
Abstract
Drought is a major abiotic stress factor that affects the yield of maize (Zea mays L.) during the grain filling period. Therefore, it is critical to reveal the molecular mechanisms underlying maize drought stress tolerance. To analyze the characteristics and functions of differentially abundant proteins (DAPs) in response to drought stress during the maize kernel-filling stage, here, two maize hybrid varieties with contrasting drought tolerance (tolerant ND476 and sensitive ZX978) were subjected to water sufficient (control) and water deficit (drought) treatment conditions for 12 days at the grain filling stage. Physiological assays and leaf proteome analysis were performed. We conducted iTRAQ analysis on 12 samples from 4 groups and obtained 1,655 DAPs. Amongst these, five essential sets of drought responsive DAPs were identified by bioinformatics techniques. Four significantly enriched metabolic pathways were identified in ND476, which were shown to be associated with the ribosome, metabolic, basal transcription factors and photosynthesis pathways. Further, the phenotypic characterization and qRT-PCR analysis confirmed our iTRAQ sequencing data. Remarkably, significantly higher drought tolerance of ND476 may be attributed to the following responses: (a) oxidoreductase, peroxidase and hydrolytic enzyme activities to promote cell redox homeostasis maintenance; (b) elevated expression of stress defense proteins; and (c) reduced synthesis of redundant proteins in order to help plants preserve energy to fight drought stress. In conclusion, these results offer more insights into the molecular mechanisms underlying maize drought tolerance at the grain filling stage and can be a foundational basis for molecular breeding of drought tolerant maize varieties.
Introduction
Plants are subjected to a variety of environmental pressures throughout their life cycles, for example, cold, heat, drought, salt, etc., which have a significant impact on plant activities and the formation of crop yields [Citation1–5]. Recent studies have shown that drought is the major environmental determinant of plant productivity; the drought-induced crop losses nearly equal to those caused by all other natural disasters combined [Citation6–8]. In order to adapt to drought stress, plants perform a series of molecular, cellular, physiological and metabolic responses. It has been proved that cultivating drought-resistant varieties is an economical and effective way to reduce drought stress [Citation9]. Therefore, understanding plant responses to drought is the main objective of breeders [Citation10]. Thus, understanding the drought tolerance mechanisms of crops under such climatic and environmental changes is of great significance for crop drought tolerance breeding [Citation11]. Therefore, the development of drought-tolerant maize varieties has become more important to breeders to ensure food security.
Known to have originated in the Mexican highlands around 8,700 years ago, maize is one of the most common cereal crops in the world, along with wheat and rice [Citation2,Citation12]. Maize is recognized as one of the most important food crops in the world and has high yield potential and nutritional value as food, feed and fuel [Citation13,Citation14]. Although maize production is considerably affected by many biotic and abiotic stresses, drought is the major one. In maize, drought has a profound effect on grain formation during the filling period. The loss of maize yield is caused by water deficit in the reproductive stage. During the early stages of the kernel growth, water deficit strongly leads to a short grain-filling period with an ultimate decrease in grain yield [Citation15–17]. In the north China plain, drought during the maize filling stage, can not only contribute to poor kernel development, but can also weaken the defense capabilities of maize against pathogen attack [Citation9,Citation18]. To elucidate the drought response is therefore very important for maize growth and development during the seed filling period.
Physiological, biochemical and phenotypic identification have contributed towards making great progress in the research on drought tolerance of maize at the seedling stage and reproductive stages [Citation19–21]. However, researches on drought tolerance of maize are mainly focused on physiological, biochemical and basic molecules, and transcriptome analysis has been mostly adopted [Citation22–24], but the mechanisms of drought stress tolerance in maize at the kernel filling stage are still unclear [Citation25–28]. The kernel filling period is a critical phase along the life cycle of the maize plant, during which drought stress will significantly reduce the yield and quality of the crop. Therefore, investigation of the changes in the plant proteome is highly important since proteins, unlike transcripts, are direct effectors of plant stress response [Citation29–31].
To better understand these changes, isobaric tags for relative and absolute quantification (iTRAQ) were used to detect statistically significant changes in the maize proteome. ITRAQ is considered to be one of the most stable differential quantitative proteomics techniques, which uses isotope-based labeling to produce very small coefficients of variation in quantitative measurements [Citation32]. As a powerful technique to perform quantitative proteome analysis, iTRAQ allows identification of more numerous proteins and can provide more reliable quantitative information than the traditional 2-DE analysis [Citation33].
Hence, in order to understand the drought stress response during maize kernel filling, this study used the iTRAQ technique for proteome analysis of two maize hybrids (ND 476 and ZX 978). After pollination of the two varieties, drought treatment was carried out for 12 days, during which the panicle traits were measured and bioinformatics analysis was performed. This will lay a certain theoretical foundation and practical significance for the proteome analysis of maize drought stress in the future.
Materials and methods
Plant materials and drought stress treatment
Two maize hybrids with contrasting drought sensitivity [tolerant NongDan 476 (ND476) and sensitive ZhongXin 978 (ZX978)] were used in this experiment. Seeds of the two hybrids were provided by the North China Key Laboratory for Crop Germplasm Resources of Education Ministry, Hebei Agricultural University, China. The detailed criteria on our selection of these hybrids are described in our recent study [Citation34].
The field experiment was conducted at the Qing Yuan Experiment Station of Hebei Agricultural University, Baoding, China (115 E, 38 N) under a fully automated rain-proof shelter. Randomized block design was used in the experiment, which was repeated three times in the control group and the drought stress treatment group. The area of each experimental unit/plot is 5 m × 5 m, and the plant spacing is 0.6 m × 0.3 m, with 128 plants in each plot. Conventional irrigation was used in both treatments until the grouting period. The soil moisture content of the plots with sufficient water was maintained between 70 and 80%, while the soil moisture content of the plots with insufficient water was maintained between 15% and 20% [Citation35]. The relative soil water content of one meter underground was monitored by TZS-1 soil moisture measurement instrument (Zhejiang Tuopu Technology Co. Ltd., Zhejiang, China).
In order to manage the self-crossing during the flowering period, strict bagging management was carried out one week before the flowering period. We started the drought treatment on August 11 about 45 days after seed germination, and continued the artificial drought for 12 days until the afternoon of August 23. For each treatment, we took the middle grain of each ear of corn for experimental analysis. During each sample collection, we ensured that the samples collected were frozen in liquid nitrogen immediately before being stored at extremely low temperature (−80 °C) for subsequent use. Each measurement was repeated three times.
Phenotypic characterizations
The phenotypic characteristics of the seeds of two hybrids were determined under both water sufficient (control) and water deficit (drought) conditions. Ten panicles were randomly selected from each group, and the number of kernel rows per ear and the number of kernel per row were measured and averaged.
Protein extraction
Kernels were sonicated three times on ice using a high intensity ultrasonic processor (Scientz) in lysis buffer (8 mmol/L urea, 1% Protease Inhibitor Cocktail). The remaining debris was removed by centrifugation at 12,000g at 4 °C for 10 min. Finally, the supernatant was collected and the protein concentration was determined with a BCA kit according to the manufacturer’s instructions (23225, Thermo Fisher Scientific, Shanghai, China). Protein quality was confirmed with SDS-PAGE (tricine-sodium dodecyl sulfate polyacrylamide gel electrophoresis) [Citation36].
Protein digestion and iTRAQ labeling
For digestion, the protein solution was reduced with 5 mmol/L dithiothreitol for 30 min at 56 °C and alkylated with 11 mmol/L iodoacetamide for 15 min at room temperature in darkness. The protein sample was then diluted by adding 100 mmol/L TEAB to urea concentration of less than 2 mol/L. Finally, trypsin (Promega, Madison, WI, USA) was added at 1:50 trypsin-to-protein mass ratio for the first digestion overnight and 1:100 trypsin-to-protein mass ratios for a second 4-h digestion. After trypsin digestion, the peptide fraction was desalted by Strata X C18 SPE column (Phenomenex) and vacuum-dried. The peptide fraction was reconstituted in 0.5 mol/L TEAB and processed according to the manufacturer’s protocol for TMT kit/iTRAQ kit. Briefly, one unit of TMT/iTRAQ reagent was thawed and reconstituted in acetonitrile. The peptide mixtures were then incubated for 2 h at room temperature and pooled, desalted and dried by vacuum centrifugation.
