Abstract
A series of chiral 1,3,4-oxadiazole-5-thiols incorporating 2-substituted-benzenesulfonamide moieties has been prepared from amino acids, via the ester and carbohydrazide intermediate, followed by cyclization with carbon disulfide. Some of these compounds have been investigated for the inhibition of three physiologically relevant carbonic anhydrase (CA, EC 4.2.1.1) isoforms, the human cytosolic hCA I and II, and the human, transmembrane, tumor-associated isozyme hCA IX. All these compounds showed weak (millimolar) affinity for the three isozymes, except two carbohydrazides and two heterocyclic thiols which selectively inhibited the tumor-associated isozyme with inhibition constants around 10 μM. Such compounds constitute interesting lead molecules for the possible design of CA IX-selective inhibitors.
Introduction
The α-carbonic anhydrases (CAs, EC 4.2.1.1) are widespread metalloenzymes in all life kingdoms Citation1-5. In higher vertebrates, including humans, 16 isoforms have been described up to now, of which at least CA II, IV, VA, VB, VII, IX, XII, XIII and XIV constitute interesting targets for the development of novel antiglaucoma, antitumor, antiobesity or anticonvulsant drugs Citation6-13. Indeed, by catalysing a simple but fundamental reaction, the reversible hydration of CO2 to bicarbonate and a proton, these metalloenzymes are involved in a multitude of physiological and pathological processes, and their inhibition leads to responses that may be exploited therapeutically Citation1-5. Three other genetically distinct classes of CAs have been described in various organisms, the β-CA - δ-CA families, proving that such a critical catalyst for life processes has been invented by nature at least in four different occasions Citation1-6. Among them, very recently, representatives of the α- or β-CA class have been cloned and characterized in Plasmodium falciparum [Citation14], Mycobacterium tuberculosis [Citation15,Citation16], Cryptococcus neoformans [Citation17] or Candida spp. [Citation18], some of them being also investigated from the inhibition aspect [Citation14,Citation17] as it has been proved that these enzymes are critical for the growth or virulence of the pathogens Citation14-18.
Tumor cells over-express at least two α-CA isoforms, the transmembrane CA IX and XII Citation1-5. As tumors have a lower extracellular pH (pHe) than normal cells, and this acidic medium contributes to tumor growth and metastasis through various mechanisms, it has been thought that cancer-associated CAs may play a role in the acidification processes [Citation19]. Indeed, recently CO2 (in addition to lactic acid) was demonstrated to be a significant source of acidity in tumors [Citation20], pointing to the involvement of CA IX and XII in tumor biology [Citation19,Citation20]. Noteworthy, CA IX-mediated acidification of tumor cells was reversed by sulfonamide CA inhibitors not only in transfected cells, but also in tumor cell lines that naturally express the CA IX protein [Citation19,Citation20]. This effect was observed only under hypoxia (the cellular stress triggering CA IX over-expression) [Citation21] and not in normoxic conditions [Citation19,Citation20]. A potent fluorescent sulfonamide CA IX inhibitor was also shown to bind only to hypoxic cells expressing CA IX and not to their normoxic counterparts, or to control cells lacking CA IX [Citation19,Citation20]. Such experiments represented the proof-of-concept that inhibitors accumulate only in cells containing CA IX with an intact catalytic domain, and may thus become useful tools for reducing tumor acidosis or for imaging of hypoxic tumor cells [Citation19,Citation20]. CA IX inhibitors may thus prevent the decrease of pHe observed in many tumors and might be used in combinations with other antitumor drugs to increase the efficacy or the uptake of weakly basic anticancer drugs [Citation19,Citation20]. Continuing our investigations in this field, the aim of the present study was the design, synthesis and in vitro evaluation of novel CA IX inhibitors, belonging to different classes of derivatives, other than the widely used sulfonamides and sulfamates Citation22-26.
Materials and methods
Chemistry
Melting points were determined on a Gallenkamp digital melting point apparatus and are uncorrected. Optical rotation data were recorded on a Perkin-Elmer 241 polarimeter. IR spectra were recorded in KBr discs on a FT-IR model FTS 3000 MX spectrometer. Elemental analysis was performed on a Carlo Erba 1106 elemental analyzer. 1H NMR (400 and 500 MHz) spectra were recorded on a Bruker NMR spectrophotometer. The chemical shifts of proton signals are in parts per million (ppm) downfield from tetramethylsilane (TMS) as an internal standard. EI-MS spectra were recorded on a MAT 312 and MAT 311A mass spectrometer. Thin layer chromatography (TLC) was performed on precoated silica gel 60 F254 aluminum sheets (Merck). The purity of the synthesized compounds was established by TLC in two different solvent systems (ethyl acetate: petroleum ether 1:2 and acetone: petroleum ether 1:1). The clinically used sulfonamide CA inhibitors (CAIs) acetazolamide AAZ, methazolamide MZA, ethoxzolamide EZA, dichlorophenamide DCP and indisulam IND, employed as standard inhibitors in the enzyme assays, were commercially available from Sigma-Aldrich or have been prepared as previously described [Citation26]. Recombinant human CA isoforms hCA I, II and IX have been prepared as reported earlier by our group Citation28-31, and their activity measured by a stopped flow CO2 hydration assay [Citation32].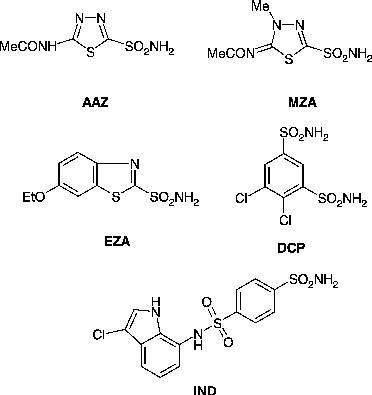
The investigated 1,3,4-oxadiazole-5-thiol derivatives 5a-o were prepared from commercially available (from Sigma-Aldrich) amino acids 1a-e whereas the corresponding esters 3a-o were obtained by literature procedures Citation33-35. Compound 2 g (yield 72%, m.p. 149°C, lit. 149°C) was previously reported in the literature [Citation36].
General procedure for the synthesis of compounds 4a-o
A mixture of 3a (10 mmole) and hydrazine hydrate (80%) in absolute ethanol (50 mL) was refluxed for 9 h. The excess solvent was distilled off and the residue was filtered, washed with water and recrystallized from 60% aqueous ethanol. Other hydrazides 4b-o were prepared in a similar fashion. The spectral data for 4a-o are given below.
