
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
As the biological datasets accumulate rapidly, computational methods designed to automate protein function prediction are critically needed. The problem of protein function prediction can be considered as a multi-label classification problem resulting in protein functional annotations. Nevertheless, biologists prefer to discover the correlations between protein attributes and functions. We introduce a multi-label supervised topic model into protein function prediction and investigate the advantages of this approach. This topic model can not only work out the function probability distributions over protein instances effectively, but also directly provide the words probability distributions over functions. To the best of our knowledge, this is the first effort to apply a multi-label supervised topic model to the protein function prediction. In this paper, we model a protein as a document and a function label as a topic. First, a set of protein sequences is formalized into a bag of words. Then, we perform inference and estimate the model parameters to predict protein functions. Experimental results on yeast and human datasets demonstrate the effectiveness of this multi-label supervised topic model on protein function prediction. Meanwhile, the experiments also show that this multi-label supervised topic model delivers superior results over the compared algorithms. In summary, the method discussed in this paper provides a new efficient approach to protein function prediction and reveals more information about functions.
Introduction
Proteins are a kind of macromolecules and the main component of a cell, and thus it is the most essential and versatile material of life. The research on protein functions is of great significance in the development of new drugs, better crops, and even the development of synthetic biochemicals [Citation1]. Along with the fast development of computational methods, machine learning algorithms designed to predict the functional annotations of proteins by using known information, such as sequence, structure, and functional behaviour, have become important long-standing research works in post-genomic era.
Protein function annotation has the nature of subjectivity. At present, there are two popular schemes in protein function annotation: FunCat [Citation2]and gene ontology (GO) [Citation3]. On the basis of solid computer science and biological principles, GO is rapidly being regarded as the most common scheme of functional annotation [Citation1]. GO provides a set of terms for describing genes functions and the relationships between functions, which are classified into three categories: biological process (BP), molecular function (MF) and cellular component (CC) [Citation4]. The GO terms are organized in a direct acyclic graph (DAG), where the function described by a descendant GO term is more specific than its ancestor terms. In this article, the words ‘function’ and ‘GO term’ are used synonymously.
Protein function prediction has become an active research area for bioinformatics in recent years. The most traditional approach is to utilize the protein sequence or structure similarity to transfer functional information. As an example, BLAST [Citation5] is one of the most widely used sequence-based approaches. In addition, there are two kinds of methods in protein function prediction according to the types of input data: feature-based approaches and graph-based approaches. Feature-based approaches process the dataset whose instances have a feature space composed by a fixed set of attribute values. These features can be extracted from amino acid sequence, textual repositories, motifs, the isoelectric point and post-translational modifications. Using these constructed attribute features and annotated proteins, a classifier can be trained first and then be used to predict function annotations for unannotated proteins. On the other hand, graph-based approaches [Citation6–8] assume that the closely related proteins (or genes) share similar functional annotations on the basis of network structure information. In [Citation9], protein interactions were measured by several computational approaches.
From the view of machine learning, the feature-based approach belongs to a classification problem essentially: it processes a set of samples, where each sample is described by a feature space and labelled by one or more labels. Meanwhile, from the view of protein function prediction, a protein annotated with several GO terms can be viewed as a sample and each GO term corresponds to a label. Thus, the solutions of multi-label classification have great potential to protein function prediction [Citation10–14]: Celine Vens et al. [Citation15] discussed three hierarchical multi-label classifiers, which are based on the decision tree algorithm; then, the experiments were carried on 24 datasets from functional genomics; Yu [Citation16] proposed a multiple kernels (ProMK) method to process multiple heterogeneous protein data sources for predicting protein functions; a novel ant colony algorithm was proposed in [Citation17] for hierarchical multi-label classification. Although good results of multi-label classification are achieved by methods mentioned above, the outputs of them are the corresponding label sets for samples in testing set, and the relationships between individual attributes and their most appropriate labels are unknown. In particular, as the number of terms per protein increases, it is difficult for algorithms mentioned above to distinguish the features which are critical for a particular label.
