Abstract
Quantitative structure-activity relationships (QSARs) within a series of cytochrome P450 2C9 (CYP2C9) and cytochrome P450 2C19 (CYP2C19) inhibitors are reported. In particular, it is noted that compound lipophilicity, in the form of log P values (where P is the octanol/water partition coefficient), is an important factor in explaining the variation in inhibitory potency within these series of compounds, many of which also act as substrates for the respective enzymes. In addition, there is a role for hydrogen bonding and π-π stacking interactions within the P450 active site which represent secondary factors in the binding processes of these compounds.
Introduction
The cytchromes P450 (CYP) are a superfamily of heme-thiolate enzymes which are present in species from all five biological kingdoms Citation1-3, and about 4500 individual members are currently known (D.R. Nelson, personal communication). In mammalian systems, it is apparent that enzymes of the CYP1, CYP2 and CYP3 families are primarily associated with the Phase 1 metabolism of drugs and other xenobiotics Citation3-5. This situation holds true for human drug metabolism where CYP1A2, CYP2C9, CYP2D6 and CYP3A4 enzymes metabolize the majority of known drug substrates [Citation3]; there are smaller contributions from CYP2A6, CYP2B6, CYP2C8, CYP2C19 and CYP2E1, however. The CYP2C subfamily overall plays a significant role in the Phase 1 metabolism of many drug substrates, including:- omeprazole (CYP2C19), S-warfarin (CYP2C9), S-mephenytoin (CYP2C19), diclofenac (CYP2C9), phenytoin (CYP2C9 and CYP2C19), taxol (CYP2C8) and carbamazepine (CYP2C8), for example [Citation6]. Selective inhibitors are known for CYP2C8 (quercetin), CYP2C9 (sulfaphenazole) and CYP2C19 (N-benzylnirvanol and N-benzylphenobarbital) with the latter exhibiting stereoselectivity towards this enzyme Citation7-8.
Two human CYP2C subfamily enzymes have been resolved crystallographically Citation9-11 and a rabbit CYP2C isoform (namely, CYP2C5) has also had its crystal structure determined, with and without the presence of a bound substrate Citation12-14. Structural models of CYP2C8 and CYP2C9 built by homology with CYP2C5 have been shown to possess conformations close to that of the CYP2C8 and CYP2C9 crystal structures [Citation15] with average root mean square distances for the α-carbons of 1.2Å and 1.5Å, respectively. Consequently, it would appear that modelling by homology from an optimal crystallographic template within the same subfamily, where primary sequence identities are between 75% and 80%, represents a satisfactory method for investigating enzyme-substrate and enzyme-inhibitor interactions in P450s.
Methods
Using a multiple sequence alignment of CYP2 family enzymes, models of CYP2C9 and CYP2C19 were constructed via homology with substrate-bound CYP2C5 (pdb code: 1n6b), which is of known structure [Citation12] and the methodology employed has been published previously [Citation6]. These models were used for the docking of inhibitors of both CYP2C9 and CYP2C19, in order to establish their likely mode of binding to the respective enzymes. The procedures involved an interactive docking process in Sybyl (Tripos Associates) which also utilizes a dynamic hydrogen bond facility. In this way, the hydrogen bond forming possibilities can be monitored as the substrate is docked within the active site, and the presence of a bound substrate, dimethyl-2-phenyl-2H-pyrazol-3-ylbenzenesulfonamide (DMZ), in the active site region of the CYP2C5 crystal structure aids in this process [Citation12].
The CYP2C9 crystal structure produced by the Astex group [Citation9] represents a useful template for homology modelling of CYP2C19, where there is a 91% sequence identity. In addition, this crystal structure of a mutant form of the enzyme contains a proline residue at position 220, which is the same as that encountered in CYP2C19, and is thought to play a crucial role in substrate recognition and selectivity, as has been reported from site-directed mutagenesis experiments [Citation16]. Consequently, it would appear that the Astex CYP2C9 crystal structure (pdb code: 1og5) is the most appropriate for constructing a homology model of CYP2C19, and this was our preference in the present study. However, for the docking of CYP2C9 substrates and inhibitors, the recently published crystal structure [Citation11] was employed (pdb code: 1r9o) although some missing sections of peptide required loop-searching of the pdb for their addition, followed by energy minimization using molecular mechanics (Tripos force field) to provide a satisfactory protein geometry. These three-dimensional structures of CYP2C9 and CYP2C19 were employed for substrate and inhibitor docking in order to identify the possibility of hydrogen-bonded and π-π stacking contacts between the substrate/inhibitor and enzyme active site residues.
