
ABSTRACT
Cloning of coding sequence (CDS) is an important step for gene function research. Here, we reported a simple and efficient strategy for assembling multiple-exon into an intron-free CDS from genomic DNA (gDNA) by an isothermal recombination reaction-based PCR (IRR-PCR) method. As an example, a 2067-bp full-length CDS of the anther-specific expression gene OsABCG15, which is composed of seven exons and six introns, was generated by IRR-PCR using genomic DNA of rice leaf as the template. Actually, this approach can be wildly applied to any DNA sequences assembly to achieve CDS cloning, gene fusion and multiple site-directed mutagenesis in functional genomics studies in vitro.
Introduction
Isolation of coding sequence (CDS) of a target gene is an initial step to study the function of the corresponding protein in molecular biology. For most of eukaryotic genes are interrupted by non-coding introns, the genetic coding information is only stored in the exons. Therefore, the removal of all introns is necessary to acquire a gene CDS. This goal is usually achieved by conventional cDNA cloning method which is involved in total RNA extraction and reverse transcription. In addition to the setback of time-consumption, RNA is prone to degradation, which makes cDNA cloning experiments problematic. Even worst, the extraction of total RNA or mRNA are extremely difficult in the case of some tissue-specific or low-level expression genes in rare tissues. In addition, many genes are tissue-specific or condition-inducible expression, thus they may not be expressed in the available fresh tissues. Compared with RNA, genomic DNA (gDNA) is stable and easy to handle. Thus, gene splicing of CDS or cDNA using gDNA as template has been a proven method in functional genomics.[Citation1–4] Several strategies including the overlap extension PCR-based method (OE-PCR) [Citation1–3] and the Golden Gate ligation-based method (GG-PCR) [Citation4] have demonstrated the advantages of isolation of cDNA from gDNA. However, the current methods are of high-cost, cumbersome and inefficient. For example, the gDNA Splicing [Citation2] and SPLICE technique,[Citation3] all based on OE-PCR, require complex PCR procedures and multiple rounds of PCR. When assembling more than four DNA molecules simultaneously, these methods are likely to produce false positive and nonspecific amplification results. The possible presence of the type IIs restriction enzyme site(s) within the exon fragments limit applications of the GG-PCR method.[Citation4] Additionally, purification procedures of the first-round PCR products (different exon fragments) are required in most current methods.[Citation1–4] Thus, a simple, rapid and low-cost method for CDS isolation is highly desired in molecular biology.
The recently developed in vitro isothermal recombination technique, also called Gibson Assembly, is a powerful tool for DNA molecule assembly.[Citation5–7] Overlapping DNA fragments can be assembled into a large and complete DNA molecule in a single reaction at 50 °C based on the reaction of three enzymes (the 5′-T5 exonuclease, the Phusion polymerase and the Taq DNA ligase). Based on this method, Jiang et al. and Zhu et al. have developed strategies for one-step assembling hairpin RNA interference vectors and one-step multi-type plasmid modifications, respectively.[Citation8,Citation9]
Here, we report a simple and efficient isothermal recombination reaction-based PCR method (IRR-PCR) for cloning of any eukaryotic CDS or cDNA from gDNA. When no gene expression data and/or correct tissue samples are available, this procedure will be very useful for straightforward CDS cloning of genes identified after analysis of sequences of genome clones (from genomic libraries, e.g. BAC clones) or characterized genomic regions (e.g. after application of genome walk). It is also very effective for CDS cloning using rare materials (e.g. herbarium samples). To exhibit the feasibility of the IRR-PCR in in-vitro splicing, a full-length CDS for a seven-exon anther-specific transporter OsABCG15 gene [Citation10] was generated from gDNA of rice leaves.
Materials and methods
Genomic DNA extraction
The gDNA was extracted from rice young leaves according to previous protocol.[Citation11]
Preparation of master mixture
A home-made 2× master mixture for in vitro isothermal recombination reaction was slightly modified according to a previous publication.[Citation5] Briefly, it consists of 160 μL 5× isothermal buffer (25% PEG-8000, 500 mmol/L Tris-HCl pH 7.5, 50 mmol/L MgCl2, 50 mmol/L DTT, 1 mmol/L each of dNTPs, 5 mmol/L NAD, stored at −20°C), 1.5 units of T5 exonuclease (Epicentre), 20 units of Phusion polymerase (NEB), 2000 units of Taq ligase (NEB) and deionized water to a final volume of 0.4 mL.