A topic model is a kind of probabilistic generative model that has been widely applied to the domains of text mining and computer vision. Probabilistic latent semantic analysis (PLSA) [Citation18] and latent Dirichlet allocation (LDA) [Citation19] are two typical topic models. In these methods, each document is a mixture of ‘topics’, where each ‘topic’ is a mixture of ‘words’ in a vocabulary. The aim of topic modelling is to discover a topic distribution over each document and a word distribution over each topic, which actually reflects its capacity to cluster for the corpus. This is because the documents with a similar topic probability distribution can be grouped together. Nonetheless, a topic model is not only a clustering algorithm [Citation20]. Specially, the characteristics of LDA have been discussed for multi-label problem in [Citation21]: (1) it transforms the word-level statistics of each document to its label-level distribution; (2) it models all labels at the same time rather than treating each label independently. In multi-label classification problem, LDA can also incorporate a label set into its learning procedure and become a supervised model. Therefore, some variations of LDA that focus on text field have been proposed for multi-label classification [Citation22–24]; one of them has been applied in bioinformatics in the latest studies [Citation25,Citation26]. Especially in [Citation27–29], they introduced PLSA and LDA to predict GO terms of proteins using available GO annotations previously without any protein feature.
Nevertheless, to the best of our knowledge, LDA or its variation has not yet been integrated into protein function prediction based on the multi-label classification. Therefore, we extend a supervised topic model (e.g. Labelled-LDA [Citation22]) to the protein function prediction in this paper.
For multi-label classification, Labelled-LDA associates each label with a corresponding topic directly. Naturally, we consider each protein to be a document [Citation25], and GO terms (topics) are shared by a corpus. Thus, three phases are involved in the prediction process. In the first phase, we organize the protein sequences into a bag of words (BoW); the word can be any basic building block of a protein sequence. In the second phase, Labelled-LDA takes a training protein set with known function as an input of training model. Finally, the unannotated proteins composed of testing set are classified by the fully trained classification model. This model provides the GO term probability distribution over proteins as an output, and each term is interpreted as a probability distribution over words. Therefore, Labelled-LDA provides a new efficient approach for predicting protein functions and revealing more information about functions.
The rest of the article is structured as follows. In the ‘Materials and methods’ section, we describe the computational tools and datasets, with a focus on the BoW of protein sequences and a Labelled-LDA model adopted in this paper; the ‘Results and discussion’ section presents the classification results; we also show the usefulness of Labelled-LDA in two protein function prediction experiments: yeast dataset [Citation30] and human dataset; experimental results show that Labelled-LDA delivers superior results over the compared algorithms; finally, the conclusions are drawn.
Materials and methods
In this section, a computational approach of protein function prediction is discussed. First, we organize the protein sequences into a BoW, and then give a brief description of Labelled-LDA to explain the process of protein function prediction.
The BoW of protein sequence
In feature-based approaches, each protein is characterized as a feature vector. For inferring annotation rules, a machine learning algorithm takes these feature vectors and known annotations as an input to train model [Citation31]. In order to adapt to topic model that has been developed for text mining, we set up a parallelism between text documents and proteins in our framework. A single protein sequence, considering only the amino acid sequence without any header, like for instance in the FASTA format, represents a document [Citation32]. Thus, a dataset of sequences can be considered as a corpus. First, the protein sequence should be organized to a BoW. This article mainly uses the following BoW method:
A protein sequence is composed of one text string, defined on a fixed 20 amino acids alphabet (G,A,V,L,I,F,P,Y,S,C,M,N,Q,T,D,E,K,R,H,W).
We suppose that a protein P is represented as a sequence of L amino acid residues:
(1)
(1)
Where
denotes the residue at chain position 1, and the rest can be done in the same manner.
For example, there is a protein sequence as follows:
Words can be extracted from protein sequences following the so-called k-mers decomposition. A window of size k (suppose k = 2 in Equation (2)) is sliding from the N-terminal to C-terminal, one amino acid at a time. According to the BoW model used in text analysis, the position of a k-mer in the original sequence is not taken into account.