General procedure for the synthesis of compounds 5a-o
Compound 4a (5.5 mmole) was dissolved in 80 ml absolute ethanol, and 6.6 mmole of carbon disulfide was added followed by 5.5 mmole of potassium hydroxide dissolved in 10 ml of water. The reaction mixture was stirred for 15 min and then refluxed for 16.5 h. The progress of the reaction was monitored by TLC. The excess ethanol was distilled off. The reaction mixture was diluted with water and acidified with 4N HCl to pH 2-3 (Congo red). The solid obtained was filtered, washed with water and recrystallized from 60% aqueous ethanol. Other compounds 5b-o were synthesized by the same procedure. Micro analytical, optical rotation, IR, 1H-NMR and mass spectral data of compounds 5a-o are given below.
General procedure for the synthesis of compounds 6a-b, 6k
An amount of 0.75 mmole (250 mg) of 5a, 0.22 mmole (0.3 ml) of Et3N and a catalytic amount (25 mg) of 4-dimethylamino-pyridine (DMAP) were stirred in 25mL of dry CH3Cl for 15 min. 0.8 mmole (0.1 mL) of benzyl bromide was added and the mixture was stirred for 5 h at 30– 70°C. The reaction mixture was washed with dilute HCl, brine, water and dried over anhydrous Na2SO4. The excess solvent was distilled off and the product was recrystallized from aqueous ethanol. The synthesized compounds 6a-b and 6k were characterized by IR, 1H-NMR and MS data.
2b: Micro crystals (aq. ethanol) m.p. 135–137°C, Yield = 71%; IR (KBr)(ν, cm− 1) 3311– 2750(NH, OH), 1707(C = O), 1375 and 1157(SO2).
2c: Micro crystals (aq. ethanol) m.p. 140–142°C, Yield = 72%, IR (KBr)(ν, cm− 1) 3289– 2680 (NH, OH), 1716(C = O), 1376 and 1167(SO2).
2e: Micro crystals (aq. ethanol) m.p. 145–147°C, Yield = 77%, IR (KBr)(ν, cm− 1) 3307– 2731 (NH, OH), 1725(C = O), 1365 and 1159(SO2).
2f: Crystalline (aq. ethanol) m.p. 139–142°C, Yield = 74%, IR (KBr)(ν, cm− 1) 3351– 2690 (NH, OH), 1733(C = O), 1375 and 1166(SO2).
2j: Micro crystals (aq. ethanol) m.p. 135–137°C, Yield = 75%, IR (KBr)(ν, cm− 1) 3315– 2695(NH, OH), 1717(C = O), 1376 and 1165(SO2).
2 l: Micro crystals (aq. ethanol) m.p. 133–135°C, Yield = 71%, IR (KBr)(ν, cm− 1) 3285– 2711(NH, OH), 1714(C = O), 1375 and 1166(SO2).
3b: Needles (aq. ethanol) m.p. 47–48°C, Yield = 71%, IR (KBr)(ν, cm− 1) 3237(NH), 1729(C = O), 1375 and 1165(SO2).
3c: Needles (aq. ethanol) m.p. 51–53°C, Yield = 67%, IR (KBr)(ν, cm− 1) 3255 (NH), 1717(C = O), 1376 and 1166(SO2).
3e: Needles (aq. ethanol) m.p. 87–89°C, Yield = 81%, IR (KBr)(ν, cm− 1) 3256(NH), 1721(C = O), 1355 and 1159(SO2).
3f: Micro crystals (aq. ethanol) m.p. 87–89°C, Yield = 67%, IR (KBr)(ν, cm− 1) 3277(NH), 1726(C = O), 1377 and 1167(SO2).
3 g: Crystalline (aq. ethanol) m.p.79°C, Yield = 78%, IR (KBr)(ν, cm− 1) 3256 (NH), 1717 (C = O), 1355 and 1159 (SO2).
3 h: Crystalline (aq. ethanol) m.p. 81°C, Yield = 62%, IR (KBr)(ν, cm− 1) 3309(NH), 1709(C = O), 1357 and 1171(SO2).
3i: Crystalline (aq. ethanol) m.p. 89°C, Yield = 62%, IR (KBr)(ν, cm− 1) 3309(NH), 1709(C = O), 1357 and 1171(SO2).
3j: Micro crystals (aq. ethanol) m.p. 77–78°C, Yield = 80%, IR (KBr)(ν, cm− 1) 3287(NH), 1717(C = O), 1372 and 1165(SO2).
3 l: Micro crystals (aq. ethanol) m.p. 76–78°C, Yield = 71%, IR (KBr)(ν, cm− 1) 3285(NH), 1716(C = O), 1372 and 1166(SO2).
4b: Crystalline (aq. ethanol) m.p. 91–93°C, Yield = 75%, IR (KBr)(ν, cm− 1) 3377, 3285(NH), 1671(C = O), 1376 and 1155(SO2). 1H NMR (acetone-d6) δ 1.66–1.75(m, 2H, CH2), 2.55(t, 2H, CH2, J = 7.4 Hz), 2.81(t, 2H, CH2, J = 6.9 Hz), 7.45(d, 2H, ArH J = 8.0 Hz), 7.89(d, 2H, ArH J = 8.0 Hz), 9.61(bs, 2H, NH), 11.29(bs, 1H, NH). EI-MS (%) 291 (M+, 19), 175(100), 111(87), 76(9), 57(11).
4c: Crystalline (aq. ethanol) m.p. 110–112°C, Yield = 62%, IR (KBr)(ν, cm− 1) 3332, 3260(NH), 1656(C = O), 1372 and 1161(SO2). 1H NMR (acetone-d6) δ 1.69–1.78(m, 2H, CH2), 2.78(t, 2H, CH2, J = 6.9 Hz), 2.84(t, 2H, CH2, J = 7.8 Hz), 3.89 (s, 3H, OCH3), 7.25(d, 2H, ArH J = 8.1 Hz), 7.59(d, 2H, ArH J = 8.2 Hz), 9.57(bs, 2H, NH), 10.75(bs, 1H, NH). EI-MS (%) 287 (M+, 11), 223(10), 185(100), 107(57), 76(8), 57(23).
4e: Crystalline (aq. ethanol) m.p. 161–163°C, Yield = 85%, IR (KBr)(ν, cm− 1) 3319, 3292(NH), 1653(C = O), 1365 and 1149(SO2). 1H NMR (acetone-d6) δ 0.98(dd, 3H, CH3, J = 7.2 Hz, J = 7.2 Hz), 1.82–1.92(m, 2H, CH2), 4.38(t, 1H, CH, J = 7.5 Hz), 7.65(d, 2H, ArH J = 8.0 Hz), 8.01(d, 2H, ArH J = 8.0 Hz), 9.72(bs, 2H, NH), 11.25(bs, 1H, NH). EI-MS (%) 291 (M+, 14), 175(25), 111(100), 76(19), 57(9).