Primer design
Primers for amplification and assembly of exons were designed referring to Zhu et al.[Citation9] Briefly, the primer comprises two regions from 5′ to 3′: a 20- to 25-bp 5′-end overlapping homologous sequence with the adjacent exon and a 3′-end exon-specific sequence. The outermost forward or reverse primers (F1 or R7) have 20-bp 5′-end sequences overlapping with the plasmid pGEX-4T-2. All primers used in this research are listed in Table S1, and the detailed locations of them in the plasmid construct are shown in Figure S1.
PCR amplification of exons
Exon amplification was carried out using gDNA as template and with different primer pairs (Figure ). For each exon, the PCR was performed using a high fidelity KOD FX Polymerase (KFX-101, TOYOBO) in standard reaction set-up, under the two-step process: 30 cycles (98°C for 10 s and 68°C for 30 s).
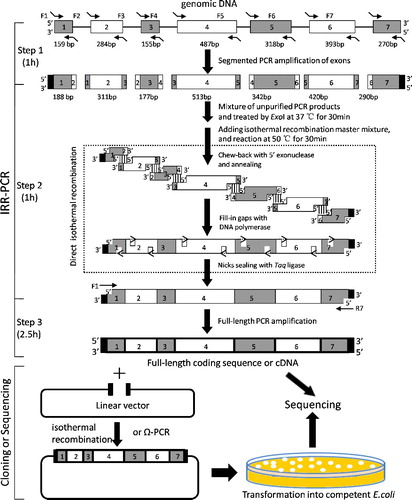
Figure 1. A schematic diagram of the IRR-PCR strategy. Step 1, DNA fragments preparation: the segmented seven exons were amplified using gDNA templates and primers containing 5′-end overlapping homologous sequence with the adjacent exon by normal PCR. Step 2, isothermal assembly of fragments: after ExoI treatment and heat inactivation, the first-round PCR products were mixed and subjected to isothermal assembly. Step 3, PCR amplifying splicing products: the assembled full-length CDS was amplified by PCR with the outermost primers (F1 and R1). The PCR products of splicing DNA molecules could be directly sequenced or cloned into a linear functional plasmid by isothermal recombination or Ω-PCR method. The black blocks indicate the overlapping homologous sequences with the multiple cloning sites of prokaryotic expression plasmid pGEX-4T-2. The dashed box in step 2 indicates the detailed process of isothermal in vitro recombination reaction.
Isothermal recombination reaction
The concentrations of unpurified PCR products of exons were approximately determined by agarose gel electrophoresis, and then they were directly mixed into 10 μL (about 0.05∼0.1 pmol each), followed by adding 1 μL (20 units) exonuclease I (ExoI, NEB) to digest the remanent primers at 37°C for 1 hour, and finally heat inactivation of ExoI at 80°C for 10 min. Then, 10 μL of 2× master mixture was added in the mixed PCR products, and assembly reaction carried out at 50°C for 30 min.
IRR-PCR amplification
The assembled product (0.5--1 μL) was amplified by PCR using the two outermost primers (F1 and R7). The reaction was carried out in 50 μL of PCR mixture, using 1 μL (1 unit) of KOD FX Polymerase, under the following process: 98°C for 1 min, followed by 30 cycles (98 °C for 10 s and 68 °C for 2 min), followed by a final extension at 68°C for 5 min. The IRR-PCR products were used directly for sequencing.
Cloning of splicing DNA into a functional plasmid
For IRR-PCR strategy, one IRR-PCR splicing full-length DNA and the same linear vector were mixed in a molar ratio of 2:1 to complete isothermal recombination reaction. For direct assembly, above seven exons and one linear prokaryotic expression vector pGEX-4T-2 vector (about 0.05 pmol each) were mixed. The two assembled products (about 0.03 pmol each) were transformed separately into 50-μL E. coli DH5α competent cells by heat shock method, followed by pipetting 950-μL SOC into the mixture at 37 °C for 1 h, respectively. Finally, 100 µL of each mixture was plated and incubated overnight at 37 °C, and then the number of colonies were calculated as transformation efficiency. Colony PCR was performed to screen 12 randomly picked colonies in order to calculate positive rate. Positive clones were identified by restrict enzyme digestion. Then, the clones with the correct insert were selected for sequence analyses.
