
Abstract
The walnut is an excellent food product with abundant nutrients. However, the inference of its population structure has been hindered by the intricate phylogenetic relationships among Juglans species. In this study, RAD sequencing was conducted to investigate the genetic variation and population structure among 41 walnut cultivars from Southwestern Sichuan (SS, n = 10), Eastern Sichuan (ES, n = 26) and Northern China (NC, n = 5). The resulting 6357 single-nucleotide polymorphisms divided the 41 walnut cultivars into two major groups corresponding to the ES and SS gene pools, and NC was clustered with the ES1 subgroup. Additionally, two cultivars, WB01 and SMJ, were reclassified to correct their previous morphological classifications. The migration rate from ES to SS was greater than that in the reverse direction, and the genetic differentiation between the ES and SS populations was high. Moreover, the estimated expected heterozygosity (He = 0.308) and polymorphism levels (Pi (π) = 0.030) of the SS group were greater than those of the ES group. Similarly, the average genetic distance of the SS population (0.563) was higher than that of the ES population (0.522), and the rate of linkage disequilibrium decay in the SS population was faster. All the results indicate that the SS population has greater genetic diversity and is more primitive than the ES group. The present study provides a theoretical basis for the division, conservation, and utilization of walnut germplasm resources, and these findings can be applied in breeding programs to obtain high-quality cultivars of Juglans.
Introduction
The walnut (Juglans) has health-related and economic importance worldwide [Citation1,Citation2]. In China, walnut species are distributed between 21.48° and 44.90° north latitude and 75.25° and 124.35° east longitude. There are six species in the walnut genus (Juglans), namely, J. regia, J. sigillata, J. cathayensis, J. mandshurica, J. cordiformis, and J. hopeiensis. However, J. regia L. (the Persian or English walnut) and J. sigillata Dode (the Chinese iron walnut) are the most commercially important species for nut production in China [Citation3–5]. The introduction of J. regia to China, Russia and Eastern Europe is believed to have occurred after the domestication by ancient tribes in Iran and Afghanistan [Citation6], but recent research suggests that Xinjiang Province in China is probably another origin of J. regia [Citation7]. With widespread cultivation around the world, this species grows well in virtually any region with a temperate climate. J. sigillata, with original unique features of semi-domestication, is mainly distributed in southwestern China, Vietnam, and Burma [Citation8]. In Sichuan Province, as a result of the influence of geographical latitude and topography, the zonal and vertical directions of climate change are pronounced, and the difference between the East and the West is very large. According to the records of walnut distribution, the Sichuan area is mainly divided into the Western Sichuan region and the Eastern Sichuan region, with the Minjiang River and Daxiangling Mount forming the boundary. J. sigillata accounts for most of the walnut population in Western Sichuan, whereas J. regia predominates in Eastern Sichuan.
In addition, because of the foehn effect and topographical enclosure, the valleys in the mountainous areas of Southwestern Sichuan (including Liangshan Yi Autonomous Prefecture and Panzhihua) have an especially dry and hot climate, and many original forests of J. sigillata populate this region [Citation9]. To date, many cultivars have been bred for agricultural production. However, the genetic diversity of Juglans is ambiguous and complex, and the inference of its population structure has been hindered by the intricate phylogenetic relationships among Juglans species. Thus, discovering an effective molecular marker is critical for revealing the genetic diversity and breeding high-quality walnut germplasms.
Although walnut species have been cultivated for centuries, their breeding is recent, and only a few systemic studies on their population structure have been reported [Citation10,Citation11]. Furthermore, the genetic similarity among walnut cultivars has increased, and classification based on phenotypic performance has become increasingly difficult because several elite breeding parents have been widely used. Taxonomists have particular difficulties clarifying additional species that are not widely accepted and show morphological diversity because of the considerable variation between J. regia and J. sigillata under different biogeoclimatic conditions. Accurate information on the population structure and genetic variation in Sichuan is important for scientific management measures of walnuts, but the previous studies have provided discordant patterns of its genetic evolution [Citation3–5,Citation10,Citation12]. These conflicting results mean that the population structure and evolution of walnut species in Sichuan are uncertain, thus, additional markers throughout the entire genome (including coding regions and non-coding regions) need to be discovered. New markers might be useful for detecting the walnut population structure, quantifying the extent of spatial demographic changes, and discovering imprints of variation, all of which are important for an effective breeding program.
Next-generation sequencing technologies have significantly reduced the sequencing cost, and SNP (single-nucleotide polymorphism) markers have become increasingly popular for their abundance in the genome and very simple genetic mode [Citation13]. The walnut genome was recently sequenced and reported, and more SNP markers have been developed by whole-genome or transcriptome re-sequencing [Citation4,Citation14]. By the end of 2019, GenBank (National Center for Biotechnology Information, NCBI), the public repository for DNA sequence data in the USA, had listed 43,454 nucleotide sequences for J. regia, including nuclear and chloroplast genes, and expressed sequence tags (ESTs). Restriction site-associated DNA sequencing (RAD-Seq), a sequencing-by-synthesis technique based on next-generation sequencing (NGS), has proved to be particularly useful in non-model species because it combines the advantages of low cost and high throughput [Citation15,Citation16]. In species with available reference genomes or close relative genomes, RAD-Seq is a cost-effective alternative to whole-genome resequencing (WGRS) for population genomic scans [Citation16–18].