(2)
(2)
Then, the conjoint triad representation of protein P is obtained as follows:
For each protein, the statistics denotes the frequency of amino acid block
in the protein sequence,
. In the end, a 400-dimension vector is obtained to represent this protein, which also is the BoW of this protein.
In addition to the above method, we also adopt the sequential feature extraction (SFE) method in [Citation32]. In the ‘Results and discussion’ section, we take the experimental comparative study between these two methods.
Labelled latent Dirichlet allocation
As mentioned above, a protein is viewed as a document in this paper; a GO term is viewed as a label or topic. By that analogy, the amino acid block of protein sequence composes the vocabulary as in Equation (2),
is a word in BoW. After the description of BoW, the process of protein function prediction in Labelled-LDA is discussed in this section.
Labelled-LDA makes the explicit assumption that the correspondence between the label sets and the shared topics of whole protein collection is one-to-one. Meanwhile, a topic only appears in a protein whose observed labels link to this topic.
From a global perspective, the protein collection corresponds to a set of global shared topics, and each of topics can be represented as a multinomial distribution on the vocabulary with a size of
. Thus, the shared
topics correspond to a
multinomial parameter matrix
.
From a local perspective, the training protein set including proteins can be represented as , the protein
is composed of
observed samples and can be expressed as
. The word index of each sample
comes from the vocabulary
, and thus the protein
can also be represented as
. In addition, the protein
has
observable labels (GO term)
, where the index of each label comes from a label set
. The observable label
can be selected from the global label set
by a sparse binary vector
, where
is defined as
(3)
(3)
The topic-assignment of sample in the protein
is a hidden variable
that needs to be allocated. In the classical LDA model, each document
shares the global topics. But in Labelled-LDA model, on the basis of its
-dimension topic weight vector
, observed label set of document(protein)
limits the topic weight vector to dimension
that is the size of
. In this way, topic weight vector of protein
can be expressed as
, where
is the sign of dot product. Therefore, when a symmetric Dirichlet prior distribution is applied on the topic weight vector
, the hyperparameters of the Dirichlet prior is also limited to
.
The generative process of Labelled-LDA can be described as follows. The corresponding graphical model is shown in .
Figure 1. The graphical model of labelled LDA. The box indicates repeated contents; the number in the lower right corner is the number of repetitions; the grey nodes represent observations; the white nodes represent hidden random variables or parameters; the arrows denote the dependencies.
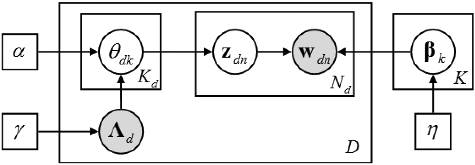
For each topic :
Generate
For each protein
:
For each topic
Generate
Generate
Generate
For each word
Generate
Generate
Learning and inference
As the generative process of Labelled-LDA described above, the only difference between Labelled-LDA and the traditional LDA model is that topic weight vector and its prior hyperparameters are both limited by the prior label set of document. What is more, the relation of labels in global label set and the global shared topics is one-to-one mapping. Put another way, Labelled-LDA introduces supervised learning process to the traditional LDA, and, therefore, can deal with documents and their multiple labels at the same time.
In a Labelled-LDA model, the unknown parameters to be estimated are the global topic multinomial parameters , local topic weight
and local hidden variables
; the known data are the observed word samples
and their joint distribution. As shown in Equation (Equation4
(4)
(4) ),
(4)
(4)
Equation (4) has some implicit relationship such as ,
. For the convenience to compare with traditional LDA, we omit the prior distribution
in Equation (4).
, which is the posterior distribution of unknown model parameters, can be estimated through the joint distribution. It is the core learning task of Labelled-LDA.