The biological data on inhibitors of CYP2C9 and CYP2C19 has been taken from the literature Citation17-18 and the Km or Ki values converted into the negative logarithmic form, or into the related free energies, ΔGbind, for the generation of QSARs and lipophilicity relationship analysis (see ). We have found previously Citation19-21 that this is a useful method for exploring the lipophilicity relationships of P450 substrates in general. In particular, the intercept on the y axis of such a relationship enables a direct estimation of the enzyme-substrate interaction energy. This is presumed to be composed of a combination of hydrogen bond and π-π stacking energies, as distinct from the desolvation component to the binding energy estimated from compound lipophilicity. In fact, findings so far appear to support this approach by agreement with the molecular modelling, QSAR and experimental approaches.
Table I. Dataset for CYP2C9 inhibitors.
Results and discussion
presents a dataset for 15 CYP2C9 inhibitors with biological data (in the form of affinity for CYP2C9) expressed as either Ki or Km values taken from the literature [Citation17]. These raw data were converted into their logarithmic form, − log Ki or − log Km, for the purposes of correlation, and they were also expressed as ΔGbind values where ΔGbind = RTlnK, where K may refer to either Ki or Km in this context, R is the gas constant and T is the absolute temperature, taken as 310K. We have shown previously Citation20-21 that the employment of such conversions into energy values facilitates the application of lipophilicity relationships in P450-substrate interactions, in addition to that of other forms of QSAR analysis which tend to employ logarithmic forms of the biological activity. Such transformations of the raw data seem to work well in establishing the likely contributions to the overall binding energy, including those not related to lipophilic character, such as hydrogen bonds and π-π stacking interactions.
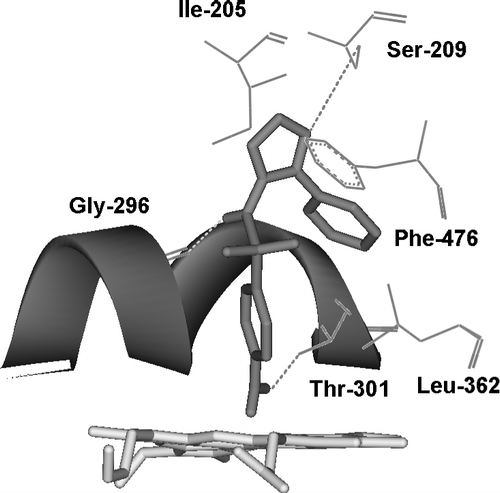
It is apparent that linear relationships exist between compound lipophilicity and CYP2C9 inhibition/binding for the 15 compounds including several NSAIDs listed in , despite the fact that some of the values are for inhibition (Ki) and some are for metabolism (Km). Although the slopes are almost identical, the two sets of equations are of different intercept and, in graphical terms, these appear as two parallel lines of essentially equivalent gradient (namely, 0.369 and 0.328). The likely mode of binding for a typical CYP2C9 inhibitor, such as sulfaphenazole, is shown in , where potential hydrogen-bonded contacts with active site residues are indicated.
Figure 1 The active site region of CYP2C9 containing the bound inhibitor, Sulfaphenazole (pink), with close contact residues labelled and hydrogen bonds shown as dashed lines.
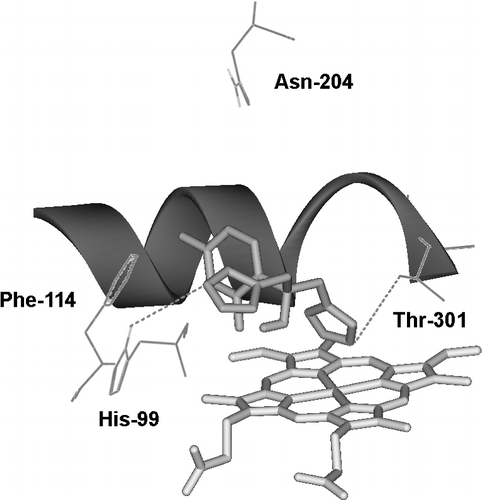
presents data for 10 inhibitors of CYP2C19 with Ki values taken from the published literature Citation22-24. The ΔGpart values generated from log P data on the 10 compounds shows a good correlation with ΔGbind for the inhibitory activity, although it is apparent that the numbers of non-lipophilic active site interactions varies within the dataset. This has been compensated for by evaluating the baseline lipophilicity relationship graphically to reveal outliers and then correcting the binding energy data accordingly, based on the energy differences between outlier and baseline relationship (data not shown). The particular compounds involved are noted in , together with the energy corrections made for π-π stacking and hydrogen bonding. By this procedure, an overall correlation of 0.98 can be achieved (R2 = 0.96) thus illustrating the utility of this approach to lipophilicity analysis. The probably mode of binding for a typical CYP2C19 inhibitor, such as fluconazole, is shown in , where potential hydrogen-bonded contacts with active site residues are indicated.