In the present study, we focused on three walnut populations in Eastern Sichuan province, Southwestern Sichuan province and Northern China. These populations were independently derived from J. regia and J. sigillata populations with some direct gene flow. On the basis of their history, we hypothesize that most of their adaptive evolution in Sichuan is the result of selection on standing genetic variation in their RAD sequences. Thus, the genomes were sequenced to more than 10-fold coverage using RAD-Seq to construct paired-end (PE) libraries. Then, all clean reads were mapped against the refined 700 Mb reference genome from ‘Chandler’ [Citation19]. After that, SNPs were identified for the genomic analysis of 41 samples. Various analyses of the samples were carried out, including nucleotide diversity, allele frequencies, maximum-likelihood tree, genotype structure, linkage disequilibrium, and gene flow. The objectives of the present study were to (1) gain knowledge of RAD markers’ reproducibility across populations at a medium sequencing depth, (2) create genomic resources for walnut research, (3) use SNP markers to determine the genetic structure and gene flow among the Eastern and Southwestern Sichuan regions, with Northern China cultivars used as a control group, and (4) compare walnut species’ genotypic variation and population genetic structure among geographical climates.
Materials and methods
Plant materials
A total of 41 fresh tender leaf samples were collected from the healthy walnut cultivars used for RAD-Seq (Supplemental Table S1, ), including 5 samples (J. regia) from Northern China (NC group, including Liaoning, Henan, Shandong, and Xinjiang province), 26 samples (J. regia) from Eastern Sichuan province of China (ES group) and 10 samples (J. sigillata, except for YYZ) from Southwestern Sichuan province of China (SS group). Representative nuts were selected from each region to more intuitively and concretely observe the phenotypic differences among the three regional samples ().
Figure 1. Detailed information for 41 Juglans cultivars collected in the present study. (a) Geographic distribution of the Juglans populations in the present study. The solid circles in black, red and blue represent the origin of cultivars in the NC, ES and SS regions, respectively. (b) Phenotypic differences between the representative varieties of the three groups. MY has deep pits or seal-like depressions on the surfaces of the nuts and dark-colored kernels with tough septa, whereas XX2 has wrinkled nut surfaces, light-colored kernels and papery septa. The nut surface of CZ1 is relatively more similar to that of XX2. Source: Prof. Wei Gong.
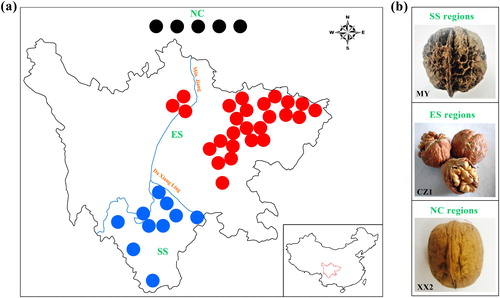
Table 1. P-distance among samples in three genetic groups of Juglans.
DNA extraction, RAD-seq library preparation and sequencing
Genomic DNA (gDNA) was extracted from fresh tender leaves using a DNA extraction kit following the manufacturer’s instructions (TAKARA, Dalian). Since high-quality gDNA is required in the RAD genotyping technique, its concentration and purity, as determined by the ratios of absorbance at 260/280 nm and at 260/230 nm, were quantified by a NanoDrop ND-2000 spectrophotometer (Thermo Fisher Scientific, Waltham, Massachusetts, USA) and Qubit 2.0 Fluorometer (Invitrogen, Thermo Fisher Scientific, Waltham, Massachusetts, USA). This procedure ensured high-quality samples and comparable DNA concentrations for further work.
New restriction-site-associated DNA (RAD) tag generation and typing were carried out using the method described by [Citation20], which greatly simplifies and enhances genetic analysis through the use of an Illumina Genome Analyzer sequencer and the incorporation of nucleotide barcodes for sample tracking. RAD libraries were generated using the restriction enzyme Taq I using a method described by [Citation21]. Paired-end (PE 150) sequencing with an average insert size of 410 bp was carried out using an Illumina HiseqTM 4000 platform for a total throughput of four lanes at the Majorbio Genomics Sequencing Laboratory (Shanghai, China). Finally, 41 multiplexed sequencing libraries were constructed, and each DNA sample was assigned a unique nucleotide multiplex identifier (MID) for barcoding.
Inferring genotypes and discovering markers
Raw sequence reads were filtered in the following steps. Reads with a barcode that did not match one of the expected barcodes (i.e. a sequencing error in the barcode) and sequence reads of poor overall quality were removed from the dataset. The clean reads were then sorted by barcode and aligned to the walnut genome using Bowtie [Citation22] with a maximum of two mismatches within the first 28 bases and a summed base quality for all mismatches in the read that was no greater than 70. Following alignment, the read counts of four possible nucleotides at each nucleotide site were tallied for each individual. The sequence reads were trimmed to 85 nucleotides from the 3′-end to ensure that more than 90% of the nucleotides had a quality value above Q30 (equal to 0.1% sequencing error) and more than 99% were above Q20 (equal to 1% sequencing error). The trimmed reads were clustered into read tags (RAD-tags) by sequence similarity using the software program Stacks version 1.13 [Citation23] to produce unique candidate alleles for each RAD locus. Reads were further trimmed by removing the portion of the sequence within the restriction enzyme recognition site (i.e. a maximum base-pair mismatch of two, and only one was allowed) since any nucleotide polymorphism in this area would result in the absence of RAD tags, and the inclusion of these data would underestimate the total nucleotide diversity.
The quality of the sequence distributions deduced from the reference J. regia sequence ‘Chandler’ (https://www.ncbi.nlm.nih.gov/genome/?term=Juglans%20regia) was controlled by BWA (parameter:mem -t 4 -k 32 -M) [Citation24], and the consistency between observed and expected patterns was checked before performing the actual genome scan. Then, the comparison results were filtered by SAMTOOLS [Citation25] (parameter: 11 rmdup), with more than 10-fold coverage and Qual values greater than 50.0 for both samples. The quality of the analysis was ensured by filtering all of the SNPs with the following standards: the loci whose Maximum Aerobic Function (MAF) was less than 0.05 must be removed, and screening genotype coverage must be above 100%. Finally, polymorphism SNPs were applied in the follow-up analysis. According to the reference genome annotation information from the ANNOVAR software, the SNP annotations were obtained for each locus in exonic regions that would cause a mutation in protein translation annotation.
