba0.4Ce0.2SrNiO4 > La0.4Ba0.4Ce0.2SrNiO4 > La0.7Ce0.3SrNiO4. Physical and chemical properties of La-based for NO decomposition are improved by substituting with Pr.
NO decomposition efficiencies achieved with La0.4Ba0.4Ce0.2SrNiO4 and Pr0.4Ba0.4Ce0.2SrNiO4, respectively, decrease with the addition of 5% O2 or 5% CO2 because high basicity of the catalysts facilitates adsorption of O2 and CO2, reducing the active sites for NO decomposition.
NO decomposition efficiencies achieved with catalysts modified slightly decrease as 10% H2O(g) is introduced the gas stream. Possibly, H2O(g) can be adsorbed on the catalyst surface, resulting in the reduction of active sites.
Development of the catalysts that can tolerate relatively high O2, H2O(g), and CO2 contents of the typical flue gases is essential for successful NO decomposition application.
Acknowledgment
The authors thank Dr. G.T. Pan for valuable discussion.
Funding
The authors would like to thank the Industrial Technology Research Institute for financial support.
Additional information
Funding
Notes on contributors
Kuan Lun Pan
Kuan Lun Pan is a Ph.D. student at the Graduate Institute of Environmental Engineering, National Central University, Chungli, Taiwan, Republic of China.
Mei Chung Chen
Mei Chung Chen is a master’s degree student at the Graduate Institute of Environmental Engineering, National Central University, Chungli, Taiwan, Republic of China.
Sheng Jen Yu
Sheng Jen Yu is a researcher at the Industrial Technology Research Institute (ITRI), Hsinchu, Taiwan, Republic of China.
Shaw Yi Yan
Shaw Yi Yan is a researcher at the Industrial Technology Research Institute (ITRI), Hsinchu, Taiwan, Republic of China.
Moo Been Chang
Moo Been Chang is Distinguished Professor at the Graduate Institute of Environmental Engineering, National Central University, Chungli, Taiwan, Republic of China.
References
- Belessi, V.C., C.N. Costa, T.V. Bakas, T. Anastasiadou, P.J. Pomonis, and A.M. Efstathiou. 2000. Catalytic behavior of La–Sr–Ce–Fe–O mixed oxidic/perovskitic systems for the NO+CO and NO+CH4+O2 (lean-NOx) reactions. Catal. Today 59:347–363. doi:10.1016/S0920-5861(00)00300-X
- Belessi, V.C., Ladavos, A.K., and Pomonis, P.J. 2001. Methane combustion on La–Sr–Ce–Fe–O mixed oxides: Bifunctional synergistic action of SrFeO3−x and CeOx phases. Appl. Catal. B Environ. 31:183–194. doi:10.1016/S0926-3373(00)00279-4
- Bowker, M. 2008. Automotive catalysis studied by surface science. Chem. Soc. Rev. 37:2204–2211. doi:10.1039/b719206c
- Ferri, D., and Forni, L. 1998. Methane combustion on some perovskite-like mixed oxides. Appl. Catal. B Environ. 16:119–126. doi:10.1016/S0926-3373(97)00065-9
- Gao, L., Chua, H.T., and Kawi, S. 2008. The direct decomposition of NO over the La2CuO4 nanofiber catalyst. J. Solid State Chem. 181:2804–2807. doi:10.1016/j.jssc.2008.06.051
- Hong, W.J., Lwamoto, S., and Lnoue, M. 2011. Direct NO decomposition over a Ce–Mn mixed oxide modified with alkali and alkaline earth species and CO2-TPD behavior of the catalysts. Catal. Today 164:489–494. doi:10.1016/j.cattod.2010.10.063
- Iwamoto, M. 1996. Heterogeneous catalysis for removal of NO in excess oxygen. Progress in 1994. Catal. Today 29:29–35. doi:10.1016/0920-5861(95)00256-1
- Iwamoto, M., and Hamada, H. 1991. Removal of nitrogen monoxide from exhaust gases through novel catalytic processes. Catal. Today 10:57–71. doi:10.1016/0920-5861(91)80074-J
- Ishihara, T., Ando, M., Sada, K., Takiishi, K., Yamada, K., Nishiguchi, H., and Takita, Y. 2003. Direct decomposition of NO into N2 and O2 over La(Ba)Mn(In)O3 perovskite oxide. J. Catal. 220:104–114. doi:10.1246/cl.2003.1176