Similar to Markov chain Monte–Carlo (MCMC) sampling technique in LDA, we use the Collapsed Gibbs sampling (CGS) to train a Labelled-LDA model. By marginalizing the model parameters from the joint distribution in Equation (4), the collapsed joint distribution of
is obtained, and the dependency between model parameters
and hidden variables
is retained. Then, the prediction probability distribution of a hidden variable
can be computed from that collapsed joint distribution as a transition probability of state-space in the Markov chain. Through Gibbs Sampling iteration, the Markov chain converges to the target stationary distribution after the burn-in time. Finally, by means of collecting sufficient statistic samples from the converged Markov chain state-space and averaging among the samples, we can get posteriori estimates of corresponding parameters.
Due to the limitation of document's observable labels, the predictive probability distribution for the topic-assignment of sample is
(5)
(5)
is the number of samples that were assigned to the topic
and word
, and the current sample is
.
is the number of samples that were assigned to the topic that links to label
in protein
, and the current sample is
.
Datasets
In order to test and verify prediction effect of the proposed method, we utilize two types of datasets. The first one is yeast dataset (Saccharomyces cerevisiae), and the second one is human dataset constructed by ourselves.
Saccharomyces cerevisiae (SC) dataset
The yeast dataset is proposed in [Citation15]. This dataset includes several aspects of the yeast genome, such as sequence statistics, phenotype, expression, secondary structure, and homology. We mainly use the sequence statistics that depends on the amino acid sequence of protein. The feature vector includes amino acid frequency ratios, molecular weight, sequence length and hydrophobicity.
Meanwhile, we adopt the method proposed in [Citation33]: choose a part of terms from the protein functions, where the selected GO terms must annotate at least 30 proteins.
Human dataset
The human dataset is constructed from the Universal Protein Resource (UniProt) databank [Citation34] (released in April 2015). The search string is {‘organism name: Human’ AND ‘reviewed: yes’}, and then the function annotations of proteins are obtained from downloaded UniProt format text file. We exclude the GO terms annotated as ‘obsolete’ or with evidence code ‘IEA’ (Inferred from Electronic Annotation) from the original protein function annotations. Meanwhile, we download the GO file [Citation35] (released in December 2015). There is a DAG relationship between GO terms. According to the true path rule [Citation36], a protein annotated with a descendant term is also annotated with its ancestor terms. By this way, we process our dataset to expand GO terms of a protein. Finally, the same strategy as S. cerevisiae (SC) dataset is adopted for preparing label vectors (choose a part of GO terms from original function set).
The statistics of above two datasets is listed in . ‘Labels’ is the sum of GO annotations of all the proteins; ‘≥30’ denotes the number of terms associated with at least 30 proteins.
Table 1. The statistics of two datasets.
Results and discussion
In contrast to other methods, the experimental results of Labelled-LDA are discussed in this section, and show a better performance.
Predictive performance measures
There are several evaluation criteria for measuring the accuracy of protein function prediction. The result of Labelled-LDA is a list of topics score for each test protein, which means the probability of each term over this protein. Therefore, this list of score can be transformed into two formats: binary and ranking. We use three representative multi-label learning evaluation criteria on the basis of these two formats. For binary results, we use Hamming loss; for ranking results, we use Average precision (AP) and One Error. The definitions of these criteria can be found in [Citation37] and are described below.
Hamming loss evaluates how many times on average a bag label pair is incorrectly predicted (including predicts the incorrect label and the correct label is not been predicted). When the value is 0, the classification performance is achieving the optimal state of ideal. Thus, the smaller the value of Hamming loss is, the better the performance.
AP is a very popular performance measure in information retrieval. In the prediction ranking results, AP calculates the average ratio of the front ranking labels that belong to the truth labels for every label. For multi-label classification, AP measures the performance of learned model in each label. The higher the value of AP is, the better the performance.
One Error is used to compute the number of samples that the first label is incorrect predict among all the test samples. If the value is smaller, the better the performance of multi-label classification is.