Construction of walnut phylogeny and population structure analyses
The reads were further trimmed by removing the portion of the sequence within the restriction enzyme recognition site, since any nucleotide polymorphism in this area results in the absence of RAD tags. Pairwise genetic distances were calculated using the software PowerMarker 3.25 [Citation26]. Diploid genotypes at each nucleotide site for each individual were determined in a maximum-likelihood statistical framework [Citation18]. The maximum-likelihood tree was generated by MEGA5 using genetic distance data [Citation27]. The program STRUCTURE v2.3.4 was used to infer genotype structures [Citation28]. A population number (K value) ranging from 1 to 10 was assumed to identify the optimal K with the highest log-likelihood [Citation29]. Principal Coordination Analysis (PCoA) was performed using GCTA software [Citation30], and SNPs with >20% of missing data and a minor allele frequency (MAF) of <0.05 were excluded from further analysis. Because the number of members of the NC group was not sufficient to exactly estimate the linkage disequilibrium (LD), the correlation coefficient values (r2) of the alleles were only calculated using TASSEL [Citation31] and VCFtools [Citation32] to measure the LD level in the ES and SS populations. The average r2 value was calculated for each length of distance, and LD decay figures were drawn for the two populations of walnut genotypes.
Genetic diversity between the ES and SS populations
A sliding window approach was applied to quantify the polymorphism levels (Pi (π), pairwise nucleotide variation as a measure of variability), selection statistics (Tajima’s D, a measure of selection in the genome), and genetic differentiation between the ES and SS populations (FST). In total, window sliding of 88,121 bp was performed in 22,030 bp steps, the SNPs for each population are described in the subsection ‘SNP calling’. Additionally, we developed a series of PERL scripts that consider genotype frequencies in the two groups and calculate the values of Pi (π) and Tajima’s D for both groups and FST between the two populations following the formulas for these statistics [Citation33–35]. The expected heterozygosity (He) was also estimated using Arlequin 3.5.1.3 with a cutoff of 0.2 for the marker missing data rate [Citation36].
Migration patterns among the populations
The coalescence-based program MIGRATE-N 3.2.6 [Citation37,Citation38] was used to investigate the gene flow between populations with the AIC model of maximum likelihood and estimate the mutation-scaled effective population size θi = 4 Neμ, where Ne is the effective population size, and μ is the mutation rate per generation per locus, as well as mutation-scaled migration rates M = m/μ, where m is the immigration rate per generation among populations.
Results and discussion
RAD sequencing and SNP calling
The genetic, geographic and morphological diversity of walnuts were represented by 41 samples (Supplemental Table S1), and ‘Chandler’ (J. regia) was used as the reference sequence. All the walnut samples were used together for genome sequencing by using the RAD strategy to exclude species or restriction enzyme interference and ensure the quality of sequencing. As a result, the average percentage of sequence reads that mapped to the reference genome was 88.51% (Supplemental Table S2), and 7,360,659 putative SNPs were obtained. The number of clean reads for each individual drastically varied between 7,018,308 and 22,424,696. After applying successive filters, 160,309 (2.18%) SNPs were deemed adequate. The 160,309 SNPs were aligned against the 700-Mb reference genome of J. regia ‘Chandler’, the Project number of this dataset is PRJEB36778 in EVA database. All of the SNPs were mapped across 4743 of the 105,811 contigs in ‘Chandler’. The highest representation was obtained for LIHL01055144 (1116 SNPs), and 629 contigs were detected with only 1 SNP. Moreover, 158,049 (98.59%), 928 (0.58%), 479 (0.30%), 764 (0.48%) and 89 (0.05%) SNPs were located in the intergenic, exonic, downstream, upstream and ncRNA-exonic regions, respectively. Of the 928 exonic SNPs, 319 (34.38%), 375 (40.41%), 10 (1.08%) and 224 (24.13%) were synonymous SNVs (single-nucleotide variants), nonsynonymous SNVs, stop-gain SNVs and unknown, respectively. Finally, 6357 SNPs with the highest scores in ADT (assay design tool) were used for genotyping, with an average density of one SNP per 4.4 kp. These loci had more than 10-fold coverage, and the mean coverage of each sample ranged between 23× and 64×, which is higher than the average RAD-Seq depth in studies on cucurbit [Citation16], sweet potato [Citation39], Liriodendron [Citation40] and several other species. All these results suggested that good reliability and stability could be obtained from the subsequent genetic analyses of the 41 samples in the present study.
Table 2. Genetic diversity of the ES and SS groups of Juglans.
The use of NGS to explore population genomic features in non-model species is still in its infancy, and some researchers have achieved this goal by WGRS, GBS, or RAD-Seq. In this regard, bioinformatic pipelines for model species can be used in data processing [Citation41]. However, this approach is still prohibitively expensive or difficult for the majority of non-model species. Thus, highly important and urgently needed research is carried out on population genomics by using RAD-Seq to reduce complexity. In a previous study, [Citation16] identified 3226 SNPs and supported the wide use of RAD-Seq in population genomic studies for non-model species, even with low-to-medium sequencing efforts. [Citation42] collected J. hopeiensis to construct phylogenetic trees for Asian Juglans species using RAD-seq. Owing to restriction fragment length bias and the highly variable read depths at RAD loci, increasing the sequencing coverage is not always effective or necessary [Citation20,Citation43]. Several reports have postulated that low-depth RAD-Seq can be very useful for dissecting the population genomics of non-model species as long as the SNP markers used are evenly distributed, in general [Citation16]. Walnuts are well-known to be dioecious and multi-outcrossing species that naturally hybridize, so the genotypic diversity of the population is maintained [Citation44]. For studying genetic diversity, SNPs that are established throughout the colony DNA pool are better than markers developed from a single individual [Citation45].