- Iwakuni, H., Shinmyou, Y., Yano, H., Goto, K., Matsumoto, H., and Ishihara, T. 2008. Effects of added CO2 and H2 on the direct decomposition of NO over BaMnO3-based perovskite oxide. Bull. Chem. Soc. Jpn. 81:1175–1882. doi:10.1246/bcsj.81.1175
- Iwakuni, H., Shinmyo, Y., Yano, H., Matsumoto, H., and Ishihara, T. 2007. Direct decomposition of NO into N2 and O2 on BaMnO3-based perovskite oxides. Appl. Catal. B Environ. 74:299–306. doi:10.1016/j.apcatb.2007.02.020
- Ivanov, D.V., Pinaeva, L.G., Isupova, L.A., Nadeev, A.N., Prosvirin, I.P., and Dovlitova, L.S. 2011. Insights into the reactivity of La1−xSrxMnO3 (x = 0–0.7) in high temperature N2O decomposition. Catal. Lett. 141:322–331. doi:10.1007/s10562-010-0503-0
- Imanaka, N., and Masui, T. 2012. Advances in direct NOx decomposition catalysts. Appl. Catal. A Gen. 431– 432:1–8. doi:10.1016/j.apcata.2012.02.047
- Khan, H.R., and Frey, H. 1993. R.f. plasma spray deposition of LaMOx (M=Co, Mn, Ni) films and the investigations of structure, morphology and the catalytic oxidation of CO and C3H8. J. Alloy Compd. 190:209–217. doi:10.1016/0925-8388(93)90401-8
- Kirchnerova, J., and Klvana, D. 1994. Preparation and characterization of high surface perovskite electrocatalysts. Int. J. Hydrogen Energy 19:501–506. doi:10.1016/0360-3199(94)90004-3
- Klvana, D., Vaillancourt, J., Kirchnerova, J., and Chaouki, J. 1994. Combustion of methane over La0.66Sr0.34Ni0.3Co0.7O3 and La0.4Sr0.6Fe0.4Co0.6O3 prepared by freeze-drying. Appl. Catal. A Gen. 109:181–193. doi:10.1016/0926-860X(94)80117-7
- Kirchnerova, J., Alifanti, M., and Delmon, B. 2002. Evidence of phase cooperation in the LaCoO3–CeO2–Co3O4 catalytic system in relation to activity in methane combustion. Appl. Catal. A Gen. 231:65–80. doi:10.1016/S0926-860X(01)00903-6
- Kumar, S., Vinu, A., Subrt, J., Bakardjieva, S., Rayalu, S., Teraoka, Y., and Labhsetwar, N. 2012. Catalytic N2O decomposition on Pr0.8Ba0.2MnO3 type perovskite catalyst for industrial emission control. Catal. Today 198:125–132. doi:10.1016/j.cattod.2012.06.015
- Lisi, L., Pirone, R., Russo, G., and Stanzione, V. 2009. Cu-ZSM5 based monolith reactors for NO decomposition. Chem. Eng. J. 154:341–347. doi:10.1016/j.cej.2009.04.025
- Nitadori, T., and Misono, M. 1985. Catalytic properties of La1−xA′xFeO3 (A′ = Sr, Ce) and La1−xCexCoO3. J. Catal. 93:459–466. doi:10.1016/0021-9517(85)90193-9
- National Association of Securities Dealers Automated Quotations System (NASDAQ), USA. 2014. http://www.nasdaq.com/ (accessed June 15, 2014).
- Pechini, M.P. 1967, 11 July. Method of preparing lead and alkaline earth titanates and niobates and coating method using the same to form a capacitor. US Patent 3,330,697.
- Pérez-Ramírez, J., Kapteijn, F., Schöffel, K., and Moulijn, J.A. 2003. Formation and control of N2O in nitric acid production: Where do we stand today?. Appl. Catal. B Environ. 44:117–151. doi:10.1016/S0926-3373(03)00026-2
- Pietraszek, A., Da Costa, P., Marques, R., Kornelak, P., Hansen, T.W., Camra, J., and Najbar, M. 2007. The effect of the Rh–Al, Pt–Al and Pt–Rh–Al surface alloys on NO conversion to N2 on alumina supported Rh, Pt and Pt–Rh catalysts. Catal. Today 119:187–193. doi:10.1016/j.cattod.2006.08.009
- Ran, R., Wu, X., Quan, C., and Weng, D. 2005. Effect of strontium and cerium doping on the structural and catalytic properties of PrMnO3 oxides. Solid State Ionics 176:965–971. doi:10.1016/j.ssi.2004.11.018
- Russo, N., Mescia, D., Fino, D., Saracco, G., and Specchia, V. 2007. N2O decomposition over perovskite catalysts. Ind. Eng. Chem. Res. 46:4226–4231. doi:10.1021/ie0612008
- Sugisawa, T., Shiraishi, J. Machihara, D., Irokawa K., Miki, H., Kodama, C., Kuriyama, T., Kubo, T., and Nozoye, H. 2001. Adsorption and decomposition of NO on Pt (112). Appl. Surface. Sci. 169– 170:292–295. doi:10.1016/S0169-4332(00)00675-9