Above all, these evaluation criteria reflect the different aspects of model performance. In our experiments, the training set is 70% proteins of the entire dataset, and the left 30% proteins are taken as the testing set. The annotated GO terms of testing proteins are used in the testing process to evaluate the performance of algorithms. Nevertheless, in the training process, these annotated GO terms of testing proteins are treated as unknown. To avoid randomness, the partition and evaluation processes are repeated in 10 independent rounds. Finally, the average results are computed and shown in the next section.
Parameters configurations
In order to ensure the convergence of this model, we set the maximum number of iterations as 1000 times. Besides, the Labelled-LDA learning framework involves two different parameters: and
. The performance of Labelled-LDA under different parameter configurations is shown in .
Figure 2. The values of AP on two datasets under different values of and
. (a) The AP value when
varies from 0.01 to 0.1 with a fixed
; (b) the AP value when
increases from 0.01 to 0.1 with a fixed
.
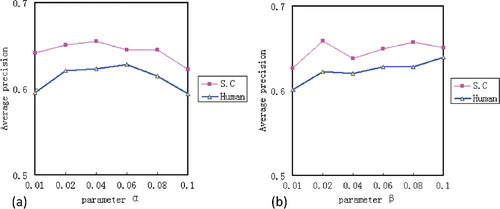
Here, when is fixed to 0.1,
varies from 0.01 to 0.1 with an interval of 0.02; likewise, when the fixed
equals to 0.02,
increases from 0.01 to 0.1 with an interval of 0.02. It shows that the performance of Labelled-LDA reaches the peak in SC dataset by setting the parameter
to 0.04, and in human dataset by setting
to 0.06. Nonetheless, we find that the value of One Error is not best in the above values. As shown in , when we set
to 0.02 in SC dataset and set
to 0.04 in human dataset, the One Error value is better. Meanwhile, it shows that the performance of the Labelled-LDA reaches the peak in SC dataset by setting the parameter
to 0.08, and in human dataset by setting
to 0.1.
Figure 3. The values of One Error on two datasets under different values of . Contrary to AP, the value of One Error is smaller, the better the performance.
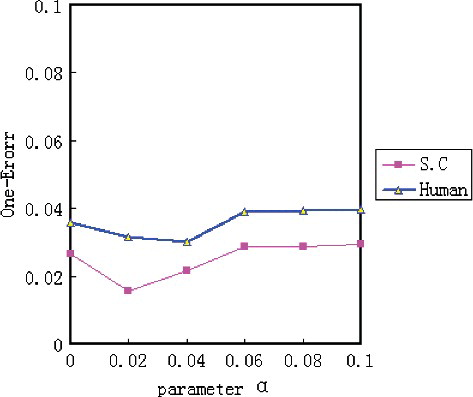
Therefore, in Labelled-LDA experimental setup, we set the parameter to 0.02 and the parameter
to 0.08 for SC dataset. For human dataset, we set
to 0.04 and
to 0.1.
Performance of the Labelled-LDA model
We compare Labelled-LDA with BP-MLL (backpropagation for multi-label learning) [Citation38], HOMER (hierarchy of multi-label classifiers) [Citation39] and IBLR_ML (instance-based logistic regression for multi-label learning) [Citation40] in two datasets, and the BoW method is also 2-mers. These algorithms are classic multi-label classifiers and open source tools including Mulan [Citation41] library.
shows the Hamming loss, AP and One Error values of the four algorithms in SC and human dataset, respectively. For the three evaluation criteria, ↑(↓) indicates the relationship between criterion value and model performance, i.e. the larger (smaller) the value, the better (worse) the performance. The values showed in bold are the best results in the four algorithms.
Table 2. The comparison results (mean ± SD) with three baseline methods on two datasets.