Genetic distance and diversity
The genetic diversity of a population is the product of long-term evolution and a prerequisite for its adaptation and evolution, and related research has become increasingly in-depth in recent years [Citation45]. In the present study, the genetic diversity among the natural collections was investigated using 6357 SNPs with ≥100% successful calls (anchored and unanchored), and the P-distances—the proportion of nucleotide sites at which two compared sequences are different—were calculated. The genetic dissimilarity coefficient varied between genotypes, and the 41 cultivars had an average P-distance of 0.554, with the greatest value (0.856) found between TH0035 and MY and the minimum value (0.053) found between XX2 and QQS (Supplemental Table S3). Additionally, the 5 cultivars in the North China (NC) group, the 26 cultivars in the Eastern Sichuan (ES) group and the 10 cultivars in the Southern Sichuan (SS) group had average P-distances of 0.309, 0.522 and 0.563, respectively. The average genetic distance (P-distance) between samples in the SS population is higher than that in the ES population, indicating a greater difference between samples in SS. For the reference genome (‘Chandler’), P-distance comparisons revealed less divergence among cultivars in the NC group (0.397) than among cultivars in the ES group (0.542) and SS group (0.681). For the NC, ES, and SS groups (), the minimum P-distance (0.464) was between NC and ES, and the maximum P-distance (0.665) was between NC and SS. The total mean expected heterozygosity (He) was 0.286, and the He values for the ES gene pool (n = 26) and SS gene pool (n = 10) were 0.274 and 0.308, respectively. The SS (n = 10) group, which is composed of cultivars, breeding lines, and landraces, had a higher value (0.030) of polymorphism (Pi (π)) than the ES (n = 26) group, whose Pi (π) was 0.027. Fewer samples of the SS population were used relative to those of the ES population, but both of them have high He and Pi (π), indicating that the genetic diversity is higher in the SS population. For the entire set of samples, the Pi (π) and Tajima’s D indices were estimated to be 0.298 and 1.648, respectively. The genetic differentiation (FST) between the ES and SS populations was estimated to be 0.285 (p < 0.01) (). Similarly, a high genetic differentiation (0.217) was obtained among the natural walnut populations across the Eurasian continent [Citation46]. These results indicated that a strong phylogeographic pattern was formed in the walnut evolutionary process.
Population structure and geographic differentiation
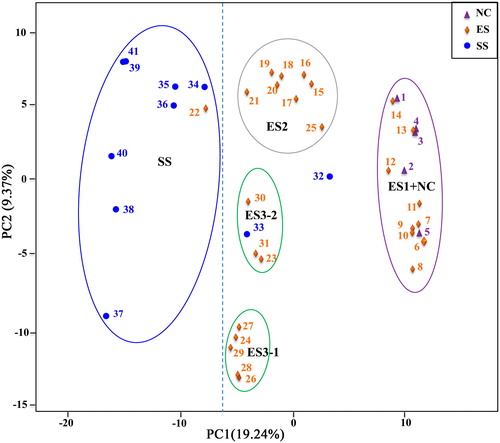
With the aim of elucidating the phylogeny of walnut species beyond the results of previous studies [Citation3,Citation9], the genetic relationships between walnut cultivars were derived using the panel of 6357 SNPs with ≥100% successful calls. The results show a large genetic dissimilarity between gene pools and relatively small genetic dissimilarity within each gene pool, as verified by the maximum-likelihood tree built using the rate of base-pair substitution (). A phylogenetic tree was generated for the group of 41 genotypes (aspect ‘Chandler’), and the genotypes were clustered into two major groups. Group I was mainly formed by the Southwestern Sichuan group (SS) and could be further divided into SS1 and SS2 subfamilies. Additionally, Group II contained the Eastern Sichuan (ES) and Northern China (NC) groups, and the ES group could be clustered into ES1, ES2 and ES3 subfamilies. Moreover, ‘Chandler’ was grouped together with ES1 but distinguished from ES2 and ES3. Then, the first two principal components (PCs) were calculated to further understand the genetic relationships among the 41 walnut cultivars (). The first axis (PC1), which explained the largest molecular variation in the data, consistently separated the cultivars by gene pool with a value of 19.21%. Similar to the cluster analysis, principal components analysis (PCoA) revealed a clear structuring of the genotypes based on the ES and SS genotypes, which were separated by PC1. The ES group could also be divided into three subfamilies, and the ES1 and NC genotypes were still clustered together. However, ES3 was further separated into ES3-1 and ES3-2 genotypes by PC2.
Figure 2. Maximum-likelihood trees representing the relationships among Juglans germplasms based on 6357 SNPs.
Note: The solid circles represent the SS genotypes, which are shown in dark blue (SS2) and light blue (SS1), diamonds represent the ES genotypes, which are shown in purple (ES1), orange (ES2) and green (ES3), triangles represent the NC genotypes, which are shown in purple.