High performance liquid chromatography fractionation and liquid chromatography-tandem mass spectrometry
The tryptic peptides were dissolved in 0.1% formic acid (solvent A), directly loaded onto a home-made reversed-phase analytical column (15-cm length, 75 μm i.d.). The gradient was comprised of an increase from 6% to 23% solvent B (0.1% formic acid in 98% acetonitrile) over 26 min, 23% to 35% in 8 min and an increase to 80% in 3 min then a hold at 80% for the last 3 min, all at a constant flow rate of 400 nL/min on an EASY-nLC 1000 UPLC system.
The peptides were subjected to NSI source followed by tandem mass spectrometry (MS/MS) in Q ExactiveTM Plus (Thermo) coupled online to the UPLC. The electrospray voltage applied was 2.0 kV. The m/z scan range was 350 to 1800 for full scan, and intact peptides were detected in the Orbitrap at a resolution of 70,000. Peptides were then selected for MS/MS using NCE setting as 28 and the fragments were detected in the Orbitrap at a resolution of 17,500. A data-dependent procedure that alternated between one MS scan followed by 20 MS/MS scans with 15.0 s dynamic exclusion. Automatic gain control (AGC) was set at 5E4. Fixed first mass was set as 100 m/z.
DAPs functional classification, pathway enrichment
The successfully identified DAPs were used as queries to search the Interpro (https://www.ebi.ac.uk/interpro/) and Pfam (http://pfam.xfam.org/); Gene Ontology (GO) (http://www.geneontology.org/) and the KEGG (http://www.genome.jp/kegg/) databases. Additionally, by searching the maize sequence database Gramene (http://ensemble.gramene.org/Zea_mays/), we obtained the corresponding gene sequences of the DAPs. The GO terms were assigned to each DAP based on BLASTX similarity (E-value < 1.0 × 105) and known GO annotations using the Blast2GO tool (https://www.blast2go.com). For the functional annotation and classification of the identified DAPs, GO analysis was performed to categorize the DAPs into their BP, MF and CC involvement in response to drought stress. Moreover, the DAPs were assigned to various metabolic pathways using the KEGG pathway analysis. Further, significant KEGG pathway enrichment analysis was performed using the hypergeometric test, with Q (Bonferroni-corrected p value) < 0.05 set as the statistically significant threshold. We used String web-based program (version 10.5) (http://www.string-db.org/) to construct a protein interaction network for the identified DAPs.
RNA extraction, cDNA synthesis and qRT-PCR analysis
Total RNA was isolated from non-stressed and stressed kernels of the two hybrids (ND 476 and ZX 978) and prepared for qRT-PCR analysis using the Omini Plant RNA Kit (DNase I) (CWBIO, Beijing, China) based on the manufacturer’s instructions. For cDNA synthesis, 1 µg of total RNA was reverse-transcribed in a total volume of 20 µL, using HiFiscript cDNA Synthesis Kit (CWBIO, Beijing, China) according to the manufacturer’s instructions. Twenty DAPs were selected and gene-specific primers were designed for qRT-PCR using Primer Premier 5 Designer software. Each qRT-PCR reaction mixture comprised 2 µL of template cDNA, 1 µL of forward primer (50 pmol), 1 µL of reverse primer (50 pmol) and 10 µL of 2 × Fast Super EvaGreen qPCR Master mix (US Everbright Inc., Suzhou, China) in a total reaction volume of 20 µL. A steady and constitutively expressed maize gene GAPDH (accession no. X07156) was used as the internal reference gene. Each sample had three technical replicates. The relative mRNA abundance was calculated according to the 2−ΔΔCT method.
Results and discussion
Phenotypic differences in two maize hybrids under drought
We recorded the phenotypic characteristics of two maize hybrids under drought stress and normal treatment. The two varieties of corn grew under the same conditions for 12 days, except for different water treatments or drought treatments. As expected, there was no significant phenotypic difference between the two varieties under adequate moisture conditions, as both maintained normal and complete features and structures. However, the phenotypic characteristics of ZX 978 were significantly different after drought stress; and the phenotype of ND 476 did not change much. According to the statistical analysis, under drought condition, the ear length of the sensitive variety ZX 978 decreased significantly (). In addition, drought also led to an increase in the length of corn bald tip; the phenomenon of wizened kernels was obviously observed (). Under drought conditions, the panicle length of ZX 978 decreased significantly but the bald tip length increased significantly, whereas those of ND 476 did not change significantly (). In addition, drought did not reduce the number of kernel rows per ear (), but significantly reduced kernels per ear () of ZX 978, whereas these of ND 476 did not change significantly.
Figure 1. Phenotypic characterization of ears of the two maize hybrids. (a, b) Ear phenotypes. (c) Ear length. (d) Ear bare tip length. (e) Kernel rows per ear. (f) Kernels per row.
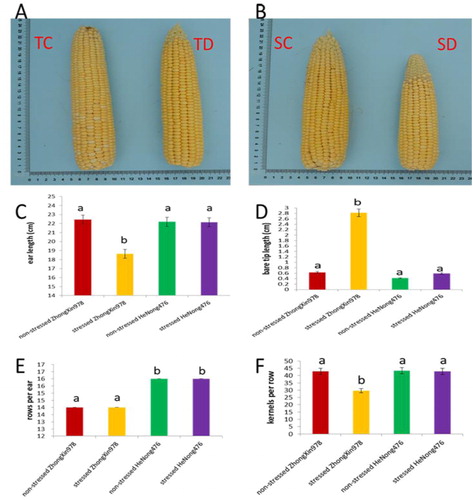
Therefore, as seen from , in terms of grain morphology and structure composition, the length, size, panicle number and grain number of ND 476 were basically the same; the sensitive hybrid ZX 978, however, was quite the opposite. Under the condition of drought, the plants of the sensitive variety ZX 978 were short, the panicle was small, and grain rows were decreased. At the same time, photosynthetic product synthesis weakened resulting in grain wrinkles; bald tip phenomenon was obvious. This result shows that drought can lead to delayed cell development, short plants and wrinkled grains, resulting in decreased yield and quality. This is the same result as we expected, so it can prove and strengthen the credibility of our paper. Based on the above analysis, we can conclude that under drought stress conditions, ZX 978 is more sensitive than ND 476 during the grouting period.
Inventory of maize kernel proteins identified by iTRAQ
According to the result of Mascot software, a total of 815,688 spectra were detected; and 219,576 peptides, accounting for 26.9% of the total number, were obtained through database comparison and analysis. There were 113,635 peptides, 87,018 unique peptides, 9895 identified proteins and 8310 quantifiable proteins. Among these 9895 identified proteins (), there were 186 less than 10 kDa (1.88%), 7892 were 10–70 kDa (79.12%), 1090 were 70–100 kDa (11.02%) and 790 were 70–100 kDa (7.98%). In addition, 5720 (84.57%) proteins were detected based on at least two unique peptides whilst the remaining 1111 (15.43%) proteins had only one identified unique peptide (). Proteins with at least one unique peptide were used for a subsequent analysis of DAPs. Protein sequence coverage was shown to be approximately below 35% (). According to the peptide length distribution of each protein, more than 85% of the peptide length was between 7 and 25 amino acids, of which 7–9 and 9–11 amino acids were modal length ().
Analysis of DAPs observed in different comparisons
Comparative proteomic analysis was used to investigate the changes of protein profiles in the kernels of ND 476 (drought-tolerant, T) and ZX 978 (drought-sensitive, S) hybrids under drought stress conditions. A pairwise comparison of before and after treatments (drought, D, and control, C) were performed in ND 476 (TD_TC) and ZX 978 (SD_SC) individually. A total of 50 DAPs were detected under TD_TC (), of which 34 proteins were up-regulated and 16 proteins were down-regulated. Under the TD_SD group, there were 891 DAPs, 474 of which were up-regulated and 417 of which were down-regulated. Under the group of SD_SC, there were a total of 42 DAPs (), of which 30 showed up-regulation and 12 showed down-regulation ().