From Table 2, we can observe that Labelled-LDA achieves the best results in AP and Hamming loss criterion. As an example, on AP, the Labelled-LDA achieves 0.5%, 5.6% and 6.3% improvements over BP-MLL, HOMER and IBLR_ML in SC dataset, and achieves 0.6%, 19.8% and 10.4% improvements over BP-MLL, HOMER and IBLR_ML in human dataset. Likewise, on Hamming loss, the Labelled-LDA achieves 30%, 4.3% and 31% improvements over BP-MLL, HOMER and IBLR_ML in SC dataset, and achieves 6.5%, 18.7% and 3.2% improvements over BP-MLL, HOMER and IBLR_ML in human dataset. Nevertheless, on One Error, BP-MLL gets better results than Labelled-LDA. These results are consistent with recent studies that a multi-label learning algorithm rarely outperforms other algorithms on all criteria. Meanwhile, we also find that the overall performance of each algorithm in SC dataset is better than performance in human dataset. This is due mainly to the smaller number of labels in SC dataset. However, the Labelled-LDA can do more to increase AP performance in the case of larger labels, and this is also the prominent feature of protein function dataset. Above all, these results indicate that Labelled-LDA is an effective method for protein function prediction.
Comparison between two BOW methods
According to the description in 'Materials and methods' section, in our prediction framework, each protein sequence that associates with several particular GO terms is treated as a ‘document’ that is composed of words. We adopt two BoW methods in our experiments. Because of the lack of protein sequence data in yeast dataset, we only perform contrast experiment of two BoW methods on human dataset. summarizes the performance of the two BoW methods in terms of AP, One Error and Hamming loss.
Table 3. The comparative results of two BOW methods at the human dataset.
As seen in , the Labelled-LDA achieves almost the same performance in two BoW approaches. The AP value based on the 2-mers is higher than SFE.
A function modelling example of a protein
According to the function prediction process described in 'Materials and methods' section, we can find that the generation of a protein sequence depends on the choice of a GO term (label/topic) and its corresponding amino acid block (words). Put another way, given the relation matrix of topics–words, a protein sequence decides its corresponding GO terms. To better explain this implementation process, we take Homeobox protein Hox-D9 of human as an example, which is shown in . The 2-mers BoW is used in this example.
Figure 4. A function modelling example of a protein. The dotted lines with arrows denote the choosing process of protein sequence, GO terms and the amino acid blocks.
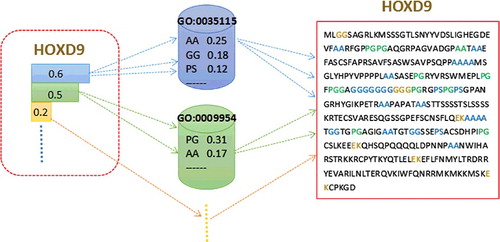
As shown in , in the sequence of protein Hox-D9, the amino acid blocks come from different GO terms, such as an amino acid blocks ‘AA’ chosen from ‘GO:0035115’ and a ‘PG’ chosen from ‘GO:0009954’. For GO term ‘GO:0035115’, the selection probability of ‘AA’ is 0.25. Similarly, the selection probability of ‘GO:0035115’ is 0.6 in protein Hox-D9. The higher the probabilities, the stronger the correlations. It is important to note that the same amino acid block may be chosen from different GO terms, such as a ‘AA’ can be chosen from either ‘GO:0035115’ or ‘GO:0009954’. Therefore, we are not able to draw a conclusion that a protein sequence with more ‘AA’ should be annotated by ‘GO:0035115’. Put another way, the functions of a protein are the results of the joint action of all amino acid blocks rather than a single one.
Above all, we can utilize various feature extraction of protein sequence to construct sequence words when the Labelled-LDA model is used, as long as the words describe the characteristics of protein effectively. Then, the relationship between functions and variety words will be disclosed. This is the greatest strength of Labelled-LDA for protein function prediction. Therefore, we can anticipate that Labelled-LDA is a potential method to reveal a deeper biological explanation for protein functions.