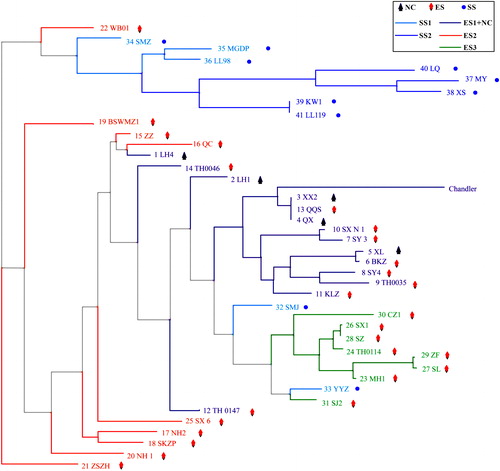
Figure 3. Principal coordinate analysis (PCoA) of the 41 walnut genotypes with 6357 SNPs.
Note: The PCoA results from the first two statistically significant components are shown (p < 0.05). The proportion of the variance explained by each PC is shown in parentheses along each axis. The groups include NC (1–5), ES (6–31) and SS (32-41).
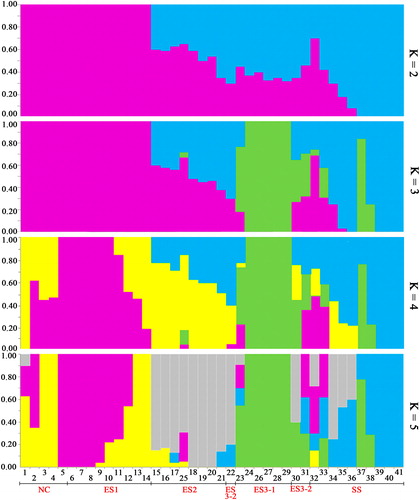
Walnuts that have grown in close association with humans have been subjected to unique evolutionary and ecological processes [Citation8]. For example, artificial selection led to morphological changes in cultivated populations, and the dispersal of humans expanded the natural range of the species, this range expansion can lead to sympatry and hybridization with other allopatric congeneric species. Various terrains and climates coexist in Sichuan province. To understand the geographic structuring of genetic diversity, the relationship between genetic structures among the 41 walnut populations was evaluated. An admixture analysis was performed, and the minimum CV-error (cross-validation error) was obtained when the value of K was 2 (). Thus, K = 2 was selected as the best partition for the origin-based subdivision of the Eastern Sichuan group (purple) and Southwestern Sichuan group (blue), and the NC group still belonged to the ES group (). This indicates that significant variation is associated with the species type and geographic origin of J. regia and J. sigillata, which is consistent with previous studies [Citation47,Citation48]. Higher K values were fitted to the true differentiation history of the populations to obtain more detailed information about the population structure of the 41 walnut cultivars. From the results, ES3 was clearly observed as an additional group at K = 3, and the five cultivars of ES3-1 (Nos. 24, 26, 27, 28, and 29) maintained the same assignment when the K value was higher than 2. This group mainly contains hybrid cultivars, although there is no evident explanation for this split, it probably resulted from breeding programs involving directed crosses. Four of these cultivars are hybrid germplasms of J. sigillata and J. regia (Supplemental Table S1), whereas No. 24 is a traditional (landrace) cultivar. At K = 4, the NC group (in pure purple) was subdivided into two clusters, represented by the genotypes of XL, which was introduced from Shandong province, and XX2, which was introduced from Xinjiang province. It is possible that the different climatic characteristics of Shandong province and Xinjiang province caused a certain variation in the offspring. At K = 5, ES2 (in gray) was obtained as an additional group, including eight landraces from the ES population with almost pure blood, which was heterozygous when K was below 5. This population was probably caused by the unique climate and geography in the ES region and formed by crosses between the native species J. sigillata and J. regia. Compared with WB01, SMJ was closely related to J. regia from the ES population, which is not consistent with the result found by the government of Sichuan Province. Our results show that SMJ does not belong to J. sigillata (Supplemental Table S1) but to J. regia, which is consistent with the previous study [Citation5]. A similar problem also exists for WB01 because of fuzzy background information. Numerous factors of subjective and external environments can influence apparent morphological classifications [Citation49]. Thus, more accurate identification results, coupled with the present molecular study, could modify some morphological classification results.
Figure 4. Population genetic structure of the 41 walnut genotypes with 6357 SNPs.
Note: The population structure inferred by the Bayesian approach based on RAD-SNPs for K values ranging from 2 to 5. Each cultivar is represented by a vertical line that is divided into colored segments according to the proportion of the division identified for 2–5 subpopulations. The groups include NC (1–5), ES (6–31) and SS (32–41).
Linkage disequilibrium and migration rate estimation
Characterizing the patterns of linkage disequilibrium (LD) is critical for the design of association studies, interpretation of association peaks, and the transfer of alleles in marker-assisted selection [Citation50]. To characterize the mapping resolution for genome scans, we quantified the average extent of linkage disequilibrium (LD) decay (). LD decays were determined by using the squared correlations of allele frequencies (R2) against the distance between polymorphic sites in the SS and ES genotypes. Generally, shorter LD decay represents faster decay, a lower linkage between SNPs, and decreased anthropogenic domestication. The calculated results show that LD decays rapidly in walnuts, with the correlation coefficient R2 decreasing to half of its maximum value at distances of 125 and 75 kb for the Eastern Sichuan and Southwestern Sichuan varieties, respectively. The rate of LD decay in the SS population was faster than that in the ES population. This suggests that the probability of linkage between SNPs in the SS population is smaller, whereas the genetic diversity decreased faster during the process of ES population domestication. The results also confirm that the SS population experienced weaker effects of domestication and selection. Furthermore, the SS population shows greater genetic diversity, so it is more primitive than the ES population. This reveals that the SS population has great potential for further development and improvement in breeding programs [Citation19]. Except for YYZ, samples from the SS population generally belong to J. sigillata, whereas the ES population mainly contains J. regia. This indicates that J. sigillata may be in a relatively primitive evolutionary state. Therefore, J. sigillata may have originated independently in the Southwest Sichuan region. The walnut perhaps survived the last glaciations in some refugia, and all the modern distribution probably was affected by humans [Citation51]. Some previous studies also suggested that J. regia and J. sigillata should be considered as a single species, and J. sigillata might be an ecotype of J. regia [Citation42,Citation52]. So further research could uncover more information about the phylogenetic relationships among them.