4f:Crystalline (aq. ethanol) m.p. 165–167°C, Yield = 65%, IR (KBr)(ν, cm− 1) 3297, 3242(NH), 1671(C = O), 1375 and 1166(SO2). 1H NMR (Acetone-d6) δ 0.89(t, 3H, CH3, J = 7.4 Hz), 1.84–1.89(m, 2H, CH2), 3.91(s, 3H, OCH3), 4.37(t, 3H, CH3, J = 7.4 Hz), 7.12(d, 2H, ArH J = 8.2 Hz), 7.72(d, 2H, ArH J = 8.2 Hz), 9.75(bs, 2H, NH), 10.75(bs, 1H, NH). EI-MS (%) 287 (M+, 100), 230(7), 211(11), 185(9), 107(21), 76(5), 57(9).
4 g: Powder (aq. ethanol) m.p. 171–172°C, Yield = 77%, IR (KBr)(ν, cm− 1) 3319, 3292 (NH), 1641 (C = O), 1323 and 1149 (SO2). 1H NMR (acetone-d6) δ = 0.65 (d, 3H, CH3, J = 6.7 Hz), 0.82 (d, 3H, CH3, J = 7.0 Hz), 2.02–2.05 (m, 1H, CH), 2.10–2.15 (m, 1H, CH), 2.40 (s, 3H, CH3), 4.25 (dd, 1H, *CH, J = 8 Hz, J = 9.0 Hz), 7.24 (d, 2H, ArH, J = 8.0 Hz), 7.60 (d, 2H, ArH, J = 8.0 Hz), 9.20 (bs, 2H, NH2), 10.41 (bs, 1H, NH). EI-MS (%) 285 (M+,10), 225(12), 197(8), 171 (20), 155 (66), 130 (11), 91 (100), 85 (26), 57 (15), 41 (11).
4 h: Crystalline (aq. ethanol), m.p. 172–174°C, Yield = 69%, IR (KBr)(ν, cm− 1) 3322, 3292(NH), 1671(C = O), 1369 and 1165(SO2). EI-MS (%) 305(M+, 19), 111(100).
4i: Crystalline (aq. ethanol), m.p. 169–171°C, Yield = 71%, IR (KBr)(ν, cm− 1) 3376, 3282(NH), 1651(C = O), 1375 and 1166(SO2). EI-MS (%) 301(M+, 11), 117(100).
4j: Micro crystals (aq. ethanol) m.p. 151–153°C, Yield = 81%, IR (KBr)(ν, cm− 1) 3366, 3251(NH), 1675(C = O), 1378 and 1165(SO2). EI-MS (%) 257(M+, 57), 91(100).
4 l: Crystalline (aq. ethanol) m.p. 160–162°C, Yield = 64%, IR (KBr)(ν, cm− 1) 3388, 3282(NH), 1672(C = O), 1375 and 1166(SO2). 1H NMR (Acetone-d6) δ 1.35(d, 3H, CH3, J = 7.4 Hz), 3.91(s, 3H, OCH3), 4.55–4.63 (m, 1H, CH), 7.12(d, 2H, ArH J = 8.0 Hz), 7.66(d, 2H, ArH J = 8.0 Hz), 9.45(bs, 2H, NH), 10.92 (bs, 1H, NH). EI-MS (%) 273(M+, 6), 214(100), 185(5), 107(67), 76(5), 57(10).
4m: Crystalline (aq. ethanol) m.p. 160–162°C, Yield = 64%, IR (KBr)(ν, cm− 1) 3266(NH), 1651(C = O), 1322 and 1151(SO2). 1H NMR (acetone-d6) δ 1.71–1.82 (m, 2H, CH2), 2.38(s, 3H, CH3), 4.37(dd, H, CH, J = 7.6 Hz, J = 7.6 Hz), 7.32(d, 2H, ArH J = 8.0 Hz), 7.66(d, 2H, ArH J = 8.0 Hz), 9.45 (bs, NH), 10.34 (bs, NH). EI-MS (%) 267 (M+, 17), 155(65), 91(100), 75(15). Anal. Calcd for C13H21O3N3: (267.33) C, 58.41; H, 7.92; N, 15.72. Found: C, 58.23; H, 7.81; N, 16.03%.
4n: Crystalline (aq. ethanol), m.p. 165–167°C, Yield = 71%, IR (KBr)(ν, cm− 1) 3331, 3210(NH), 1665(C = O), 1364 and 1155(SO2). EI-MS (%) 319(M+, 7), 111(100).
4o: Crystalline (aq. ethanol), m.p. 172–174°C, Yield = 67%, IR (KBr)(ν, cm− 1) 3369, 3241(NH), 1659(C = O), 1375 and 1165(SO2). EI-MS (%) 315 (M+, 15), 171(100).
5b: Powder (aq. ethanol), m.p 171–172°C, Yield 85%, IR (KBr)(ν, cm− 1) 3287(-NH), 2545(- C = N), 1375 and 1165(-SO2). 1H NMR (acetone-d6) δ1.92–1.98 (m, 2H, CH2), 2.80 (t, 2H, CH2, J = 7.5 Hz), 3.09 (t, 2H, CH2, J = 6.7 Hz), 7.63(d, 2H, ArH J = 8.1 Hz), 7.87 (d, 2H, ArH J = 8.2 Hz), 12.80 (bs, NH). EI-MS (%) 335(M++2), 333 (M+, 7), 260(14), 177(25), 175(69), 158(10), 141(13), 129 (91), 113(32), 112(10), 111(100), 98(24), 76(11), 75(48), 69(29), 56(22), 55(16). Anal. Calcd for C11H12O3N3S2Cl (333.7921): C, 39.58; H, 3.62; N, 12.59; S, 19.21. Found: C, 39.68; H, 3.74; N, 12.77; S, 19.20%.
5c: Powder (aq. ethanol), m.p 159–161, Yield 87%, IR (KBr)(ν, cm− 1) 3216(-NH), 2548(-SH), 1589(- C = N), 1369 and 1179(-SO2). 1H NMR (acetone-d6) δ 1.89–1.96 (m, 2H, CH2), 2.79(t, 2H, CH2 J = 6.7 Hz), 3.00(q, 2H, CH2, J = 6.8 Hz), 3.88(s, 3H, OCH3), 7.07 (d, 2H, ArH J = 8.1 Hz), 7.76 (d, 2H, ArH J = 8.2 Hz), 12.92(bs, NH). EI-MS (%) 330 (M+, 1), 329(M+, 67), 300(10), 256(14), 191(27), 173(12), 172(25), 171(100), 158(33), 155(27), 141(21), 136(23), 129(69), 123(35), 108(15), 107(27), 92(31), 77(43), 34(11), 56(6). Anal. Calcd for C12H15O4N3S2 (329.3897): C, 43.76; H, 4.60; N, 12.76; S, 19.47. Found: C, 43.44; H, 4.56; N, 12.87; S, 19.71%.