Figure 2. Number and grouping of DAPs. (a) Histogram of the number distribution of DAPs in four comparison groups. (b) Venn diagram analysis of DAPs identified in the four experimental comparisons. (c) Cluster analysis of DAPs in SD_TD comparison.
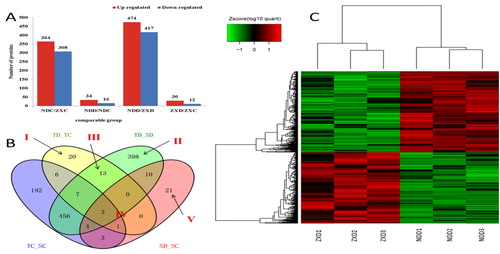
Table 1. DAPs observed specifically in tolerant hybrid ND476.
Table 2. DAPs showed differentially expressed in both the tolerant and sensitive hybrids after drought treatment.
As shown in , these five areas represent the influence of the experiment under different treatment conditions. Region I represented the specific DAPs of TD_TC, namely the specific drought response DAPs of drought-tolerant ND 476; among these 20 DAPs, 12 were up-regulated and 8 were down-regulated (). After comparative analysis, region II () showed 398 drought-response DAPs specific to TD_SD; that is, drought-sensitive hybrids and drought-tolerant hybrids shared specific DAPs after drought-treatment. Region III () shared 13 specific DAPs between TD_TC and SD_TD, namely drought-tolerant DAPs. In the TD_TC and SD_TD group, 10 of the 13 DAPs were up-regulated and 3 were down-regulated. The IV region () represented the four DAPs being shared by TD_TC and SD_SC, namely, the common (overlapping) drought response DAPs. Among them, there were two up-regulated proteins in TD_TC and down-regulated proteins in SD_SC, two down-regulated proteins in TD_TC and up-regulated proteins in SD_SC. Region V () showed 21 specific DAPs in SD_SC, of which 17 showed up-regulated proteins and 4 showed down-regulated proteins.
Table 3. Common (overlapping) drought-responsive DAPs in filling seed between ND476 and ZX 978.
Table 4. Drought-responsive maize seedling leaf proteins observed specifically in sensitive hybrid ZX 978.
Proteins associated with transcription and translation in ND476
The proteins related to transcription and translation are more expressed in ND 476 (). Ribosomal proteins make up the protein portion of the ribosome and with rRNA are essential for protein synthesis [Citation37], and they are reduced in Arabidopsis thaliana and maize under drought stress [Citation38–40]. In this study, proteins associated with transcription and translation (A0A1D6MBB0, A0A1D6NL34, B6T5W5, J7LC26,) were significantly down-regulated in response to drought stress. These results indicated that drought stress inhibits protein synthesis in plants, and ribosomal proteins had the function of reducing redundant protein synthesis under drought stress, which may help plants save energy and further combat drought stress.
Besides, nucleosome assembly protein (A0A1D6MBB0) facilitates the association with histones with DNA to form nucleosomes under drought conditions. Nucleolin (B6T5W5) functions as the first step of ribosomal RNA processing. The down-regulation of these two proteins indicated that ND 476 responded to drought conditions by inhibiting transcriptional activity and nucleosome formation to reduce the synthesis of redundant substances. Shu et al. [Citation41] believe that the decrease of L21 ribosomal protein was the main reason to maintain the osmotic potential in the root system under drought. As a consequence, this indicates that under drought stress, ND 476 can reduce the synthesis of redundant proteins by reducing transcription and translation activities, and further save energy so as to maintain its normal physiological characteristics. Overall, this may be the molecular response mechanism of drought-tolerant ND 476 under drought stress.
The molecular response of plants to abiotic stresses has been often considered as a complex process mainly based on the modulation of transcriptional activity of stress-related genes [Citation42]. In this study, the protein of transcription initiation factor TFIID subunit (C4J608) appeared to be down-regulated. There is evidence that an enhanced level of stress tolerance sometimes conferred at the expense of plant development and growth, due to metabolic costs of a disorder stress response [Citation43,Citation44]. Thus, this down-regulated protein reflects the fact that plants have developed a unique defense system.
Translocation-associated protein complex is thought to be required for efficient protein-specific translocation across the endoplasmic reticulum membrane [Citation45]. In this study, translocon-associated protein beta (TRAPB) (A0A1D6KDZ6) was the down-regulated protein. The evidence has shown that, in a reconstituted system, the absence of TRAPB results in an important decrease in translocation efficiency for proteins [Citation46]. Thus, the down-regulation of TRAPB may reduce plant metabolism and save more energy by reducing protein transmembrane transport so as to better adapt to drought stress.
Oxidoreductase related proteins in ND 476
Oxidoreductase (K7TR40, A0A1D6G3P7, A0A1D6FGJ9, B4FN73, P27788, A0A1D6N5L5) related proteins are the most prominent enzymes in the ND 476 under drought stress. When plants are stressed, the production of ROS exceeds the scavenging of ROS, so ROS oxidize proteins and nucleic acids and further cause programmed cell death [Citation47,Citation48]. Hence, the enhancement of antioxidant enzyme activity is the most effective mechanism against oxidative stress, and oxidative damage in the maize kernel is alleviated by a concerted action of enzymatic antioxidant systems. Besides, except the extradiol ring-cleavage dioxygenase (A0A1D6N5L5), all other oxidoreductases were up-regulated.
Meanwhile, ferredoxin-3 (P27788) was enriched in the ‘Photosynthesis’ KEGG pathway. Ferredoxin is recognized primarily as a component of the photosynthetic electron transport chain [Citation49]. In addition, ferredoxin modulates the activities of enzymes involved in periplasmic carbon metabolism [Citation50]. In general, we believe that when ND 476 is under drought stress, it will synthesize more proteins related to REDOX enzymes, enhance the enzyme activity, maintain the stability of the intracellular environment, and further improve the cell protection and tolerance to stress.
Other enzymes related to drought tolerance in ND 476
Drought stress induces complex molecular, cellular, physiological and developmental responses in which enzymes play a vital and indispensable role [Citation51]. Enzymes control many catalytic processes such as metabolism, nutrition and energy conversion [Citation52], and in this study, there were many types of enzymes (oxidoreductase, peroxidases, hydrolase, transferase, synthetase etc.) under drought stress, and most of them were up-regulated differentially expressed proteins, which may be the molecular mechanism of hybrid ND 476 to deal with drought stress.
In this study, synthetase (A0A1D6F072, A0A1D6L7V3, A0A1D6IBP5) related proteins also showed significant effects under drought stress. Indole-3-acetic acid (IAA) amido synthetase (A0A1D6F072) can prevent free IAA accumulation and enhance tolerance. Enhanced tolerance is maybe caused by inhibition of the expression of expansins via suppressed auxin signaling [Citation53,Citation54]. Indole-3-acetic acid amido synthetase (A0A1D6F072) is enriched in the ‘Plant hormone signal transduction’ KEGG pathway. Tryptophan synthase (A0A1D6L7V3) is an oligomeric enzyme that performs strict allosteric regulation on catalytic reactions [Citation55]. Thus, tryptophan synthase can produce increased stability with improved function for the multi-enzyme complexes. Asparagine—tRNA ligase (A0A1D6IBP5) is a down-regulated enzyme that binds aspartic amide to RNA and ACTS as glutaminase in the absence of ATP and aspartic acid [Citation56].