Moreover, it is worth noting that the estimation of migration rates has often been highly imprecise in many previous studies [Citation53]. This lack of precision is caused by differences that can be attributed to a number of factors: the variation in sample size and a lack of experimental randomization are likely candidates. Experimental randomization was not carried out for genotyping, which may have skewed the estimations of relatedness. The estimated migration rates show that the effective population size of the ES group (θi = 0.1570) is greater than that of the SS group (θi = 0.1186) (Supplemental Table S4). The trend of gene flow among the three groups was from NC to ES to SS (), which resulted in a gradual variation from north (purple) to south (blue). The highest migration rates were found among the NC and ES groups, and they tend to have a symmetrical distribution (Supplemental Table S5 and ), whereas the lowest migration rates and extremely asymmetrical distributions were observed between the NC and SS groups. Moreover, the ES group was mainly consisted of the J. regia and its hybrid offspring with J. sigillata, but the SS group was mainly the J. sigillata individuals (Supplemental Table S1). The migration from ES to SS was significantly greater than that in the reverse direction. Consistent with this result, a high asymmetric historical gene flow was detected among these two species, which occurred predominantly from J. regia into J. sigillata [Citation52]. Based on the above results, we suggest that the walnuts in Sichuan province were likely derived from the North China population. Moreover, the reference genome ‘Chandler’—an important commercial variety in California and a good representative of common walnut germplasm—was closer to the cultivars from NC and the ES1 subgroup, which confirms the comprehensive exchange of common walnuts in China (or even worldwide) throughout history, thus leading to the unclear spatial genetic structure of J. regia in China [Citation8,Citation12]. Furthermore, a higher purple proportion was obtained when the cultivar was closer to the northeast of the ES group. However, there was no obvious variation in the area of the SS group. These results indicate that the ES group exhibits significant geographical patterns and more anthropogenic domestication, whereas more primitive species might exist in the SS group. Walnuts are a wind-pollinated species with heavy seeds. Previous studies involving wind-pollinated forest trees have demonstrated that pollen can travel over 19 km [Citation54]. Additionally, Southwestern Sichuan has underdeveloped agriculture and transportation and complex terrain, which may restrain pollen movement and facilitate the preservation of a great variety of genetic resources in these walnuts [Citation55]. The results indicate that the southwest of China is one of the centers of origin of walnuts, which agrees with other studies [Citation6–8]. Moreover, this result is consistent with the structure analysis, which shows that the migration rates between ES and SS populations were highly asymmetrical, and migration from ES to SS was greater than that in the reverse direction. This suggests that greater genetic isolation is associated with increasing geographic distance.
Figure 5. Comparison of genetic structure distribution rates in 41 walnut genotypes. Source: Prof. Wei Gong.
Note: The pie chart in each location represents the proportions of cultivars belonging to the corresponding subgroups in the NC, ES and SS groups. The green arrows and the numbers in red and black are the gene flow direction and gene flow rates estimated in the present study.
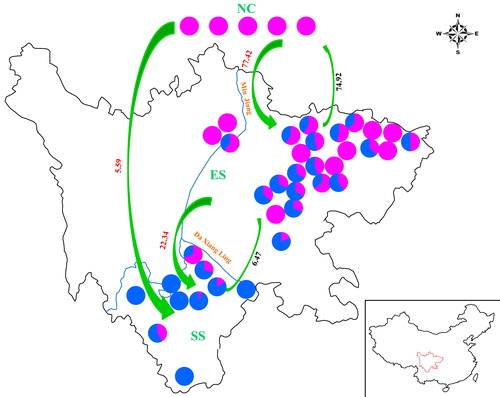
The structure of walnut populations provides insight into historical processes of crop diffusion within and across the agroclimatic zones of Eastern and Southwestern Sichuan. Research on sorghum populations has suggested that agroclimatic constraints have been as important as geographic isolation in shaping the diffusion process [Citation56]. In contrast to J. regia, which is mainly distributed in Eastern Sichuan, J. sigillata, which predominates in Southwestern Sichuan, has deep pits or seal-like depressions on the surfaces of its nuts, dark-colored kernels with tough septa, and the strongest pattern of population clustering, which ranges primarily in the ancestral region of domestication or adjacent areas with similar climates that lack freezing cold weather. This implies that these phenotypes can be maintained by selection, although it is not clear whether natural selection was imposed by the environment and adaptation demands of the species. Since the FST between the ES and SS population was 0.285, selection was likely to have been sufficiently powerful to allow for population differentiation, even in situations in which the gene flow between them was high. Notably, the J. regia types were distinctly divided, whereas those from North China were clustered together with ES1 to form a distinct subgroup. In summary, in the present study, the RAD-SNPs approximately explain the origin of differentiation of the cultivars in the Eastern and Southwestern Sichuan regions. Thus, further studies could focus on collecting more samples from different countries and regions to expand the population and reveal more underlying information about the phylogenetic relationships among walnut species.