5e: Crystalline (ethyl. acetate/ pet. ether), = +41.64 (C = 1.00 g/100 cm3, acetone); m.p 191–192°C, Yield 81%. IR (KBr)(ν, cm− 1) 3233(-NH), 2545(-SH), 1589(- C = N), 1365 and 1149(-SO2). 1NMR (acetone-d6) δ 0.94 (t, 3H, CH3 J = 7.4 Hz), 1.84–1.90 (m, 2H, CH2), 4.39 (dd, H, CH J = 7.8 Hz, J = 7.5 Hz,), 7.61 (d, 2H, ArH J = 8.0 Hz), 7.85 (d, 2H, ArH J = 8.0 Hz) 12.89(bs, NH). EI-MS (%) 313 (M+, 12), 260(14), 177(25), 175(68), 129(91), 111(100), 76(11), 69(29). Anal. Calcd for C11H12O3N3S2Cl (333.7921): C, 39.58; H, 3.62; N, 12.59; S, 19.21. Found: C, 39.59; H, 3.65; N, 12.20; S, 19.48.
5f: Crystalline (ethyl. acetate/ pet. ether), = +44.32 (C = 1.05 g/100 cm3, acetone); m.p 211–213°C, Yield 91%. IR (KBr)(ν, cm− 1) 3266(-NH), 2565(-SH), 1582(- C = N), 1356 and 1145(-SO2). 1H NMR (acetone-d6) δ 0.92 (t, 3H, CH3 J = 7.5 Hz), 1.82–1.89 (m, 2H, CH2), 3.86(s, 3H, OCH3), 4.36 (dd, H, CH J = 7.6 Hz, J = 7.7 Hz,), 7.01 (d, 2H, ArH J = 8.0 Hz), 7.71 (d, 2H, ArH J = 8.2 Hz). EI-MS (%) 330(M+, 12), 260(14), 191(26), 172(25), 171(100), 158(32), 155(28), 129(70), 107(86), 92(30), 77(42), 64(10). Anal. Calcd for C12H15O4N3S2 (329.3897): C, 43.76; H, 4.60; N, 12.76; S, 19.47. Found: C, 43.56; H, 4.37; N, 12.78; S, 19.57%.
5 g: Powder (aq. ethanol), = +28.64 (C = 0.69 g/100 cm3, acetone); m.p. 190–191°C, Yield = 93%, IR (KBr)(ν, cm− 1) 3287 (NH), 2555 (SH), 1597 (C = N), 1351 and 1161 (SO2), 669 (C(S). 1NMR (acetone-d6) δ = 0.87 (d, 3H, CH3, J = 6.7 Hz), 1.03 (d, 3H, CH3, J = 6.7 Hz), 2.02–2.05 (m, 1H, CH), 2.1–2.12 (m, H, CH), 2.37 (s, 3H, CH3), 4.17 (dd, 1H, *CH, J = 8.4 Hz, J = 9.2 Hz), 7.24 (d, 1H, NH, J = 9.0 Hz), 7.30 (d, 2H, ArH, J = 8.0 Hz), 7.63 (d, 2H, ArH, J = 8.2 Hz), 12.80 (bs, NH+SH). EI-MS(%) 327(M+,9), 284 (16), 225 (4), 172 (10), 155 (74), 130 (19), 91 (100), 65 (29). Anal. Calcd for C13H17O3N3S2 (327.414)): C, 47.69; H, 5.23; N, 12.83; S, 19.58. Found: C, 47.36; H, 4.95; N, 13.12; S, 19.95%.
5 h: Micro crystals (aq. ethanol), = +29.32 (C = 0.70 g/100 cm3, acetone); m.p. 195–199°C, Yield = 91%, IR (KBr)(ν, cm− 1) 3287(NH), 2553(-SH), 1595(C = N), 1351and 1161(SO2), 669 (C-S). 1NMR (acetone-d6) δ 0.91(d, 3H, CH3 J = 7.1 Hz), 1.07(d, 3H, CH3 J = 7.1 Hz), 2.21– 2.25(m, H, CH), 4.25(dd, 1H, CH, J = 8.4 Hz, J = 8.4 Hz), 7.65(d, 2H, ArH J = 8.0 Hz), 7.89(d, 2H, ArH J = 8.0 Hz), 12.89 (bs, NH). EI-MS (%) 347(M+, 9), 175(100), 155(11), 111(74), 76(7). C12H14O3N3S2Cl (347.8463): HRMS: (347.7452).
5i: Micro crystals (aq. ethanol), = +44.45 (C = 1.07 g/100 cm3, acetone); m.p. 201–203°C, Yield = 65%, IR (KBr)(ν, cm− 1) 3262(NH), 1582(C = N), 1376and 1155(SO2), 702 (C-S). 1H NMR (acetone-d6) δ 0.87(d, 3H, CH3 J = 6.7 Hz), 1.03(d, 3H, CH3 J = 6.7 Hz), 2.10– 2.13(m, H, CH), 3.89(s, 3H, OCH3), 4.20 (dd, 1H, CH, J = 9.2 Hz, J = 9.2 Hz), 7.28(d, 2H, ArH J = 8.0 Hz), 7.64(d, 2H, ArH J = 8.0 Hz), 12.80 (bs, NH). EI-MS (%) 343(M+, 11), 234(21), 171(100), 107(57), 76(5). C13H17O4N3S2 (343.4278): HRMS: (343.4365).
5j: Crystalline (aq. ethanol), = +43.41 (C = 1.06 g/100 cm3, acetone); m.p.205–207°C, Yield 83%, IR (KBr)(ν, cm− 1) 3282(-NH), 2551(-SH), 1589(-C = N), 1375 and 1116(-SO2). 1H NMR (acetone-d6) δ 1.46 (d, 3H, CH3), 2.4 (s, 3H, CH3), 4.57–4.65 (m, H, CH), 7.34 (d, 2H, ArH J = 8.2 Hz), 7.68 (d, 2H, ArH J = 8.2 Hz), 12.85(bs, NH). EI-MS (%) 299(M+, 45), 235(12), 198(21), 155(66), 91(100), 65(18). Anal. Calcd for C11H13O3N3S2 (299.3633): C, 44.13; H, 4.38; N, 14.04; S, 21.42. Found: C, 44.07; H, 4.43; N, 14.31; S, 21.13%.