Conclusions
In this paper, protein function prediction problem can be modelled as a multi-label classification task, and Labelled-LDA as a multi-label topic model introduced from natural language processing is successfully used in protein function prediction. For setting up a parallelism between text documents and protein sequences, the following are considered: a set of protein sequences is considered as a corpus of documents and is represented as a BoW; the words can be any basic building blocks of protein sequences. The Labelled-LDA model emphasizes the relationship between sequence words and a given space of GO terms for the purpose of classifying new proteins. The predictive performance of this model is measured by the AP, One Error and Hamming loss and experiments on two dataset show that the Labelled-LDA algorithm is superior to several baseline multi-label classification methods.
There are many problems in the biology domain that can also be formulated as a multi-label classification task. Thus, multi-label topic model is a potential method in many applications of modern proteomics.
Disclosure statement
The authors declare that they have no competing interests.
Additional information
Funding
References
- Pandey G, Kumar V, Steinbach M. Computational approaches for protein function prediction: a survey. Twin Cities: Department of Computer Science and Engineering, University of Minnesota; 2006.
- Ruepp A, Zollner A, Maier D, et al. The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res. 2004;32(18):5539–5545.
- Gene Ontology Consortium. The gene ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32(Suppl 1):D258–D261.
- Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–29.
- Altschul SF, Madden TL, Schäffer A, et al. Gapped BLAST and PSIBLAST: a new generation of protein database search programs. Nucleic Acids. 1997;25:3389–3402.
- Yu G, Fu G, Wang J, et al. Predicting protein function via semantic integration of multiple networks. IEEE/ACM Trans Comput Biol Bioinf. 2016;13(2):220–232.
- Piovesan D, Giollo M, Leonardi E, et al. INGA: protein function prediction combining interaction networks, domain assignments and sequence similarity. Nucleic Acids Res. 2015;43(W1):W134–W140.
- Piovesan D, Giollo M, Ferrari C, et al. Protein function prediction using guilty by association from interaction networks. Amino Acids. 2015;47(12):2583–2592.
- Pellegrini M, Haynor D, Johnson JM. Protein interaction networks. Expert Rev Proteomics. 2004;1(2):239–249.
- Cerri R, Barros RC, de Carvalho AC. A genetic algorithm for hierarchical multi-label classification. Proceedings of the 27th annual ACM symposium on applied computing. Trento: ACM; 2012. p. 250–255.
- Clark WT, Radivojac P. Analysis of protein function and its prediction from amino acid sequence. Proteins Struct Funct Bioinf. 2011;79(7):2086–2096.
- Zhang ML, Zhou ZH. A review on multi-label learning algorithms. IEEE Trans Knowl Data Eng. 2014;26(8):1819–1837.
- Wu JS, Huang SJ, Zhou ZH. Genome-wide protein function prediction through multi-instance multi-label learning. IEEE/ACM Trans Comput Biol Bioinf. 2014;11(5):891–902.
- Wu JS, Hu HF, Yan SC, et al. Multi-instance multilabel learning with weak-label for predicting protein function in electricigens. BioMed Res Int. 2015; 2015(1):1–9.
- Vens C, Struyf J, Schietgat L, et al. Decision trees for hierarchical multi-label classification. Mach Learn. 2008;73(2):185–214.
- Yu G, Rangwala H, Domeniconi C, et al. Predicting protein function using multiple kernels. IEEE/ACM Trans Comput Biol Bioinf. 2015;12(1):219–233.
- Otero FEB, Freitas AA, Johnson CG. A hierarchical multi-label classification ant colony algorithm for protein function prediction. Memetic Comput. 2010;2(3):165–181.
- Dumais ST. Latent semantic analysis, Ann Rev Inf Sci Technol. 2004;38(1):188–230.
- Blei DM, Ng AY, Jordan MI. Latent Dirichlet allocation. J Mach Learn Res. 2003;3:993–1022.
- Lin L, Lin T, Wen D, et al. An overview of topic modeling and its current applications in bioinformatics. SpringerPlus. 2016;5(1):1608.
- Rubin TN, Chambers A, Smyth P, et al. Statistical topic models for multi-label document classification. Mach Learn. 2012;88(1–2):157–208.