Results and discussion
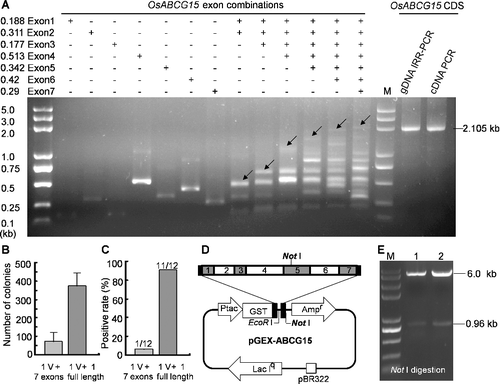
In this report, we showed that the IRR-PCR is a simple and efficient strategy to generate a CDS by isothermal assembly of exons from gDNA and PCR amplification. This method includes three steps (): (i) preparation of exon fragments; (ii) isothermal assembly of exons; (iii) PCR amplification of full-length products. The seven-exon fragments of OsABCG15 gene were amplified from rice gDNA (). After mixed and treated by ExoI, these unpurified PCR products were directly used as a template to perform isothermal in vitro recombination reaction. An expected band of about 2 kb for the assembled seven exons (Exon1–Exon7) was distinctly detected by agarose gel electrophoresis (). Subsequently, the full-length assembled seven-exon sequence was further amplified by PCR using the two outermost primers (F1 and R7) (). The results showed that the CDS product of IRR-PCR has the anticipated size in conformity to the PCR product derived from conventional rice anther-specific cDNA (). Sequencing confirmed that the sequence of the IRR-PCR product is identical to the cDNA sequence of OsABCG15.
Figure 2. Splicing full-length CDS of OsABCG15 from gDNA by the IRR-PCR strategy. (a) PCR-amplified exon products and assembled products of different exons by isothermal in vitro recombination were displayed on agarose gel electrophoresis. The expected band joined by seven exons and other intermediate combination products were demonstrated. Arrows indicate the predicted target products of the exon combinations. The full-length CDS of OsABCG15 assembled by IRR-PCR had the same size with the anther-specific cDNA derived from conventional cDNA cloning. The amplified full-length sequence (2105 bp) contained a 2067-bp coding sequence and two short flanking sequences. (b) and (c) Comparison of transformation efficiency and positive rate between direct assembly and IRR-PCR strategy. Seven exons and one IRR-PCR splicing full length were assembled separately into the linear pGEX-4T-2 vector. (d) The map of recombinant plasmid pGEX-ABCG15 and (e) identification of positive clone by Not I digestion.
For some tissue-specific or low-level expression genes in rare tissues, the in vitro gene splicing of CDS or cDNA from gDNA by OE-PCR, or modified OE-PCR are widely used methods. However, splicing more than four DNA fragments by these methods are with low efficiency and high false positives.[Citation4] Moreover, some intermediate by-products may reduce the yield of full-length DNA. Our IRR-PCR strategy enables isolation of full-length CDS from gDNA based on isothermal assembly-based PCR reactions. Compared to current in vitro gene splicing methods,[Citation1-4] the IRR-PCR method has the following advantages: first, the IRR-PCR is simpler and more efficient for splicing of multiple-exon genes using a gDNA template; second, the IRR-PCR is more convenient, for first-round PCR products of exon fragments can be directly used in assembly and full-length CDS amplification without purification; third, the exon fragments for IRR-PCR are of restriction endonuclease-free therefore seamless and finally, if necessary, any types of mutations (base substitution, insertion, deletion) can be introduced into the CDS sequence by designing the targeted mutation(s) in the primers. Thus, this method is superior to the GG-PCR method that is only suitable for linking fragments without the used type IIs restriction enzyme site.[Citation4] The use of home-made in vitro isothermal recombination reaction mixture reduces the cost for the IRR-PCR. High-fidelity DNA polymerase is strongly recommended in the PCR amplification to reduce the possible base mutation. In addition, to prevent miss-priming and frame shift in some of the obtained CDS clones resulted from the longer stretch of one nucleotide chain, or simple sequence repeats, a careful design of the PCR primers (covering the regions flanking the repeated sequence) and sequencing check of the selected CDS clone(s) should be accomplished.