5 l: Powder (aq. ethanol), = +22.67(C = 0.54 g/100 cm3, acetone); m.p. 177–179°C, Yield 79%. IR (KBr)(ν, cm− 1) 3266(-NH), 2550(- C = N), 1586(-C = N), 1371 and 1162(-SO2). 1H NMR (acetone-d6) δ1.53 (d, 3H, CH3, J = 7.2 Hz), 3.89 (s, 3H, OCH3), 4.74– 4.65(m, H, CH), 7.52(d, 2H, ArH J = 8.1 Hz), 7.76 (d, 2H, ArH J = 8.1 Hz), 12.85(bs, NH). EI-MS (%) 315(M+, 9), 241(11), 180(9), 175(69), 135(100), 133(21), 107(57), 76(11). Anal. Calcd for C11H13O4N3S2 (315.3627): C, 41.90; H, 4.21; N, 13.32; S, 20.33. Found: C, 42.13; H, 4.20; N, 13.22; S, 20.46%.
5m: Crystalline (aq. ethanol), = +41.27 (C = 1.00 g/100 cm3, acetone); m.p 171–173°C, Yield 85%, IR (KBr)(ν, cm− 1) 3283(NH), 1595(C = N), 1375 and 1156(SO2). 1H NMR (acetone-d6) δ 0.83–0.88 (m, 6H, 2CH3), 1.62–1.65 (m, 2H, CH2), 2.39 (s, 3H, CH3), 4.35 (dd, H, CH, J = 7.5 Hz, J = 7.5 Hz), 7.31(d, 2H, ArH J = 8.0 Hz), 7.64(d, 2H, ArH J = 8.0 Hz), 10.70 (bs, NH). EI-MS (%) 341(M+, 48), 284(63), 186(33), 155(100), 91(82), 112(45), 111(23), 65(11), 57 (5). C14H19O3N3S2 (341.4553): HRMS: (341.4678).
5n: Micro crystals (aq. ethanol), = +23.47 (C = 0.51 g/100 cm3, acetone); m.p. 185–187°C, Yield = 91%, IR (KBr)(ν, cm− 1) 3291(NH), 1592(C = N), 1356and 1142(SO2), 667 (C-S). 1H NMR (acetone-d6) δ 0.89–0.95(m, 6H, 2CH3), 1.82–1.89(m, 2H, CH2), 4.39 (dd, 1H, CH, J = 7.6 Hz, J = 7.6 Hz), 7.65(d, 2H, ArH J = 8.1 Hz), 7.81(d, 2H, ArH J = 8.1 Hz). EI-MS (%) 361(M+, 7), 304(5), 186(6), 175(100), 111(66), 76(5), 57 (12). C13H16O3N3S2Cl (361.8731): HRMS: (361.8620).
5o: Micro crystals (aq. ethanol), = +39.37 (C = 0.91 g/100 cm3, acetone); m.p. 177–178°C, Yield = 73%, IR (KBr)(ν, cm− 1) 3282(NH), 1583(C = N), 1377and 1166(SO2), 703 (C-S). 1NMR (acetone-d6) δ 0.82–0.87(m, 6H, 2CH3), 1.61–1.64(m, 2H, CH2), 1.75–1.82(m, 1H, CH), 4.33(dd, 1H, CH, J = 7.5 Hz, J = 7.5 Hz), 7.31(d, 2H, ArH J = 8.0 Hz), 7.65(d, 2H, ArH J = 8.0 Hz), 12.80(bs, NH).EI-MS (%) 357(M+, 12), 300(4), 281(3), 251(11), 186(10), 71(100), 107(55), 77(6), 57 (19). C14H19O3N3S2 (357.4547): HRMS: (357.4588).
6a: Micro crystals (aq. ethanol), m.p. 117–119°C, Yield = 74%, IR (KBr)(ν, cm− 1) 3262 (NH), 1582(C = N), 1355 and 1165(SO2), 672(C-S). 1H NMR (acetone-d6) δ 1.88–1.96 (m, 2H, CH2), 2.38 (s, 3H, CH3), 2.87 (t, 2H, CH2, J = 6.61 Hz), 2.99 (t, 2H, CH2, J = 6.7 Hz), 4.40 (s, 2H, CH2-Ph), 7.21(bs, NH, replaceable with D2O), 7.23–7.27(m, 5H, ArH), 7.40 (d, 2H, ArH J = 8.4 Hz), 7.75 (d, 2H, ArH J = 8.4 Hz). EI-MS (%) 403(M+, 7), 312 (3), 186(2), 155(47), 129(42), 91(100), 77(13) 76(3).
6b: Crystalline (aq. ethanol), m.p. 133 − 135°C, Yield = 73%, IR (KBr)(ν, cm− 1) 3266 (NH), 1582 (C = N), 1328 and 1156 (SO2), 662 (C-S). 1H NMR (acetone-d6) δ 1.91–1.97 (m, 2H, CH2), 2.81 (t, 2H, CH2, J = 7.5 Hz), 3.10 (t, 2H, CH2, J = 6.7 Hz), 4.35(s, 2CH2, S-CH2-Ar), 7.28 (bd, NH, replaceable with D2O, J = 8.0 Hz), 7.33–7.38 (m, 5H, ArH), 7.62 (d, 2H, ArH, J = 8.0 Hz), 7.88 (d, 2H, ArH, J = 8.2 Hz). EI-MS (%) 423 (M+, 2), 359 (4), 346(2), 332(12), 260(16), 177(25), 175 (66), 112(9), 111(69), 91 (100), 77(47).
6k: Needles (aq. ethanol), = +43.32 (C = 1.05 g/100 cm3, acetone); m.p 116–117°C, Yield 75%. IR (KBr)(ν, cm− 1) 3289(-NH), 1592(-C = N), 1371 and 1166(-SO2), 679(C-S). 1NMR (acetone-d6) δ 1.51 (d, 3H, CH3 J = 7.0 Hz), 4.35 (s, 2H, CH2), 4.64–4.71(m, 1H, CH), 7.22 (bs, NH, replaceable with D2O), 7.27–7.36 (m, 5H, ArH) 7.66 (d, 2H, ArH J = 8.4 Hz), 7.89(d, 2H, ArH J = 8.0 Hz). EI-MS (%) 409 (M+, 7), 218 (20), 208 (3), 177 (37), 175(100), 139 (28), 111(39), 91(88), 75 (20).