Conclusions
The walnut data represent a large body of genome sequences for Juglans species and offer sources of near relatives in this clade for comparative genomic analysis. RAD-Seq analysis develops more information about molecular markers and provides an effective strategy for researching the phylogenetics and genetic structure of walnuts. The ES and SS populations were selected naturally and had high differentiation and considerable gene exchange. J. sigillata independently originated in Southwestern Sichuan, and it has rich genetic diversity and great potential for development and improvement. The SNP panels could be applied in breeding and genetic analyses of walnut germplasms. Other researchers can acquire these markers, which will benefit their further studies. The present study only represents a snapshot of the population genetics of the established trees. The gene flow of successive walnut generations and population structure will probably be affected by potential population fragmentation in the future. Current management policies should consider accommodating differences in allele variation in different biogeoclimatic zones. It is suggested that the habitats of Southwestern Sichuan Province in China be capable of supporting greater walnut diversity and given special consideration in future breeding programs.
Authors’ contributions
Wei Gong formulated and designed the experiments, Si-Yu Yan, Jing-Yan Wang performed the experiments, Jing-Yan Wang, Si-Yu Yan draw the figures and analyzed the data, Jing-Yan Wang wrote the paper, Wei Gong and Wen-Kai Hui revised and proofread the paper. All the authors read and approved the final manuscript.
Supplemental Material
Download PDF (267.4 KB)Supplemental Material
Download PDF (109.4 KB)Disclosure statement
The authors declare no conflict of interest.
Availability of data and materials
The data supporting the results presented in this article are included as additional files. All RAD-seq data in this study are archived in European Variation Archive database with the project number PRJEB36778.
Additional information
Funding
Related Research Data
References
- Bae W, Kim J, Chung J. Production of granular activated carbon from food-processing wastes (walnut shells and jujube seeds) and its adsorptive properties. J Air Waste Manag Assoc. 2014;64(8):879–886.
- Baghkheirati EK, Bagherieh-Najjar MB. Modelling and optimization of Ag-nanoparticle biosynthesis mediated by walnut green husk extract using response surface methodology. Mater Lett. 2016;171:166–170.
- Chen L, Ma Q, Chen Y, et al. Identification of major walnut cultivars grown in China based on nut phenotypes and SSR markers. Sci Hortic-Amsterdam. 2014;168:240–248.
- Ciarmiello LF, Piccirillo P, Pontecorvo G, et al. A PCR based SNPs marker for specific characterization of English walnut (Juglans regia L.) cultivars. Mol Biol Rep. 2011;38(2):1237–1249.
- Quan S, Qianwen X, Yongfei L, et al. Research on the main economic characters of the giant walnut in Shimian. North Hortic. 2011;18:15–18. (In Chinese)
- Bayazit S, Kazan K, Gulbitti S, et al. AFLP analysis of genetic diversity in low chill requiring walnut (Juglans regia L.) genotypes from Hatay, Turkey. Sci Hortic-Amsterdam. 2007;111(4):394–398.
- Pollegioni P, Woeste KE, Chiocchini F, et al. Landscape genetics of Persian walnut (Juglans regia L.) across its Asian range. Tree Genet Genom. 2014;10(4):1027–1043.
- Gunn BF, Aradhya M, Salick JM, et al. Genetic variation in walnuts (Juglans regia and J. sigillata; Juglandaceae): species distinctions, human impacts, and the conservation of agrobiodiversity in Yunnan, China. Am J Bot. 2010;97(4):660–671.
- Aradhya MK, Potter D, Gao F, et al. Molecular phylogeny of Juglans (Juglandaceae): a biogeographic perspective. Tree Genet Genom. 2007;3(4):363–378.
- Marrano A, Martinez-Garcia PJ, Bianco L, et al. A new genomic tool for walnut (Juglans regia L.): development and validation of the high-density Axiom™ J. regia 700K SNP genotyping array. Plant Biotechnol J. 2019;17(6):1027–1036.
- Sun YW, Hou N, Woeste K, et al. Population genetic structure and adaptive differentiation of iron walnut Juglans regia subsp. sigillata in southwestern China. Ecol Evol. 2019;9(24):14154–14166.
- Pollegioni P, Woeste KE, Chiocchini F, et al. Ancient humans influenced the current spatial genetic structure of common walnut populations in Asia. PLoS One. 2015;10(8):e0135980.
- Pecoraro C, Babbucci M, Villamor A, et al. Methodological assessment of 2B-RAD genotyping technique for population structure inferences in yellowfin tuna (Thunnus albacares). Mar Genom. 2016;25:43–48.
- You FM, Deal KR, Wang J, et al. Genome-wide SNP discovery in walnut with an AGSNP pipeline updated for SNP discovery in allogamous organisms. BMC Genomics. 2012;13(1):354–354.
- Van Wyngaarden M, Snelgrove PVR, DiBacco C, et al. Identifying patterns of dispersal, connectivity and selection in the sea scallop, Placopecten magellanicus, using RADseq-derived SNPs. Evol Appl. 2017;10(1):102–117.
- Xu P, Xu S, Wu X, et al. Population genomic analyses from low-coverage RAD-Seq data: a case study on the non-model cucurbit bottle gourd. Plant J. 2014;77(3):430–442.
- Bruneaux M, Johnston SE, Herczeg G, et al. Molecular evolutionary and population genomic analysis of the nine-spined stickleback using a modified restriction-site-associated DNA tag approach. Mol Ecol. 2013;22(3):565–582.
- Hohenlohe PA, Bassham S, Etter PD, et al. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genet. 2010;6(2):e1000862.
- Martínez-García PJ, Crepeau MW, Puiu D, et al. The walnut (Juglans regia) genome sequence reveals diversity in genes coding for the biosynthesis of non-structural polyphenols. Plant J. 2016;87(5):507–532.