Hydrolase (A0A1D6MD20, A0A1D6Q5N4) related proteins have two proteins under drought stress. Ubiquitin (Ub) carboxyl-terminal hydrolase (A0A1D6MD20) catalyzes the hydrolysis, at the Ub-carboxyl terminus, of a wide variety of C-terminal Ub derivatives [Citation57]. It has the correct specificity to function in regenerating ubiquitin from small ubiquitin peptides and during ubiquitin-dependent protein breakdown [Citation58,Citation59]. Thus, the up-regulated ubiquitin carboxyl-terminal hydrolase greatly influences maize growth and development by modulating the activity, localization and stability of proteins. Nitrilase (A0A1D6Q5N4) consists of amidase with distinct specificities [Citation60,Citation61].
Transferase(C0PLU9, A0A1D6EM72)related proteins have two proteins and were both down-regulated. The enzyme hypoxanthine phosphoribosyltransferase (C0PLU9) catalyzes the metabolic salvage of the purine bases hypoxanthine and guanine [Citation62]. It not only participates in the remedial synthesis of purine bases, but also relates to toxic reactions. Hypoxanthine phosphoribosyltransferase is enriched in the ‘Purine metabolism’ KEGG pathway [Citation63]. Lecithin-cholesterol acyltransferase (A0A1D6EM72) plays a role in the transport, esterification, and transfer of cell-derived cholesterol [Citation64]. The down-regulation of these two enzymes indicated that hybrid ND 476 may respond to drought stress with reduced purine metabolism and sterol metabolism to save energy.
In summary, synthases, hydrolases and transferases can improve catalytic reactions, enzyme complex stability, signal molecule synthesis and cellular detoxification. Therefore, further understanding of their function will elucidate the drought tolerance mechanism of ND 476.
Proteins related to ‘stress defense response’ in ND 476
In the process of stress, the organism has a series of stress defense mechanisms, which are necessary for survival [Citation65]. It is reported that drought is the example of conditions that induce expression of genes with the stress protein [Citation66]. In this study, there are two proteins (B6TF92, A0A1D6INR0) associated with stress defense response, and both of them are up-regulated proteins. Stress reponsive proteins are produced in large quantities under stress conditions and help organisms survive through a variety of mechanisms [Citation67]. The ability of a stress protein to increase drought tolerance of plants has been demonstrated in tomatoes [Citation68]. Therefore, if we increase the up-regulated expression of the stress proteins, it would improve the drought tolerance of hybrid ND 476 during the grain-filling stage.
Key epigenetic regulation mechanisms in ZX 978
From , we can see that the protein regulated by epigenetic regulation is more expressed in ZX 978. Epigenetic regulation involves reversible changes in DNA methylation and histone modification patterns [Citation69]. The combinations of histone variants and post-translational modifications can be considered a ‘histone code’, which plays a key role in determining the transcriptional state and expression level of genes [Citation70,Citation71]. Here we show that histone modifications, particularly methylation of histone H3 (B4FCH2), have been reported to activate transcription [Citation72–74].
Studies have found that ubiquitination of histone H2B (P30755) is involved in transcriptional regulation [Citation69]. Evidence shows that H2B is important for efficient methylation of histone H3 but ubiquitination of H2B does not require methylation of H3. It is a phenomenon termed trans-regulation [Citation75,Citation76]. By promoting mitosis and transcriptional activity, these up-regulated proteins involved in epigenetic regulation may be crucial to the drought tolerance of maize kernels.
Domain-containing proteins in ZX 978
The number of expressed proteins with a domain was also large in ZX 978 (). Methyl-CpG-binding domain-containing protein (MBD, A0A1D6GLD8) is important in differential control of gene expression, and is involved in the alteration of chromatin structure and genome stability [Citation77]. The previous analysis of protein domains suggests a role in transcriptional regulation of the epigenome for most of them [Citation78,Citation79]. In addition, as a transcriptional suppressor, MBD protein implements a complete transcriptional program by coordinating histone modifications and chromatin organization [Citation79]. Hence, the drought prompted the up-regulation of MDB, and its up-regulation inhibited the transcriptional activity.
The TPR family of proteins is composed of proteins of very diverse function including organelle-targeting proteins involved in mitosis, immunophilins and a nuclear phosphatase [Citation80]. In this study, a TPR domain containing protein (B6TAE3) was up-regulated. The TPR domain is found in a variety of proteins that regulate the cell cycle, protein folding, transcription, protein transport, ubiquitin-proteomes, hormone receptor signaling, and several other pathways [Citation81,Citation82]. Thus, the up-regulation of TPR- domain- containing proteins can adopt a variety of mechanisms to respond to drought.
There is strong evidence that the SWIB and the MDM2 domains share a common evolutionary origin and thus adopt similar structures [Citation83]. The homology may suggest a similar functional mechanism for SWIB in chromatin remodeling machinery. SWIB/MDM2 domain containing protein (B6SRJ0) plays key roles in cell death, cyclin, transcription and stress pathway involvement [Citation84,Citation85]. Furthermore, it is postulated that B6SRJ0 may have a stage-specific function and participate in the drought stress response.
The CBS domain as such has no defined function(s) but plays a regulatory role for many enzymes and thus helps in maintaining the intracellular redox balance. However, the transcript levels of some members of this family are altered in response to various stresses such as drought, salinity, cold and high temperature [Citation86]. Depending on the CBS-domain-containing proteins (A0A1D6EAZ3), CBS domains have been proposed to affect multimerization and sorting of proteins, channel gating and ligand binding. In some cases, CBS domains may act as sensors of the intracellular metabolites by being activated by ATP [Citation87,Citation88]. Thus, it suggests that down-regulation of the CBS domain-containing proteins may inhibit stress response/tolerance and development in plants.
Nucleic acid repair proteins in ZX 978
Proteins related to nucleic acid repair are also thought to be significant in ZX 978. The DNA molecules of an organism are constantly subjected to a variety of damage caused by exposure to endogenous and environmental genotoxicity [Citation89]. DNA-damage-repair/toleration protein (B4FLF6) is an important pathway of mutation avoidance [Citation90]. DNA damage repair and recombinational repair are both important for cell survival of replication stress [Citation91]. Various DNA damage repair/tolerance pathways govern how a specific lesion is processed [Citation92]. Therefore, it revealed that up-regulation of protein B4FLF6 may promote DNA damage repair, mismatch repair, etc., in response to drought stress.
MMS19 nucleotide excision repair protein (NER, A0A1D6HTH7) is part of one of the most important DNA repair processes responsible for removal of damages that significantly distort the double-helix conformation [Citation93–95]. The unique feature of NER is its ability to detect and correct a wide spectrum of DNA modifications of different sizes and chemical structures [Citation96]. The NER mechanism involves a variety of proteins involved in damage identification, demarcation, gap filling and DNA synthesis [Citation97]. However, NER pathway repairs are limited to one DNA strand and MMS19 nucleotide excision repair protein was down-regulated. Hence, in this study, MMS19 nucleotide excision repair protein-like protein may reduce recognition of damaged nucleic acids, repair and stability of the double helix as well.
Gene ontology annotation and functional classification
We used Blast2GO web-based application (https://www.blast2go.com) to perform gene ontology (GO) functional annotation. We could divide the differentially expressed proteins into three categories, namely biological processes (BP), molecular functions (MF) and cellular component (CC).
For the tolerant hybrid ND 476-specific DAPs (), nucleobase-containing compound catabolic process (GO:0034655) was the most significantly enriched term in the BP category; electron carrier activity (GO:0009055), DNA binding (GO:0003677) and nucleic acid binding (GO:0003676) were significantly categorized in the MF category ().
Figure 3. GO functional classification of drought responsive proteins. (a) ND 476 specific DAPs. (b) ZX 978 specific DAPs. The y-axis represents the number of proteins in each function; x-axis displays the protein functions, categorized into three broad biological functional groups.