Generally, the splicing CDS need to be cloned into a functional vector for further research. Using the 5′-end overlapping homologous sequences with vectors in the outermost forward and reverse primers (F1 and R7), the IRR-PCR product could be seamlessly cloned into any vectors by isothermal recombination-based plasmid construction[Citation9] or by Ω-PCR method[Citation12] for further functional study. For comparison of cloning transformation efficiency and positive rate between direct splicing and indirect IRR-PCR strategies, simultaneously assembling total eight fragments (seven exons and one linear pGEX-4T-2 vector) and assembling two fragments (one IRR-PCR splicing full-length DNA and the same linear vector) were achieved by isothermal recombination reaction, respectively. Although both strategies could assemble DNA fragments into a functional plasmid, the IRR-PCR strategy has higher transformation efficiency and positive clones than direct splicing ((b--d)). This means that the IRR-PCR strategy is an effective way, especially when more DNA fragments were spliced simultaneously into a functional vector.
Conclusions
We have described a simple and effective IRR-PCR method for direct assembly of multiple exon CDS or cDNA from gDNA for some tissue-specific or low-level expression genes in rare tissues. This method does not need to prepare specific tissues, extract RNA and perform reverse transcription, and efficiently generates full-length cDNA or CDS. To further optimize this technique, we will endeavour to simplify the first step of IRR-PCR using multiplex PCR to amplify multiple fragments simultaneously in a single tube. In addition, this approach could be widely applied to any gene or DNA sequences assembly to achieve CDS cloning, gene fusion and multiple site-directed mutagenesis in functional genomics studies, even for multiplex genome editing of CRISPR/Cas9 system, IRR-PCR may also be effective to combine multiple sgRNA expression cassettes.
TBEQ_A_1191374_SM2131.docx
Download MS Word (747.9 KB)Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
Related Research Data
References
- Vandenbroeck K, Dijkmans R, van Aerschot A, et al. Engineering by PCR-based exon amplification of the genomic porcine interferon-gamma DNA for expression in Escherichia coli. Biochem Biophys Res Commun. 1991;180:1408–1415.
- An X, Lu J, Huang JD, et al. Rapid assembly of multiple-exon cDNA directly from genomic DNA. PLoS One. 2007;2:e1179.
- Davies WL, Carvalho LS, Hunt DM. SPLICE: a technique for generating in vitro spliced coding sequences from genomic DNA. Biotechniques. 2007;43:785–789.
- Zhou L, Lin Q, Zhang J, et al. A rapid DNA assembling strategy mediated by direct full-length polymerase chain reaction. Gene. 2013;523:122–125.
- Gibson DG, Young L, Chuang RY, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–345.
- Gibson DG, Smith HO, Hutchison CR, et al. Chemical synthesis of the mouse mitochondrial genome. Nat Methods. 2010;7:901–903.
- Gibson DG. Enzymatic assembly of overlapping DNA fragments. Method Enzymol. 2011;498:349–361.
- Jiang Y, Xie M, Zhu Q, et al. One-step cloning of intron-containing hairpin RNA constructs for RNA interference via isothermal in vitro recombination system. Planta. 2013;238:325–330.
- Zhu Q, Yang Z, Zhang Q, et al. Robust multi-type plasmid modifications based on isothermal in vitro recombination. Gene. 2014;548:39–42.
- Niu B, He F, He M, et al. The ATP-binding cassette transporter OsABCG15 is required for anther development and pollen fertility in rice. J Integra Plant Biol. 2013;55:710–720.
- Wang H, Chu Z, Ma X, et al. A high through-put protocol of plant genomic DNA preparation for PCR. Acta Agronomica Sinica. 2013;39:1200–1205.
- Chen L, Wang F, Wang X, et al. Robust one-tube Ω-PCR strategy accelerates precise sequence modification of plasmids for functional genomics. Plant Cell Physiol. 2013;54:634–642.