- Baird NA, Etter PD, Atwood TS, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. 2008;3(10):e3376.
- Liu C, Chen H, Ren Z, et al. Population genetic analysis of the domestic Bactrian camel in China by RAD-seq. Ecol Evol. 2019;9(19):11232–11242.
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359.
- Catchen J, Bassham S, Wilson T, et al. The population structure and recent colonization history of Oregon threespine stickleback determined using restriction-site associated DNA-sequencing. Mol Ecol. 2013;22(11):2864–2883.
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760.
- Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAM tools. Bioinformatics. 2009;25(16):2078–2079.
- Liu K, Muse SV. PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics. 2005;21(9):2128–2129.
- Tamura K, Peterson D, Peterson N, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–2739.
- Zhang X, Zhang Z, Gu X, et al. Genetic diversity of pepper (capsicum spp.) germplasm resources in China reflects selection for cultivar types and spatial distribution. J Integr Agric. 2016;15(9):1991–2001.
- Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol. 2005;14(8):2611–2620.
- Yang J, Lee SH, Goddard ME, et al. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88(1):76–82.
- Bradbury PJ, Zhang Z, Kroon DE, et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23(19):2633–2635.
- Danecek P, Auton A, Abecasis G, et al. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–2158.
- Lee S, Nguyen TX, Kim J, et al. Cytological variations and long terminal repeat (LTR) retrotransposon diversities among diploids and B-chromosome aneuploids in Lilium amabile Palibin. Genes Genomics. 2019;41(8):941–950.
- Nei M, Tajima F. Maximum likelihood estimation of the number of nucleotide substitutions from restriction sites data. Genetics. 1983;105(1):207–217.
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123(3):585–595.
- Excoffier L, Lischer HEL. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10(3):564–567.
- Beerli P. Comparison of Bayesian and maximum-likelihood inference of population genetic parameters. Bioinformatics. 2006;22(3):341–345.
- Beerli P. Estimation of the population scaled mutation rate from microsatellite data. Genetics. 2007;177(3):1967–1968.
- Feng JY, Li M, Zhao S, et al. Analysis of evolution and genetic diversity of sweet potato and its related different polyploidy wild species I. trifida using RAD-seq. BMC Plant Biol. 2018;18(1):181.
- Zhong Y, Yang A, Liu S, et al. RAD-Seq data point to a distinct split in Liriodendron (Magnoliaceae) and obvious east–west genetic divergence in L. chinense. Forests. 2018;10(1):1–13.
- Wang Y, Li X, Mao Y, et al. Genome-wide dynamic transcriptional profiling in Clostridium beijerinckii NCIMB 8052 using single-nucleotide resolution RNA-Seq. BMC Genomics. 2012;13(1):102–102.
- Mu XY, Sun M, Yang PF, et al. Unveiling the identity of Wenwan walnuts and phylogenetic relationships of Asian Juglans species using restriction site-associated DNA-sequencing. Front Plant Sci. 2017;8:1708.
- Davey JW, Cezard T, Fuentes P, et al. Special features of RAD sequencing data: implications for genotyping. Mol Ecol. 2013;22(11):3151–3164.
- Pollegioni P, Olimpieri I, Woeste KE, et al. Barriers to interspecific hybridization between Juglans nigra L. and J. regia L species. Tree Genet Genomes. 2013;9(1):291–305.
- Valdisser P, Pappas GJ, De Menezes IPP, et al. SNP discovery in common bean by restriction-associated DNA (RAD) sequencing for genetic diversity and population structure analysis. Mol Genet Genomics. 2016;291(3):1277–1291.
- Roor W, Konrad H, Mamadjanov D, et al. Population differentiation in common walnut (Juglans regia L.) across major parts of its native range-insights from molecular and morphometric data. J Hered. 2017;108(4):391–404.
- Han H, Woeste KE, Hu Y, et al. Genetic diversity and population structure of common walnut (Juglans regia) in China based on EST-SSRs and the nuclear gene phenylalanine ammonia-lyase (PAL. Tree Genet Genomes. 2016;12(6):111.
- Wang H, Pan G, Ma Q, et al. The genetic diversity and introgression of Juglans regia and Juglans sigillata in Tibet as revealed by SSR markers. Tree Genet Genomes. 2015;11(1):1–11.
- Meng P, Zhao S, Niu X, et al. Involvement of the Interleukin-23/Interleukin-17 axis in chronic Hepatitis C virus infection and its treatment responses. IJMS. 2016;17(7):1070.
- Huang X, Wei X, Sang T, et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet. 2010;42(11):961–967.
- Aradhya M, Velasco D, Ibrahimov Z, et al. Genetic and ecological insights into glacial refugia of walnut (Juglans regia L.). PLoS One. 2017;12(10):e0185974.
- Yuan XY, Sun YW, Bai XR, et al. Population structure, genetic diversity, and gene introgression of two closely related walnuts (Juglans regia and J. sigillata) in southwestern China revealed by EST-SSR markers. Forests. 2018;9(10):646.
- Bradic M, Beerli P, Garcia-de LF, et al. Gene flow and population structure in the Mexican blind cavefish complex (Astyanax mexicanus). BMC Evol Biol. 2012;12:9.
- Ward M, Dick C, Gribel R, et al. To self, or not to self. A review of outcrossing and pollen-mediated gene flow in neotropical trees. Heredity (Edinb). 2005;95(4):246–254.
- Nielsen R. Molecular signatures of natural selection. Annu Rev Genet. 2005;39:197–218.
- Morris GP, Ramu P, Deshpande SP, et al. Population genomic and genome-wide association studies of agroclimatic traits in sorghum. Proc Natl Acad Sci USA. 2013;110(2):453–458.