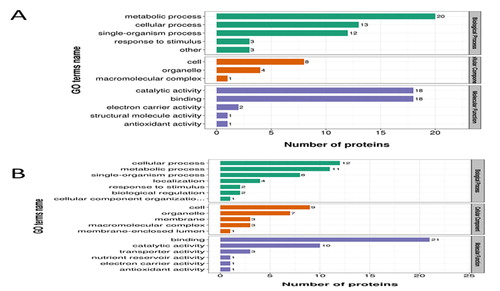
Among the significant GO terms in the sensitive hybrid ZX 978 (SC_SD, ), secretion (GO:0046903) and phospholipid metabolic process (GO:0006644) were in the BP category; nucleic acid binding (GO:0003676), protein transporter activity (GO:0008565), protein heterodimerization activity (GO:0046982), DNA binding (GO:0003677) and protein dimerization activity (GO:0046983) were in the MF category; whereas nucleosome (GO:0000786), chromatin (GO:0000785), protein-DNA complex (GO:0032993), DNA packaging complex (GO:0044815), part (GO:0044427), chromosome (GO:0005694) and nucleus (GO:0005634) were overrepresented by the CC function ().
KEGG pathway enrichment analysis
To further to analyze the functional consequences of the drought-responsive DAPs, we mapped them to the Kyoto Encyclopedia of Genes and Genomes (KEGG: https://www.genome.jp/kegg/) database and the DAPs were assigned to various biological pathways. Additionally, significant KEGG pathway enrichment analysis was performed using the hyper geometric test.
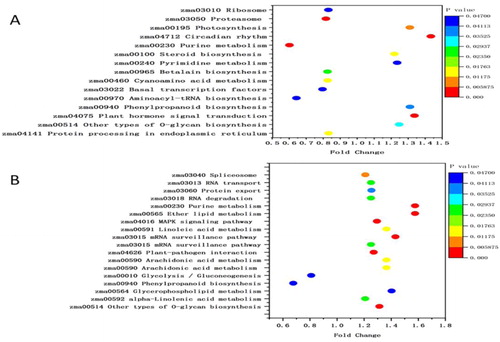
Metabolic pathways (5 proteins), Biosynthesis of secondary metabolites (4), Basal transcription factors (1), Ribosome (1), Circadian rhythm – plant (1) and Photosynthesis (1) were the important pathways in TD_TC (). Metabolic pathways (5 proteins), biosynthesis of secondary metabolites (4), arachidonic acid metabolism (2), spliceosome (2) and glycerophospholipid metabolism (2) were the significant pathways of SD_SC ().
These results show that more proteins were observed in the enriched pathways of ND 476 than ZX 978 and that the two hybrids diverge significantly in the pathway responses to drought stress. Using a hyper geometric test, the KEGG pathways that had a p value of less than 0.05 were considered to be significantly affected by drought stress. We observed that three pathways (Photosynthesis 0.0442, Basal transcription factors 0.0460 and Ribosome 0.0446) were considerably enriched among the ND 476 pathways (), whist three KEGG pathways, mRNA surveillance pathway (0.024521), Arachidonic acid metabolism (0.00068) and Glycerophospholipid metabolism (0.016) were significantly enriched among ZX 978 pathways ().
Figure 4. KEGG pathway enrichment analysis of DAPs. (a) Most significantly enriched pathways in TD_TC. (b) Most significantly enriched pathways in SD_SC. The size of the point represents the number of DAPs. TC, tolerant cultivar (ND476) under control conditions; TD, tolerant cultivar under drought conditions; SC, sensitive cultivar (ZX978) under well-watered (control) conditions; SD, sensitive cultivar under drought treatment conditions. An underscore between two genotype-treatment combinations represents comparison between those combinations, for example, TD_TC.
Overlapping DAPs between ND 476 and ZX 978 under drought conditions
According to the Venn diagram, there were four significant DAPs in the overlap between TD_TC and SD_SC. Among them, two proteins (A0A1D6JWG9 and A0A1D6EAZ3) were up-regulated and two ones (C0PLU9 and A0A1D6FCB8) were down-regulated in hybrid ND 476, whereas in the sensitive hybrid ZX 978 it was the opposite. CBS domain-containing protein (A0A1D6EAZ3) participates in transcription levels in response to drought. This protein was up-regulated in the hybrid ND 476, suggesting that it can respond to drought stress by promoting the increase of transcription level. However, this protein appeared to be down-regulated and not to promote plant growth and development under drought conditions in the sensitive hybrid ZX 978. An uncharacterized protein (A0A1D6JWG9) was up-regulated in ND 476 and down-regulated in ZX 978. This suggests that this unknown protein may play a role in promoting drought tolerance of maize varieties under drought conditions. Therefore, it can be used as a key protein for functional annotation and analysis, which will provide a theoretical basis for better elucidation of the drought response mechanism of these two maize hybrids.
Hypoxanthine phosphoribosyltransferase (C0PLU9) and rRNA processing protein-related (A0A1D6FCB8) were down-regulated in ND 476 and up-regulated in ZX 978. Both proteins are involved in base synthesis and ribosomal RNA processing. It is possible that ND 476 has less redundant protein synthesis under drought conditions, while ZX 978 does not.
Unknown proteins identified under drought stress in ND 476
We have introduced and described the known functional proteins of hybrid ND 476 in detail, but there are still many proteins whose function is unknown under drought stress. All these proteins (B6UI58, B6SHM3, B6UIG0, A0A1D6NTW2, A0A1D6JWG9, A0A1D6N294) appeared to be up-regulated. This suggests that these unknown proteins may also have some function under drought stress, but they have not been discovered yet. In addition, there was an uncharacterized protein (A0A1D6NTW2) enriched in the ‘Other types of O-glycan biosynthesis’ KEGG pathway. Therefore, if we further explore and analyze the functions of these unknown proteins, it will lay a certain theoretical basis and practical significance for further understanding of the molecular mechanism of hybrid ND 476 in response to drought stress.
qRT-PCR analysis
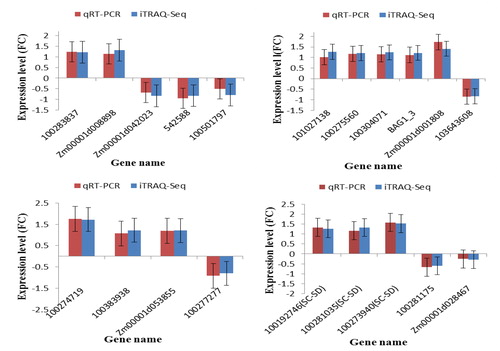
To confirm our findings based on iTRAQ sequencing data mentioned above, we conducted a supporting experiment by using qRT-PCR. A sample of 20 genes (Supplemental Table S5) was selected from the drought responsive DAPs across different groups. The results of the qRT-PCR analysis confirmed our findings obtained based on iTRAQ seq data. In particular, the patterns of iTRAQ seq expression on all 20 genes were replicated by the qRT-PCR approach (; Supplemental Table S6). A correlation coefficient (R2) (of the fold changes between qRT-PCR and iTRAQ seq) of 83.51% was obtained (), supporting the reliability of our iTRAQ sequencing data.
Figure 5. Confirmation of the iTRAQ-seq results by qRT-PCR. (a) DAPs specific to TD_TC; (b) DAPs specific to SD_TD; (c) DAPs shared between TC_TD and SD_SC; and (d) Common DAPs shared between TD_TC and TD_SD. The y-axis represents expression levels (fold change). Maize gene GAPDH (accession no. X07156) was used as the internal reference. Error bars represent the SE (n = 3). TC, tolerant cultivar (ND 476) under control conditions; TD, tolerant cultivar under drought conditions; SC, sensitive cultivar (ZX 978) under well-watered (control) conditions; SD, sensitive cultivar under drought treatment conditions. An underscore between two genotype-treatment combinations represent comparison between those combinations, for example, TD_TC.