Table II. CYP2C19 Inhibitors and their physicochemical and binding characteristics.
Figure 2 The active site region of CYP2C19 containing the bound inhibitor, Fluconazole (pale pink), with close contact residues labelled and hydrogen bonds shown as dashed lines.
The two figures represent examples of how inhibitors of CYP2C9 and CYP2C19 may bind to their respective enzymes and interact with complementary residues in the two active sites. In general, the molecular modelling tends to support the findings from the various lipophilicity relationships reported in this work. Indeed, it is possible to explain some of these findings by consideration of the likely active site interactions encountered for different inhibitors.
Furthermore, the graphical construction process leading to the lipophilicity relationships shown in Tables and enables the estimation of common interaction energies from the intercept on the y-axis, and also the treatment of outliers, in terms of additional active site interactions. As such, this would appear to be a more informative and rational approach to investigating enzyme-substrate binding interactions within the human P450s. In most cases, more than one lipophilicity line is observed and these tend to run parallel to each other with differing intercepts on the y-axis, corresponding to more than one common non-lipophilic interaction, such as an additional hydrogen bond or aromatic π-π stacking interaction. In this way, the lipophilic relationships, and their analysis in the light of substrate/inhibitor interactions within the P450 active site, help to explain the observed differences in binding affinity and relative extent of inhibition associated with enzymes in the human CYP2C subfamily.
Distinctions between compounds which are selective towards these two closely-related enzymes centre upon the generally more acidic character of CYP2C9 substrates and inhibitors, and the likely reationale for this stems from the presence of an active site basic residue, although it is probable that other residues are also involved in substrate discrimination, including those close to the periphery of this region of the enzymes concerned, as has been reported from site-directed mutagenesis studies [Citation16].
Conclusions
Inhibition of the human CYP2C subfamily enzymes, CYP2C9 and CYP2C19, can be rationalized in terms of the lipophilicity character of the compounds concerned. In particular, the construction of lipophilicity relationships facilitates an understanding of the structural determinants leading to inhibition brought about by specific interactions within the enzymes' active sites. These specific interactions may be due to hydrogen bonding, π-π stacking or a combination of the two, and this likelihood is evidenced by the different intercept values obtained in the various lipophilicity relationship expressions. Interestingly, the findings from the QSAR analyses are consistent with those obtained from molecular modelling studies of the enzymes concerned, which indicates a degree of complementarity between the two approaches.
| Abbreviations | ||
| CYP: | = | cytochrome |
| P450 NSAIDS: | = | Non-steroidal anti-inflammatory drugs |
| P: | = | octanol/water partition coefficient |
| pKa: | = | − log Ka where Ka is the acid/base dissociation constant |
| D7.4: | = | distribution coefficient at pH 7.4 |
Acknowledgements
The financial support of GlaxoSmithKline Research & Development Limited, Merck Sharp & Dohme Limited, the British Technology Group and the University of Surrey Foundation Fund is gratefully acknowledged by DFVL.