- Ramage D, Hall D, Nallapati R, et al. Labeled LDA: a supervised topic model for credit attribution in multi-labeled corpora. Proceedings of the 2009 conference on empirical methods in natural language processing. Vol. 1. Singapore: Association for Computational Linguistics; 2009. p. 248–256.
- Ramage D, Manning CD, Dumais S. Partially labeled topic models for interpretable text mining. Proceedings of the 17th ACM SIGKDD international conference on Knowledge discovery and data mining, San Diago, CA: ACM. 2011. p. 457–465.
- Yang Y, Downey D, Boyd-Graber J, et al. Efficient methods for incorporating knowledge into topic models. Conference on Empirical Methods in Natural Language Processing. September 17-21; Lisbon, Potugal; 2015. p. 308–317.
- La Rosa M, Fiannaca A, Rizzo R, et al. Probabilistic topic modeling for the analysis and classification of genomic sequences. BMC Bioinformatics. 2015;16(6):S2.
- Bisgin H, Liu Z, Fang H, et al. Mining FDA drug labels using an unsupervised learning technique-topic modeling. BMC Bioinformatics. 2011;12(10):S11.
- Pinoli P, Chicco D, Masseroli M. Enhanced probabilistic latent semantic analysis with weighting schemes to predict genomic annotations. 2013 IEEE 13th international conference on Bioinformatics and Bioengineering (BIBE).Chania, Greece: IEEE, 2013, p. 1–4.
- Masseroli M, Chicco D, Pinoli P. Probabilistic latent semantic analysis for prediction of gene ontology annotations. The 2012 international joint conference on neural networks (IJCNN). Brisbane: IEEE; 2012. p. 1–8.
- Pinoli P, Chicco D, Masseroli M. Latent Dirichlet allocation based on Gibbs sampling for gene function prediction. 2014 IEEE conference on computational intelligence in bioinformatics and computational biology. Honolulu, USA: IEEE, 2014,p. 1–8.
- Leander S, Celine V, Jan S. HMC software and datasets [ Internet]; 2009. Available from: https://dtai.cs.kuleuven.be/clus/hmc-ens/
- Krogel MA, Scheffer T. Multi-relational learning, text mining, and semi-supervised learning for functional genomics. Mach Learn. 2004;57(1–2):61–81.
- Pan XY, Zhang YN, Shen HB. Large-scale prediction of human protein–protein interactions from amino acid sequence based on latent topic features. J Proteome Res. 2010;9(10):4992–5001.
- Chua HN, Sung WK, Wong L. Exploiting indirect neighbours and topological weight to predict protein function from protein–protein interactions. Bioinformatics. 2006;22(13):1623–1630.
- Apweiler R, Bairoch A, Wu CH, et al. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2004;32(Suppl 1):115D.
- Gene Ontology Consortium. Gene ontology [ Internet]; 1999–2015. Available from: http://geneontology.org/
- Valentini G. True path rule hierarchical ensembles for genome-wide gene function prediction. IEEE/ACM Trans Comput Biol Bioinf. 2011;8(3):832–847.
- Zhang ML, Zhou ZH. A review on multi-label learning algorithms. IEEE Trans Knowl Data Eng. 2014;26(8):1819–1837.
- Zhang ML, Zhou ZH. Multilabel neural networks with applications to functional genomics and text categorization. IEEE Trans Knowl Data Eng. 2006;18(10):1338–1351.
- Tsoumakas G, Katakis I, Vlahavas I. Effective and efficient multilabel classification in domains with large number of labels. Proceedings of ECML/PKDD 2008 workshop on Mining Multidimensional Data (MMD’08) . Antwerp, Belgium: MMD2008, 2008, p. 30–44.
- Cheng W, Hüllermeier E. Combining instance-based learning and logistic regression for multilabel classification. Mach Learn. 2009;76(2–3):211–225.
- Tsoumakas G, Katakis I, Vlahavas I. Mining multi-label data. In Oded Maimonn Lior Rokach editors. Data mining and knowledge discovery handbook. New York, NY: Springer US, 2009, p. 667–685.