CA inhibition assay
An Applied Photophysics (Oxford, UK) stopped-flow instrument was used for assaying the CA-catalysed CO2 hydration activity [Citation32]. Phenol red (at a concentration of 0.2 mM) was used as indicator, working at the absorbance maximum of 557 nm, with 10 mM Hepes (pH 7.5) as buffer, 0.1 M Na2SO4 (for maintaining constant the ionic strength), following the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7–17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (1 mM) were prepared in distilled-deionized water with 10–20% (v/v) DMSO (which is not inhibitory at these concentrations) and dilutions up to 0.1 nM were done thereafter with distilled-deionized water. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E-I complex. The inhibition constants were obtained by non-linear least-squares methods from Lineweaver-Burk plots, as reported earlier, and represent the mean from at least three different determinations Citation28-31.
Results and discussion
A number of methods have been reported in the literature for the synthesis of heterocyclic compounds incorporating free mercapto and secondary sulfonamide moieties in their molecules [Citation33,Citation34]. Most of such compounds were prepared by using carboxylic acid hydrazides and sulfonic acids/sulfonylchlorides as starting materials, followed by cyclization of the sulfonylated amino acid carbohydrazide [Citation33,Citation34]. We report here a novel approach for preparing such heterocycles, using different chiral and achiral amino acids as starting material (Scheme ). The advantages of this method include easy availability of the starting materials, high yields of the final products (sulfonamides), a free mercapto group at position 5 of the oxadiazole ring, and most importantly the incorporation of chiral centres for possible stereo-selectivity in various applications of such compounds (e.g., as enzyme inhibitors). Benzyl derivatives 6a-b and 6k have also been prepared from the corresponding free thiols, by reaction with benzyl bromide (Scheme ).
Scheme 1 Reagents and conditions: i) Aq. 5% NaOH, ether, stirring at RT for 6 h; ii) H2SO4 / ethanol, refluxing for 11 h; iii) NH2-NH2.H2O / ethanol (absolute), refluxing for 9 h; iv) CS2 / KOH, ethanol, refluxing for 16.5 h. (v) Et3N, DMAP, CH3Cl(dry), benzyl bromide, stirring at 30–70°C for 5 h.
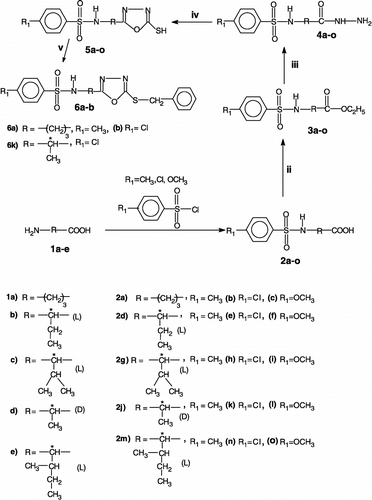
Amino acids 1a-e were converted to the corresponding sulfonamides 2a-o by reaction with 4-methylbenzenesulfonyl chloride, 4-chlorobenzenesulfonyl chloride or 4-methoxybenzenesulfonyl chloride, respectively, in alkaline medium using the standard literature procedure [Citation37]. The carboxylic acid group of these sulfonamides 2a-o was esterified with ethanol in acidic medium [Citation35], and esters 3a-o thus obtained were reacted with hydrazine hydrate (80%) to furnish the corresponding hydrazides 4a-o in good yields [Citation36]. The sulfonamides bearing the 2,5-disubstituted-1,3,4-oxadiazole moiety 5a-o were then prepared by the reaction of hydrazides 4a-o with carbon disulfide and potassium hydroxide [Citation38,Citation39]. Mass spectroscopy (see Materials and Methods for details) showed that the disubstituted 1,3,4-oxadiazoles 5 were formed by elimination of H2S from carbohydrazides 4 and CS2, and not the corresponding 1,3,4-thiadiazoles (which might be formed from the same reagents by elimination of water). The synthesis of compounds 2a, 2d, 2k, 3a, 3d, 3k, 4a, 4d, 4k, 5a, 5d and 5k has been reported earlier by our group [Citation40]. The structures of synthesized compounds was supported by physical, optical rotation data (for the chiral derivatives), microanalytical data, IR, 1H NMR and mass spectral data. The IR spectrum of the representative sulfonamide 5 g, revealed the presence of characteristic bands for -NH at 3286 cm− 1, and 2551 cm− 1 for -SH in addition to the -SO2 functional group. In the mass spectrum of 5 g, the molecular ion peak was observed at m/z 327(M+, 9). In the 1H NMR spectrum of 5 g, the following important signals were observed: δ = 0.87 (d, 3H, CH3, J = 6.7 Hz), 1.03 (d, 3H, CH3, J = 6.7 Hz) two doublets of two methyl groups attached to pro-chiral centre, 2.02–2.05 (m, 1H, CH), 2.1–2.12 (m, 1H, CH), pro-chiral centre, 4.17 (dd, H, CH, J = 8.4 Hz, J = 9.2 Hz) chiral centre, 7.24 (d, 1H, NH, J = 9.0 Hz), 7.30 (d, 2H, ArH, J = 8.0 Hz), 7.63 (d, 2H, ArH, J = 8.2 Hz), 12.80 (bs, NH+SH, replaceable with D2O) with [?] 20D = +28.64o(0.69 g / 100 cm3 of acetone). Sulfonamides 5b-c, 5e-j, 5l-o, 6a-b and 6k are new compounds not reported previously in the literature. None of these compounds has been investigated previously for their interaction with CA, although inhibition studies of these enzymes with heterocyclic thiols are available in the literature Citation41-43.