Related Research Data
References
- Lewis DFV. Cytochromes P450: Structure, Function and Mechanism. Taylor & Francis, London 1996
- Lewis DFV. Guide to Cytochromes P450 Structure and Function. Taylor & Francis, London 2001
- Rendic S. Summary of information on human CYP enzymes: Human P450 metabolism data. Drug Metabolism Reviews 2002; 34: 83–448
- Evans WE, Relling MV. Pharmacogenomics: Translating functional genomics into rational therapeutics. Science 1999; 286: 487–491
- Ingelman-Sundberg M. Implications of polymorphic cytochrome P450-dependent drug metabolism for drug development. Drug Metab Disp 2001; 29: 570–573
- Lewis DFV, Dickins M, Lake BG, Goldfarb PS. Investigation of enzyme selectivity in the human CYP2C subfamily: Homology modelling of CYP2C8, CYP2C9 and CYP2C19 from the CYP2C5 crystallographic template. Drug Metabolism and Drug Interactions 2003; 19: 257–285
- Suzuki H, Kneller MB, Haining RL, Trager WF, Rettie AE. (+)-N-3-benzyl nirvanol and ( − )-N-3-benzyl phenobarbital: New potent and selective in vitro inhibitors of CYP2C19. Drug Metabolism and Disposition 2002; 30: 235–239
- Cai X, Wang RW, Edom RW, Evans DC, Shou M, Rodrigues AD, Liu W, Dean DC, Baillie TA. Validation of ( − )-N-3-benzyl phenobarbital as a selective inhibitor of CYP2C19 in human liver microsomes. Drug Metabolism and Disposition 2004; 32: 584–586
- Williams PA, Cosme J, Ward A, Angove HC, Vinkovic DM, Jhoti H. Crystal Structure of human cytochrome P450 2C9 with bound warfarin. Nature 2003; 424: 464–468
- Schoch GA, Yano JK, Wester MR, Griffin KJ, Stout CD, Johnson EF. Srtucture of human microsomal cytochrome P40 2C8. J Biol Chem 2004; 279: 9497–9503
- Wester MR, Yano JK, Schoch GA, Yang C, Griffin KJ, Stout CD, Johnson EF. The structure of human cytochrome P450 2C9 complexed with flurbiprofen at 2.0Å resolution. Journal of Biological Chemistry 2004; 279: 35630–35637
- Wester MR, Johnson EF, Marques-Soares C, Dansette PM, Mansuy D, Stout CD. Structure of a substrate complex of mammalian cytochrome P450 2C5 at 2.3Å resolution: Evidence for multiple substrate binding modes. Biochemistry 2003a; 42: 6370–6379
- Wester MR, Johnson EF, Marques-Soares C, Dijols S, Dansette PM, Mansuy D, Stout CD. Structure of mammalian cytochrome P450 2C5 complexed with diclofenac at 2.1Å resolution: Evidence for an induced fit model of substrate binding. Biochemistry 2003b; 42: 9335–9345
- Williams PA, Cosme J, Sridhar V, Johnson EF, McRee DE. Mammalian cytochrome P450 monooxygenase: Structural adaptations for membrane binding and functional diversity. Molecular Cell 2000; 5: 121–131
- Lewis DFV. 57 varieties: The human cytochromes P450. Pharmacogenomics 2004; 5: 305–318
- Ibeanu GS, Ghanayem BI, Linko P, Li L, Pedersen LG, Goldstein JA. Identification of residues 99, 220 and 221 of human cytochrome P450 2C19 as key determinants of omeprazole hydroxylase activity. Journal of Biological Chemistry 1996; 271: 12496–12501
- Leeman TD, Transon C, Bonnabry P, Dayer P. A major role for cytochrome P450TB (CYP2C subfamily) in the actions of non-steroidal anti-inflammatory drugs. Drugs Under Experimental Clinical Research 1993; 19: 189–195
- Li X-Q, Andersson TB, Ahlstrom M, Weidolf L. Comparison of inhibitory effects of the proton pump-inhibiting drugs omeprazole, esomeprazole, lansoprazole, pantoprazole and rabeprazole on human cytochrome P450 activities. Drug Metabolism and Disposition 2004; 32: 821–827
- Lewis DFV. On the estimation of binding affinity (Gbind) for human P450 substrates based on Km and KD values. Current Drug Metabolism 2003; 4: 331–340
- Lewis DFV, Dickins M. Baseline lipophilicity relationships in human cytochromes P450 associated with drug metabolism. Drug Metabolism Reviews 2003; 35: 1–18
- Lewis DFV, Jacobs MN, Dickins M. Compound lipophilicity for substrate binding to human P450s in drug metabolism. Drug Discovery Today 2004; 9: 530–537
- Inaba T, Jurima M, Mahon WA, Kalow W. In vitro inhibition studies of two isozymes for human liver cytochrome P450. Drug Metabolism and Disposition 1985; 13: 443–488
- Wienkers LC, Wurden CJ, Storch E, Kunze KL, Rettie AE, Trager WF. Formation of (R)-8-hydroxywarfarin in human liver microsomes: A new metabolic marker for the (S)-mephenytoin hydroxylase, P4502C19. Drug Metabolism and Disposition 1996; 24: 610–614
- VandenBranden M, Ring BJ, Binkley SN, Wrighton SA. Interaction of human liver cytochromes P450 in vitro with LY307640, a gastric proton pump inhibitor. Pharmacogenetics 1996; 6: 81–91
- Hansch C, Leo A, Hoekman D. Exploring QSAR: Hydrophobic, Electronic and Steric Constants. American Chemical Society, Washington 1995
- Newton DW, Kluza RB. pKa values of medicinal compounds in pharmacy practice. Drug Intelligence and Clinical Pharmacy 1978; 12: 546–554
- Bowman WC, Rand MJ. Textbook of Pharmacology2nd edition. Blackwell, Oxford 1988