Comparative analysis of drought stress responses in maize hybrid cultivars and inbred lines at the seedling and kernel filling stages
Previously, we have conducted comparative iTRAQ proteomics analysis of divergent maize recombinant inbred lines` responses to drought stress at the seedling stage [Citation98] and kernel filling stage [Citation99]. Our findings revealed that the drought tolerance of maize inbred lines at the seedling stage was largely due to activated expression of photosynthesis proteins balancing light capture and utilization, increased stress signaling and cellular water conservation as a result of enhanced accumulation of lipid-metabolism related proteins, stimulation of chaperon activities, increased cellular redox homeostasis capacity, and reduced cellular synthesis of redundant proteins to help the plant save energy to endure drought stress [Citation98]. On the other hand, at the kernel filling stage, the drought tolerance in maize recombinant inbred lines emanated from enhanced expression of proteins involved in transcriptional elongation regulation, protein transport and epigenetic regulation mechanisms, enriched carbohydrate metabolism and secondary metabolites biosynthesis, up-regulated expression of seed storage proteins, and activation of biotic–abiotic crosstalk mechanisms [Citation99]. In the present study, the kernel filling stage drought tolerance in maize hybrids can be attributed to oxidoreductase, peroxidase and hydrolytic enzyme activities that promote cell redox homeostasis maintenance; reduced synthesis of redundant proteins in order to help plants preserve energy to fight drought stress; elevated expression of stress defense proteins; and up-regulated expression of some unknown-function proteins.
Analysis of the findings from these three studies revealed some resemblances and differences in how maize inbred lines and hybrid cultivars respond to drought stress at seedling and kernel filling stages. Notably, improved cellular homeostasis maintenance is vital in both seedling leaf cells and kernel cells, in both inbred lines and hybrid cultivars. Additionally, the reduced synthesis of redundant proteins is universally instituted in maize inbred lines seedling stage and maize hybrid kernel filling stage responses to drought stress. Moreover, activated expression of stress defense proteins is universal in the drought stress responses across maize inbred lines at the seedling and kernel filling stage, as well as in maize hybrid cultivars at the kernel filling stage, with differences only being noticed in the exact individual specific proteins involved. Further, the photosynthesis pathway was also common in the drought stress response of tolerant maize inbred lines at the seedling stage and in tolerant maize hybrid cultivars at the kernel filling stage. Meanwhile, at the kernel filling stage of maize inbred lines, the ‘protein processing in the endoplasmic reticulum’ pathway was significantly enriched. This finding may suggest that the primary processes of protein biogenesis are critical in maize cells tolerating drought stress since pathways related to photosynthesis, ribosome and protein processing were significantly enriched in inbred line seedlings, as well as kernel filling stages of both inbred lines and hybrid cultivars.
However, we noticed some differences in the drought stress responses of maize inbred lines and hybrid cultivars’ at the kernel filling, and seedlings stages. A more vivid difference was that, whereas biotic–abiotic cross tolerance mechanisms were activated at both seedling and kernel filling stages in the tolerant maize inbred line YE8112, with HC-toxin reductase being prominent, probably in response to fungus Aspergillus flavus attack; biotic–abiotic cross talk was not apparent in the drought stress response in maize hybrids at the kernel filling stage. Other differences were related to the exact metabolic pathways and number of DAPs that were significantly enriched in these pathways in response to drought stress. This eventually caused some differences in the ultimately activated drought response mechanisms between maize inbred lines and hybrid cultivars, and/or between crop growth stages. We hope that harnessing these identified similarities and differences in the drought response mechanisms will improve our understanding of the intricate molecular mechanisms underpinning maize drought tolerance and help us in developing drought resilient genotypes.
Conclusions
In this study, we performed a comprehensive comparative proteomics analysis of two maize hybrids contrasting in their drought tolerance. The results showed that the drought stress tolerance of the two hybrids was different at the kernel filling stage. An iTRAQ-based analysis method identified a total of 1655 DAPs, among which five sets of drought responsive DAPs were considered critical and further analyzed by bioinformatics techniques. Four significantly enriched metabolic pathways were identified in ND476, which were associated with ribosome, metabolic, basal transcription factors and photosynthesis pathways. Further, our qRT-PCR analysis results confirmed the iTRAQ sequencing based findings. Remarkably, significantly higher drought tolerance of ND476 may be attributed to the following responses: (a) oxidoreductase, peroxidase and hydrolytic enzyme activities to promote cell redox homeostasis maintenance; (b) reduced synthesis of excess proteins in order to help plants preserve energy to fight drought stress; (c) elevated expression of stress defense proteins; and (d) up-regulated expression of some unknown-function proteins. These results provide more insights into the molecular mechanisms of drought tolerance in maize during grain filling.
Author contributions
Conceptualization: Anyi Dong and Huijun Duan. Data curation: Anyi Dong and Yatong Yang. Funding acquisition: Huijun Duan. Investigation: Anyi Dong, Yatong Yang, Xinyue Liu, Yafei Wang, and Jiao Li. Formal analysis: Anyi Dong, Tinashe Zenda, and Huijun Duan. Methodology: Anyi Dong, Yatong Yang, and Tinashe Zenda. Project administration: Songtao Liu and Huijun Duan. Resources: Huijun Duan. Supervision: Huijun Duan and Songtao Liu. Visualization: Anyi Dong. Writing – original draft: Anyi Dong. Writing – review and editing: Anyi Dong, Tinashe Zenda, and Huijun Duan.
Supplemental Material
Download PDF (202.4 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Wang N, Liang LI, Gao WW, et al. Transcriptomes of early developing tassels under drought stress reveal differential expression of genes related to drought tolerance in maize. J Integr Agric. 2018;17(6):1276–1288.
- Gong F, Yang L, Tai F, et al. “Omics” of maize stress response for sustainable food production: opportunities and challenges. OMICS. 2014;18(12):714–732.
- Li GK, Gao J, Peng H, et al. Proteomic changes in maize as a response to heavy metal (lead) stress revealed by iTRAQ quantitative proteomics. Genet Mol Res. 2016;15(1).
- Shikha M, Pooja B, Mallikarjuna MG, et al. Structural, functional, and evolutionary characterization of major drought transcription factors families in maize. Front Chem. 2018;6:177.
- Liu Y, Zhou M, Gao Z, et al. RNA-seq analysis reveals mapkkk family members related to drought tolerance in maize. PLoS One. 2015;10(11):e0143128.
- Du CY, Duan ZY, Wang JX, et al. Drought resistance of eight maize varieties at seedling stage in Yunnan Province. Plant Dis Pests. 2015;6:28–34.
- Alam I, Sharmin SA, Kim KH, et al. Proteome analysis of soybean roots subjected to short-term drought stress. Plant Soil. 2010;333(1–2):491–505.
- Gao ZY, Liu H, Wang HL, et al. Generation of the genetic mutant population for the screening and characterization of the mutants in response to drought in maize. Chin Sci Bull. 2014;59(8):766–775.
- Yang LM, Jiang TB, Fountain JC, et al. Protein profiles reveal diverse responsive signaling pathways in kernels of two maize inbred lines with contrasting drought sensitivity. Int J Mol Sci. 2014;15(10):18892–18918.
- Zheng J, Fu J, Gou M, et al. Genome-wide transcriptome analysis of two maize inbred lines under drought stress. Plant Mol Biol. 2010;72(4–5):407–421.
- Budak H, Hussain B, Khan Z, et al. From genetics to functional genomics: improvement in drought signaling and tolerance in wheat. Front Plant Sci. 2015;6:1012.
- Zong N, Li X-J, Wang L, et al. Maize ABP2 enhances tolerance to drought and salt stress in transgenic arabidopsis. J Integr Agric. 2018;17(11):2379–2393.
- Cui LM, Wang XX, Xuan HD, et al. Changes in several physiological and biochemical indices of maize seedling roots caused by drought stress. Plant Dis Pests. 2015;6:35–37.
- Hoque MIU, Nesar Uddin M, Fakir Rasel MSA, et al. Drought and salinity affect leaf and root anatomical structures in three maize genotypes. J Bangladesh Agric Univ. 2018;16(1):47–55.
- Westgate ME. Water status and development of the maize endosperm and embryo during drought. Crop Sci. 1994;34(1):76–83.
- Tahmasebi S, Heidari B, Pakniyat H, et al. Independent and combined effects of heat and drought stress in the Seri M82 × Babax bread wheat population. Plant Breed. 2014;133(6):702–711.