Some of the compounds 4–6 reported in this study and clinically used CA inhibitors of the sulfonamide type (as standard drugs), such as acetazolamide AAZ, methazolamide MZA, ethoxzolamide EZA, dichlorophenamide DCP and indisulam IND, have been tested for the inhibition of two cytosolic, ubiquitous isozymes of human origin, i.e., hCA I and hCA II Citation1-6, as well as the human tumor-associated isoform hCA IX (). The main goal of this investigation was that of detecting CA IX inhibitors (possibly with selectivity towards this isozyme over the ubiquitous cytosolic isoforms CA I and II Citation1-6), belonging to different classes of compounds than the widely investigated sulfonamides and sulfamates Citation21-30. Data in show the following: (i) against the slow cytosolic isozyme hCA I, thiols 5a–5 g behave as weak inhibitors, with KIs in the range 129–678 μM, whereas carbohydrazides 4k and 4m, thiols 5j and 5k, as well as the S-benzylated derivative 6b showed very weak inhibitory properties (KIs in the range 1059–1675 μM); (ii) against the rapid cytosolic isoform hCA II; a weak inhibitory activity was shown only by thiols 5a, 5b and 5d (KIs in the range 552–720 μM), whereas all other investigated compounds showed very weak inhibition (KIs > 1200 μM); (iii) against the tumor-associated isozyme hCA IX, four derivatives showed good inhibition, with KIs in the range 9.9–10.1 μM. Unexpectedly, two of these compounds (4k and 4m) are carbohydrazides, a class of derivatives not previously investigated as possible CA inhibitors, whereas the remaining two are thiols (5f and 5 g). Such compounds, mainly 1,3,4-thiadiazole-5-thiol derivatives, were in fact shown to behave as micromolar CA IX inhibitors in an earlier work of our group [Citation42]. Surprisingly, the remaining thiols 5a-5e and 5j-5m, as well as the S-benzylated derivative 6b, showed quite weak CA IX inhibitory activity (KIs in the range 560–2830 μM. It should also be mentioned that although the four efficient CA IX inhibitors detected here (4k, 4m, 5f and 5 g) possess only micromolar affinity for the enzyme, these compounds on the other hand may be considered as CA IX selective inhibitors, since they have very low affinity for the ubiquitous isoforms hCA I and II. With the sulfonamide CA inhibitors, this is generally not observed, as most of such compounds usually have very high affinity both for CA I and II Citation1-7. Thus, derivatives 4k, 4m, 5f and 5 g reported here may be considered as very interesting lead molecules for the possible design of CA IX-selective inhibitors. It would also be important to understand why carbohydrazide 4k, 4m bind to the CA IX active site, whereas their binding to CA I and II is very ineffective.
Table I. Inhibition data for compounds 4-6 reported in the present paper and standard sulfonamide CA inhibitors, against isozymes I, II, and IX, by a stopped-flow, CO2 hydration assay [Citation32].
Conclusions
A series of chiral 1,3,4-oxadiazole-5-thiols incorporating 2-substitutedbenzenesulfonamide moieties has been prepared from amino acids, via the ester and carbohydrazide intermediate, followed by cyclization with carbon disulfide. Some of these compounds have been investigated for the inhibition of three physiologically relevant CA isoforms, the human cytosolic hCA I and II, and the human, transmembrane, tumor-associated isozyme hCA IX. All these compounds showed weak (millimolar) affinity for the three isozymes, except two carbohydrazides and two heterocyclic thiols which selectively inhibited the tumor-associated isozyme with inhibition constants around 10 μM. Such compounds constitute interesting lead molecules for the possible design of CA IX-selective inhibitors.
Acknowledgements
This work was supported in part by an EU grant (to CTS and AS) of the 6th framework program (EUROXY project).
References
- Supuran CT, Scozzafava A, Conway J. Carbonic anhydrase. Its inhibitors and activators. CRC Press, London 2004
- Supuran CT, Scozzafava A, Casini A. Carbonic anhydrase inhibitors. Med Res Rev 2003; 23: 146–189
- Supuran CT, Scozzafava A. Carbonic anhydrase inhibitors and their therapeutic potential. Exp Opin Ther Patents 2000; 10: 575–600
- Supuran CT. Carbonic anhydrases: Catalytic and inhibition mechanisms, distribution and physiological roles. carbonic anhydrase. Its inhibitors and activators. CRC Press, London 2004; 1–23
- Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: Current state of the art, therapeutic applications and future prospects. J Enz Inhib Med Chem 2004; 19: 199–229
- Scozzafava A, Mastrolorenzo A, Supuran CT. Modulation of carbonic anhydrase activity and its applications in therapy. Expert Opin Ther Pat 2004; 14: 667–702
- Pastoreková S, Pastorek J. Cancer-related carbonic anhydrase isozymes and their inhibition. Carbonic anhydrase. Its inhibitors and activators. CRC Press, London 2004; 255–281
- Pastorekova S, Zavadova Z, Kostal M, Babusikova O, Zavada J. A novel quasi-viral agent, MaTu, is a two-component system. Virology 1992; 187: 620–626
- Zavada J, Zavadova Z, Pastorekova S, Ciampor F, Pastorek J, Zelnik V. Expression of MaTu-MN protein in human tumor cultures and in clinical specimens. Int. J. Cancer 1993; 54: 268–274
- Zavada J, Zavadova Z, Pastorek J, Biesova Z, Jezek J, Velek J. Human tumour-associated cell adhesion protein MN/CA IX: Identification of M75 epitope and of the region mediating cell adhesion. Br. J. Cancer 2000; 82: 1808–1813
- Stubbs M, McSheehy PM, Griffiths JR, Bashford CL. Causes and consequences of tumour acidity and implications for treatment. Mol Med Today 2000; 6: 15–19
- Potter CPS, Harris AL. Diagnostics, prognostics and therapeutic implications of carbonic anhydrases in cancer. Brit J Cancer 2003; 89: 2–7
- Supuran CT, Casini A, Scozzafava A. Development of carbonic anhydrase inhibitors. Carbonic anhydrase. Its inhibitors and activators. CRC Press, London 2004; 67–147
- Krungkrai J, Scozzafava A, Reungprapavut S, Krungkrai SR, Rattanajak R, Kamchonwongpaisan S, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of Plasmodium falciparum carbonic anhydrase with aromatic sulfonamides: Towards antimalarials with a novel mechanism of action?. Bioorg Med Chem 2005; 13: 483–489
- Suarez Covarrubias A, Larsson AM, Hogbom M, Lindberg J, Bergfors T, Bjorkelid C, Mowbray SL, Unge T, Jones TA. Structure and function of carbonic anhydrases from Mycobacterium tuberculosis. J Biol Chem 2005; 280: 18782–18789