- Silveira, E.B., Perez, C.A.C., Baldanza, M.A.S., and Schmal, M. 2008. Performance of the CeZrO2 mixed oxide in the NOx decomposition. Catal. Today 133– 135:555–559. doi:10.1016/j.cattod.2007.12.057
- Tabata, K., and Misono, M. 1990. Elimination of pollutant gases—oxidation of CO, reduction and decomposition of NO. Catal. Today 8:249–261. doi:10.1016/0920-5861(90)87021-T
- Teraoka, Y., Harada, T., and Kagawa S. 1998. Reaction mechanism of direct decomposition of nitric oxide over Co- and Mn-based perovskite-type oxides. J. Chem. Soc. 94:1887–1891. doi:10.1039/a800872h
- Tofan, C., Klvana, D., and Kirchnerova, J. 2002. Decomposition of nitric oxide over perovskite oxide catalysts: Effect of CO2, H2O and CH4. Appl. Catal. B Environ. 36:311–323. doi:10.1016/S0926-3373(01)00312-5
- Voorhoeve, R.J.H., J.P. Remeika, P.E. Freeland, and B.T. Matthias. 1972. Rare-earth oxides of manganese and cobalt rival platinum for the treatment of carbon monoxide in auto exhaust. Mater. Res. Bull. 177:353–354. doi:10.1126/science.177.4046.353
- Van den Brink, R.W., S. Booneveld, J.R. Pels, D.F. Bakker, and M.J.F.M. Verhaak. 2001. Catalytic removal of N2O in model flue gases of a nitric acid plant using a promoted Fe zeolite. Appl. Catal. B Environ. 32:73–81. doi:10.1016/S0926-3373(00)00294-0
- Wang, H., Liu, J., Zhao, Z., Wei, Y., and Xu, C. 2012. Comparative study of nanometric Co-, Mn- and Fe-based perovskite-type complex oxide catalysts for the simultaneous elimination of soot and NOx from diesel engine exhaust. Catal. Today 184:288–300. doi:10.1016/j.cattod.2012.01.005
- Wood, S.C. 1994. Select the right NOx control technology. Chem. Eng. Prog. 90:32–38.
- Wu, Z., Xu, L., Zhang, W., Ma, Y., Yuan, Q., Jin, Y., Yang, J., and Huang, W. 2013. Structure sensitivity of low-temperature NO decomposition on Au surfaces. J. Catal. 304:112–122. doi:10.1016/j.jcat.2013.04.013
- Zhang, R., Alamdari, H., and Kaliaguine, S. 2006. Fe-based perovskites substituted by copper and palladium for NO + CO reaction. J. Catal. 242:241–253. doi:10.1016/j.jcat.2006.05.033
- Zhao, B., Wang, R., Yao, W., Yang, X., and Zhou Bin B. 2009. The effect of copper substitution on La2Ni1−xCuxO4 catalysts activity for simultaneous removal of NOx and diesel soot. Catal. Lett. 132:41–49. doi:10.1007/s10562-009-0041-9
- Zhao, Z., Yang, X., and Wu, Y. 1996. Comparative study of nickel-based perovskite-like mixed oxide catalysts for direct decomposition of NO. Appl. Catal. B Environ. 8:281–297. doi:10.1016/0926-3373(95)00067-4
- Zhu, J., Xiao, D., Li, J., and Yang, X. 2009. Perovskite-like mixed oxides (LaSrMn1−xNixO4+δ, 0 ≤ x ≤ 1) as catalyst for catalytic NO decomposition: TPD and TPR studies. Catal. Lett. 129:240–246. doi:10.1007/s10562-008-9807-8
- Zhu, J., Xiao, D., Li, J., Yang, X., and Wei, K. 2006. Effect of Ce and MgO on NO decomposition over La1-x-Cex-Sr-Ni-O/MgO. Catal. Commun. 7:432–435. doi:10.1016/j.catcom.2005.12.026
- Zhu, J., Xiao, D., Li, J., Yang, X., and Wu, Y. 2005. Effect of Ce on NO direct decomposition in the absence/presence of O2 over La1−xCexSrNiO4 (0 ≤ x ≤ 0.3). J. Mol. Appl. Catal. A Chem. 234:99–105. doi:10.1016/j.molcata.2005.02.015
- Zhu, J., Zhao, Z., Xiao, D., Li, J., Yang, X., and Wu, Y. 2005. CO Oxidation, NO Decomposition, and NO + CO reduction over perovskite-like oxides La2CuO4 and La2-xSrxCuO4: An MS-TPD study. Ind. Eng. Chem. Res. 44: 4227–4233. doi:10.1021/ie050317d
- Zhu, J., Zhao, Z., Xiao, D., Li, J. Yang, X., and Wu, Y. 2006. Effect of valence of copper in La2-xThxCuO4 on NO decomposition reaction. Catal. Commun. 7: 29–32. doi:10.1016/j.catcom.2005.08.008