- Bin Z, Li W, Chang X, et al. Effects of favorable alleles for water-soluble carbohydrates at grain filling on grain weight under drought and heat stresses in wheat. PLoS One. 2014;9(7):e102917.
- Wang Z, Jiang J, Liao Y, et al. Risk assessment of maize drought hazard in the middle region of farming-pastoral ecotone in northern china. Nat Hazards. 2015;76(3):1515–1534.
- Lei L, Shi J, Chen J, et al. Ribosome profiling reveals dynamic translational landscape in maize seedlings under drought stress. Plant J. 2015;84(6):1206–1218.
- Liu XD, Xh L, Wh L, et al. Analysis on difference for drought responses of maize breeds at seedling stage. J Maize Sci. 2004;3:63–65.
- Bin WU, Li XH, Mu JX, et al. Genetic variation in fifty-three maize breeds in relation to drought tolerance at seedling stage. Sci Agric Sin. 2007;4:665–676.
- Jurgens SK, Johnson RR, Boyer JS. Dry matter production and translocation in maize subjected to drought during grain fill. Agron J. 1978;70(4):678–682.
- Lu DL, Cai XM, Lu WP. Effects of water deficit during grain filling on the physicochemical properties of waxy maize starch. Starch. 2015;67(7–8):692–700.
- Jin XN, Fu ZY, Ding D, et al. Proteomic identification of genes associated with maize grain-filling rate. PLoS One. 2013;8(3):e59353.
- Zhilan X, Han K, Gu L, et al. Transcriptome analysis of heterosis in maize (Zea mays L.) hybrid longping 206. J Agric Biotechnol. 2017;25:709–721.
- Li G, Gao HY, Zhao B, et al. Effects of drought stress on activity of photosystems in leaves of maize at grain filling stage. Acta Agronom Sin. 2009;35(10):1916–1922.
- Mu XH, Chen QW, Chen FJ, et al. Within-leaf nitrogen allocation in adaptation to low nitrogen supply in maize during grain-filling stage. Front Plant Sci. 2016;7:699.
- Geng LI, Hui-Yuan G, Peng L, et al. Effects of nitrogen fertilization on photosynthetic performance in maize leaf at grain filling stage. Plant Nutr Fertil Sci. 2010;16:536–542.
- Kosová K, Vítámvás P, Prášil IT, et al. Plant proteome changes under abiotic stress—contribution of proteomics studies to understanding plant stress response. J Proteomics. 2011;74(8):1301–1322.
- Si W, Fen N, Qinbin Z, et al. Enhancing omics research of crop responses to drought under field conditions. Front Plant Sci. 2017;8:174.
- Pérez-Clemente RM, Vives V, Zandalinas SI, et al. Biotechnological approaches to study plant responses to stress. Biomed Res Int. 2013;2013:654120.
- Shuzhen W, Wenyue C, Wenfei X, et al. Differential proteomic analysis using itraq reveals alterations in hull development in rice (Oryza sativa L.). PLoS One. 2015;10(7):e0133696.
- Ma C, Zhou J, Chen G, et al. iTRAQ-based quantitative proteome and phosphoprotein characterization reveals the central metabolism changes involved in wheat grain development. BMC Genomics. 2014;15:1029.
- Jin HY, Liu ST, Zenda T, et al. Maize leaves drought-responsive genes revealed by comparative transcriptome of two cultivars during the filling stage. PLoS One. 2019;14(10):e0223786.
- Hsiao TC. Plant responces to water stress. Annu Rev Plant Physiol. 1973;24(1):519–570.
- Swägger H. Tricine-SDS-PAGE. Nat Protoc. 2006;1:16–22.
- Liu XD, Xie L, Wei Y, et al. Abiotic stress resistance, a novel moonlighting function of ribosomal protein rpl44 in the halophilic fungus aspergillus glaucus. Appl Environ Microbiol. 2014;80(14):4294–4300.
- Cui D, Wu D, Liu J, et al. Proteomic analysis of seedling roots of two maize inbred lines that differ significantly in the salt stress response. PLoS One. 2015;10(2):e0116697.
- Tai FJ, Yuan ZL, Wu XL, et al. Identification of membrane proteins in maize leaves, altered in expression under drought stress through polyethylene glycol treatment. Report. Plant Omics. 2011;4:250–256.
- Benešová M, Holá D, Fischer L, et al. The physiology and proteomics of drought tolerance in maize: early stomatal closure as a cause of lower tolerance to short-term dehydration? PLoS One. 2012;7(6):e38017.
- Shu LB, Ding W, Wu JH, et al. Proteomic analysis of rice leaves shows the different regulations to osmotic stress and stress signals. J Integr Plant Biol. 2010;52(11):981–995.
- Mazzucotelli E, Mastrangelo AM, Crosatti C, et al. Abiotic stress response in plants: when post-transcriptional and post-translational regulations control transcription. Plant Sci. 2008;174(4):420–431.
- Yamaguchi-Shinozaki K, Shinozaki K. Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu Rev Plant Biol. 2006;57:781–803.
- Rushton PJ, Somssich IE. Transcriptional control of plant genes responsive to pathogens. Curr Opin Plant Biol. 1998;1(4):311–315.
- Mesbah K, Camus A, Babinet C, et al. Mutation in the Trapalpha/Ssr1 gene, encoding translocon-associated protein alpha, results in outflow tract morphogenetic defects. Mol Cell Biol. 2006;26(20):7760–7771.
- Fons RD, Bogert BA, Hegde RS. Substrate-specific function of the translocon-associated protein complex during translocation across the ER membrane. J Cell Biol. 2003;160(4):529–539.
- Kang Y, Udvardi M. Global regulation of reactive oxygen species scavenging genes in alfalfa root and shoot under gradual drought stress and recovery. Plant Signal Behav. 2012;7(5):539–543.
- Farooq M, Wahid A, Kobayashi N, et al. Plant drought stress: effects, mechanisms and management. Agronom Sustain Agric. 2009;29:185–212.
- Nakano R, Matsumura T, Sakakibara H, et al. Cloning of maize ferredoxin iii gene: presence of a unique repetitive nucleotide sequence within an intron found in the 5'-untranslated region. Plant Cell Physiol. 1997;38(10):1167–1170.
- Alam J, Whitaker RA, Krogmann DW, et al. Isolation and sequence of the gene for ferredoxin i from the cyanobacterium anabaena sp. strain pcc 7120. J Bacteriol. 1986;168(3):1265–1271.
- Sofo A, Dichio B, Xiloyannis C, et al. Antioxidant defences in olive trees during drought stress: changes in activity of some antioxidant enzymes. Funct Plant Biol. 2005;32(1):45–53.
- Hunter T. Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell. 1995;80(2):225–236.
- Ding X, Cao Y, Huang L, et al. Activation of the indole-3-acetic acid-amido synthetase gh3-8 suppresses expansin expression and promotes salicylate- and jasmonate-independent basal immunity in rice. Plant Cell. 2008;20(1):228–240.
- Peat TS, Bottcher C, Newman J, et al. Crystal structure of an indole-3-acetic acid amido synthetase from grapevine involved in auxin homeostasis. Plant Cell. 2012;24(11):4525–4538.
- Fatmi MQ, Chang CEA, Mozzarelli A. The role of oligomerization and cooperative regulation in protein function: the case of tryptophan synthase. PLoS Comput Biol. 2010;6(11):e1000994.
- Hinchman SK, Henikoff S, Schuster SM. A relationship between asparagine synthetase A and aspartyl tRNA synthetase. J Biol Chem. 1992;267(1):144–149.
- Pickart CM, Rose IA. Mechanism of ubiquitin carboxyl-terminal hydrolase. borohydride and hydroxylamine inactivate in the presence of ubiquitin. J Biol Chem. 1986;261(22):10210–10217.
- Pickart CM, Rose IA. Ubiquitin carboxyl-terminal hydrolase acts on ubiquitin carboxyl-terminal amides. J Biol Chem. 1985;260(13):7903–7910.