- Suarez Covarrubias A, Bergfors T, Jones TA, Hogbom M. Structural mechanics of the pH dependent activity of beta -carbonic anhydrase from Mycobacterium tuberculosis. J. Biol. Chem. 2006, in press
- Klengel T, Liang WJ, Chaloupka J, Ruoff C, Schroppel K, Naglik JR, Eckert SE, Mogensen EG, Haynes K, Tuite MF, Levin LR, Buck J, Muhlschlegel FA. Fungal adenylyl cyclase integrates CO2 sensing with cAMP signaling and virulence. Curr Biol 2005; 15: 2021–2026
- Bahn YS, Cox GM, Perfect JR, Heitman J. Carbonic anhydrase and CO2 sensing during Cryptococcus neoformans growth, differentiation, and virulence. Curr Biol 2005; 15: 2013–2020
- Svastova E, Hulikova A, Rafajova M, Zat'ovicova M, Gibadulinova A, Casini A, Cecchi A, Scozzafava A, Supuran CT, Pastorek J, Pastorekova S. Hypoxia activates the capacity of tumor-associated carbonic anhydrase IX to acidify extracellular pH. FEBS Lett. 2004; 577: 439–445
- Cecchi A, Hulikova A, Pastorek J, Pastorekova S, Scozzafava A, Winum J-Y, Montero J-L, Supuran CT. Carbonic anhydrase inhibitors. Sulfonamides inhibit isozyme IX mediated acidification of hypoxic tumors. Fluorescent sulfonamides design as probes of membrane-bound carbonic anhydrase isozymes involvement in tumorigenesis. J Med Chem 2005; 48: 4834–4841
- Wykoff CC, Beasley NJ, Watson PH, Turner KJ, Pastorek J, Sibtain A, Wilson GD, Turley H, Talks KL, Maxwell PH, Pugh CW, Ratcliffe PJ, Harris AL. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res 2000; 60: 7075–7083
- Pastorekova S, Casini A, Scozzafava A, Vullo D, Pastorek J, Supuran CT. Carbonic anhydrase inhibitors: The first selective, membrane-impermeant inhibitors targeting the tumor-associated isozyme IX. Bioorg Med Chem Let 2004; 14: 869–873
- Vullo D, Innocenti A, Nishimori I, Pastorek J, Scozzafava A, Pastorekova S, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of the transmembrane isozyme XII with sulfonamides-a new target for the design of antitumor and antiglaucoma drugs?. Bioorg Med Chem Lett 2005; 15: 963–969
- Casey JR, Morgan PE, Vullo D, Scozzafava A, Mastrolorenzo A, Supuran CT. Carbonic anhydrase inhibitors. Design of selective, membrane-impermeant inhibitors targeting the human tumor-associated isozyme IX. J Med Chem 2004; 47: 2337–2347
- Supuran CT, Scozzafava A. Carbonic anhydrase inhibitors: Aromatic sulfonamides and disulfonamides act as efficient tumor growth inhibitors. J Enz Inhib 2000; 15: 597–610
- Abbate F, Casini A, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: X ray crystallographic structure of the adduct of human isozyme II with the perfluorobenzoyl analogue of methazolamide. Implications for the drug design of fluorinated inhibitors. J Enz Inhib Med Chem 2003; 18: 303–308
- Abbate F, Casini A, Owa T, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: E7070, a sulfonamide anticancer agent, potently inhibits cytosolic isozymes I and II, and transmembrane, tumor-associated isozyme IX. Bioorg Med Chem Lett 2004; 14: 217–223
- Weber A, Casini A, Heine A, Kuhn D, Supuran CT, Scozzafava A, Klebe G. Unexpected nanomolar inhibition of carbonic anhydrase by COX-2 selective Celecoxib: New pharmacological opportunities due to related binding site recognition. J Med Chem 2004; 47: 550–557
- Scozzafava A, Briganti F, Ilies M, Supuran CT. Carbonic anhydrase inhibitors: Synthesis of membrane-impermeant low molecular weight sulfinamides possessing in vivo selectivity for the membrane-bound versus cytosolic isozymes. J Med Chem 2000; 43: 292–300
- Vullo D, Franchi M, Gallori E, Pastorek J, Scozzafava A, Pastorekova S, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of the tumor-associated isozyme IX with aromatic and heterocyclic sulfonamides. Bioorg Med Chem Lett 2003; 13: 1005–1009
- Innocenti A, Casini A, Alcaro AC, Papini AM, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: The first on-resin screening of a 4-sulfamoylphenylthiourea library. J Med Chem 2004; 47: 5224–5229
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971; 246: 2561–2573
- Ladva K, Patel P, Upadhyay P, Parekh H. Synthesis and biological activities of substituted 1, 3, 4-oxadiazole derivatives. Ind. J Chem 1996; 35B: 1062
- Lai JYQ, Ferguson Y, Jones M. Preparation of substituted quinolinyl and isoquinolinyl sulfonyl chlorides for synthesis of novel sulfonamides. Synth. Commun. 2003; 33: 3427
- Furniss BS, Hannaford AJ, Rogers V, Smith PWG, Tatchell AR. Vogel Textbook of Practical Organic Chemistry4th ed. Longmann, London, a: pp 1135; b: pp 505; c: pp 1242
- Sun XW, Liang HT, Zhang ZY. Synthesis and antibacrerial activity of 4-aryl-1-(1-p-chlorophenyl-5-methyl-1,2,3-triazol-4-carbonyl)thiosemicarbazides and their related heterocyclic derivatives. Ind. J Chem 1999; 38B: 679
- Clare BW, Scozzafava A, Supuran CT. Protease inhibitors. Synthesis of a series of bacterial collagenase inhibitors of the sulfonyl amino acyl hydroxamate type. J Med Chem 2001; 44: 2253–2258
- Ahmad R, Iqbal R, Akhtar RH, Haq ZU, Duddeck H, Stefaniak L, Sitkowski J. Synthesis and structure determination of some oxadiazole-2-thione and triazole-3-thione galactosides. Nucleosides, Nucleotides and Nucleic Acids 2001; 20: 1671
- Yousif MY, Ismail AM, Elman AA, El-Kerdawy MM. Synthesis of substituted Mercapto-1,2,4-triazoles and substituted 1,2,4-triazoles-5-ylmercaptomethyl-1,3,4-oxadiazoles as Antimicrobal Agents. J Chem Soc Pak 1986; 8: 183
- Iqbal R, Zareef M, Zaidi JH, Khan KM, Arfan M, Masoudi NA. Synthesis, anti- HIV and anti-fungal activities of novel chiral and achiral sulfonamides bearing 1, 3, 4- oxadiazol-2-ylthio moiety. J Heterocyclic Chem submitted
- Supuran CT, Scozzafava A, Saramet I, Banciu MD. Carbonic anhydrase inhibitors. Inhibition of isozymes I, II and IV with heterocyclic mercaptans, sulfenamides, sulfonamides and their metal complexes. J Enz Inhib 1998; 13: 177–194
- Scozzafava A, Saramet I, Banciu MD, Supuran CT. Carbonic anhydrase activity modulators: Synthesis of inhibitors and activators incorporating 2-subtituted-thiazol-4-yl-methyl scaffolds. J Enz Inhib 2001; 16: 351–358
- Almajan GL, Innocenti A, Puccetti L, Manole G, Barbuceanu S, Saramet I, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of the cytosolic and tumor-associated carbonic anhydrase isozymes I, II and IX with a series of 1,3,4-thiadiazole- and 1,2,4-triazole-thiols. Biorg Med Chem Lett 2005; 15: 2347–2352