- Zhang Z, Li J, Liu H, et al. Roles of ubiquitination-mediated protein degradation in plant responses to abiotic stresses. Environ Exp Bot. 2015;114:92–103.
- Pace HC, Brenner C. The nitrilase superfamily: classification, structure and function. Genome Biol. 2001;2(1):REVIEWS0001.
- Brenner C. Catalysis in the nitrilase superfamily. Curr Opin Struct Biol. 2002;12(6):775–782.
- Rincón-Limas DE, Krueger DA, Patel PI. Functional characterization of the human hypoxanthine phosphoribosyltransferase gene promoter: evidence for a negative regulatory element. Mol Cell Biol. 1991;11(8):4157–4164.
- Ding H, Yue LJ, Yang CL. Progress in the study of hypoxanthine guanine phosphoribtransferase. Genetics. 2013;35:948–954.
- Francone OL, Gurakar A, Fielding C. Distribution and functions of lecithin:cholesterol acyltransferase and cholesteryl ester transfer protein in plasma lipoproteins. Evidence for a functional unit containing these activities together with apolipoproteins A-I and D that catalyzes the esterif. The J Biol Chem. 1989;264:7066–7072.
- Persson O, Valadi A, Thomas N, et al. Metabolic control of the Escherichia coli universal stress protein response through fructose-6-phosphate. Mol Microbiol. 2007;65(4):968–978.
- Isokpehi RD, Simmons SS, Cohly Ekunwe SIN, et al. Identification of drought-responsive universal stress proteins in viridiplantae. Bioinform Biol Insights. 2011;5:41–58.
- Tkaczuk KLA, Shumilin I, Chruszcz M, et al. Structural and functional insight into the universal stress protein family. Evol Appl. 2013;6(3):434–449.
- Loukehaich R, Wang T, Ouyang BZ, et al. SpUSP, an annexin-interacting universal stress protein, enhances drought tolerance in tomato. J Exp Bot. 2012;63(15):5593–5606.
- Sridhar VV, Kapoor A, Zhang K, et al. Control of DNA methylation and heterochromatic silencing by histone H2B deubiquitination. Nature. 2007;447(7145):735–738.
- Liu X, Luo M, Zhang W, et al. Histone acetyltransferases in rice (Oryza sativa L.): phylogenetic analysis, subcellular localization and expression. BMC Plant Biol. 2012;12:145.
- Chinnusamy V, Zhu J-K. Epigenetic regulation of stress responses in plants. Curr Opin Plant Biol. 2009;12(2):133–139.
- Pan LN. [Epigenetic regulation of abiotic stress response in plants to improve the stress tolerance]. Yi Chuan. 2013;35(6):745–751.
- Tariq M, Saze H, Probst AV, et al. Erasure of cpg methylation in arabidopsis alters patterns of histone h3 methylation in heterochromatin. Proc Natl Acad Sci USA. 2003;100(15):8823–8827.
- Sokol A, Kwiatkowska A, Jerzmanowski A, et al. Up-regulation of stress-inducible genes in tobacco and arabidopsis cells in response to abiotic stresses and aba treatment correlates with dynamic changes in histone h3 and h4 modifications. Planta. 2007;227(1):245–254.,
- Sun ZW, Allis CD. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature. 2002;418(6893):104–108.
- Ng HH, Xu RM, Zhang Y, et al. Ubiquitination of histone H2B by Rad6 is required for efficient Dot1-mediated methylation of histone H3 lysine 79. J Biol Chem. 2002;277(38):34655–34657.
- Ohki I, Shimotake N, Fujita N, et al. Solution structure of the methyl-CpG-binding domain of the methylation-dependent transcriptional repressor MBD1. EMBO (European Molecular Biology Organization). EMBO J. 1999;18(23):6653–6661.
- Roloff TC, Ropers HH, Nuber UA. Comparative study of methyl-CpG-binding domain proteins. BMC Genomics. 2003;4(1):1.
- Du Q, Luu PL, Stirzaker C, et al. Methyl-CpG-binding domain proteins: readers of the epigenome. Epigenomics. 2015;7(6):1051–1073.
- Callahan MA, Handley MA, Lee YH, et al. Functional interaction of human immunodeficiency virus type 1 Vpu and Gag with a novel member of the tetratricopeptide repeat protein family. J Virol. 1998;72(6):5189–5197.
- Wang H, Shen H, Wang Y, et al. Overexpression of small glutamine-rich tpr-containing protein promotes apoptosis in 7721 cells. FEBS Lett. 2005;579(5):1279–1284.
- Allan RK, Ratajczak T. Versatile TPR domains accommodate different modes of target protein recognition and function. Cell Stress Chaperones. 2011;16(4):353–367.
- Bennett-Lovsey R, Hart SE, Shirai H, et al. The SWIB and the MDM2 domains are homologous and share a common fold. Bioinformatics. 2002;18(4):626–630.
- Vieira WA, Coetzer Thérèsa L. Localization and interactions of Plasmodium falciparum SWIB/MDM2 homologues. Malar J. 2016;15:32.
- Frum R, Ramamoorthy M, Mohanraj L, et al. MDM2 controls the timely expression of cyclin A to regulate the cell cycle. Mol Cancer Res. 2009;7(8):1253–1267.
- Kushwaha HR, Singh AK, Sopory SK, et al. Genome wide expression analysis of CBS domain containing proteins in Arabidopsis thaliana (L.), Heynh and Oryza sativa L. reveals their developmental and stress regulation. BMC Genomics. 2009;10:200.
- Zhu QL, Li MY, Liu GD, et al. Molecular characterization and functional prediction of novel leaf sag encoding a CBS-domain-containing protein from coleus blumei. Chin J Biochem Mol Biol. 2007;23:262–270.
- Singh AK, Kumar R, Pareek A, et al. Overexpression of rice cbs domain containing protein improves salinity, oxidative, and heavy metal tolerance in transgenic tobacco. Mol Biotechnol. 2012;52(3):205–216.
- Salem AMH, Nakano T, Takuwa M, et al. Genetic analysis of repair and damage tolerance mechanisms for DNA-protein cross-links in escherichia coli. J Bacteriol. 2009;191(18):5657–5668.
- Karran P, Bignami M. DNA damage tolerance, mismatch repair and genome instability. Bioessays. 1994;16(11):833–839.
- Chen YH, Szakal B, Castellucci F, et al. DNA damage checkpoint and recombinational repair differentially affect the replication stress tolerance of Smc6 mutants. Mol Biol Cell. 2013;24(15):2431–2441.
- Li X, Heyer W. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008;18(1):99–113.
- Roche Y, Zhang D, Segers-Nolten GMJ, et al. Fluorescence correlation spectroscopy of the binding of nucleotide excision repair protein XPC-hHr23B with DNA substrates. J Fluoresc. 2008;18(5):987–995.
- Rodrigo G, Roumagnac S, Wold MS, et al. DNA replication but not nucleotide excision repair is required for uvc-induced replication protein a phosphorylation in mammalian cells. Mol Cell Biol. 2000;20(8):2696–2705.
- Hoogstraten D, Bergink S, Verbiest VHM, et al. Versatile DNA damage detection by the global genome nucleotide excision repair protein XPC. J Cell Sci. 2008;121(Pt 17):2850–2859.
- Min JH, Pavletich NP. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature. 2007;449(7162):570–575.
- Jaciuk M, Nowak E, Skowronek K, et al. Structure of uvra nucleotide excision repair protein in complex with modified DNA. Nat Struct Mol Biol. 2011;18(2):191–197.
- Zenda T, Liu S, Wang X, et al. Comparative proteomic and physiological analyses of two divergent maize inbred lines provide more insights into drought-stress tolerance mechanisms. IJMS. 2018;19(10):3225.
- Wang X, Zenda T, Liu S, et al. Comparative proteomics and physiological analyses reveal important maize filling-kernel drought-responsive genes and metabolic pathways. IJMS. 2019;20(15):3743.