
Abstract
Bioisosteric replacement and scaffold hopping are powerful strategies in drug design useful for rationally modifying a hit compound towards novel lead therapeutic agents. Recently, we reported a series of thienopyrimidinones that compromise dynamics at the p66/p51 HIV-1 reverse transcriptase (RT)-associated Ribonuclease H (RNase H) dimer interface, thereby allosterically interrupting catalysis by altering the active site geometry. Although they exhibited good submicromolar activity, the isosteric replacement of the thiophene ring, a potential toxicophore, is warranted. Thus, in this article, the most active 2-(3,4-dihydroxyphenyl)-5,6-dimethylthieno[2,3-d]pyrimidin-4(3H)-one 1 was selected as the hit scaffold and several isosteric substitutions of the thiophene ring were performed. A novel series of highly active RNase H allosteric quinazolinone inhibitors was thus obtained. To determine their target selectivity, they were tested against RT-associated RNA-dependent DNA polymerase (RDDP) and integrase (IN). Interestingly, none of the compounds were particularly active on (RDDP) but many displayed micromolar to submicromolar activity against IN.
Introduction
Since the beginning of the AIDS epidemic, almost 80 million people have been infected with human immunodeficiency virus, type 1 (HIV-1), and currently an estimated 38 million people are infected worldwideCitation1. Unfortunately, a treatment that eradicates the virus from infected peopleCitation2 or a vaccineCitation3 is still not available. At present, the challenge of a successful antiretroviral therapy is to convert a terminal disease into a manageable chronic infection by reducing HIV-1 levels in the blood (viral load). In this regard, the advent of the combination active antiretroviral therapy (cART) represents a major achievement in reducing mortality and morbidity of HIV-1 infected patientsCitation4. Nowadays, although 27 FDA-approved drugs are available for the treatment of AIDSCitation5, antiviral drug discovery has not waned. In fact, the selection and spread of HIV-1 variants resistant to current therapies represent the major clinical problem in the fight against AIDSCitation6–8, justifying the increasing demand for new drugs to reinforce the cART arsenal. For that reason, the design of new molecules that target key enzymes of the viral lifecycle with an innovative mode of action could have a tremendous impact on the fight against HIV/AIDS. Reverse transcriptase (RT) together with integrase (IN) and protease (PR), is one of the three viral enzymes essential for HIV-1 replication. DNA polymerase and Ribonuclease H (RNase H) are the two enzymatic activities of RT which catalyse conversion of single-stranded viral RNA into integration-competent, double-stranded linear DNA. Many site-directed mutagenesis studies have demonstrated the essential role of the RNase H domain in virus infectivity, also indicating that the retrovirus-associated activity is unlikely to be supplemented by a host enzymeCitation9,Citation10. This observation suggests RT-associated RNase H as an attractive target that may open opportunities for new systematic HIV-drug discovery effortsCitation11–15. In fact, several RNase H inhibitors have been developed over the last two decades, with promising results against drug-resistant variantsCitation16–20 the majority of which belonging to the class of active site inhibitorsCitation21. Unfortunately, many representatives of this class can be easily displaced by the nucleic acid substrateCitation22 and this drawback has made the progression of RNase H active site inhibitors towards clinical trials challenging. Consequently, attention moved to a less populated class of compounds able to allosterically tackle the viral RNase H function. Such an approach appeared extremely advantageous since the inhibition of related host enzymes, especially those of the polynucleotidyl phosphotransferase class, might be avoided. In this regard, we have recently reported a series of differently decorated thienopyrimidinones, that compromise dynamics at the p66/p51 HIV-1 RT-RNase H dimer interface and consequently interrupting catalysis by altering the active site geometry.Citation11
Interestingly, all these compounds, in contrast to active site RNase H inhibitors, were able to destabilise HIV-1 RT both in the absence and presence of the nucleic acid substrate, decreasing the melting temperature of the enzyme in some cases by more than 5 °C. In addition, by using a panel of drug-resistant and -sensitive recombinant enzymes we could identify several thienopyrimidinone derivatives which retained activity against drug-resistant variants and 2–(3,4-dihydroxyphenyl)-5,6-dimethylthieno[2,3-d]pyrimidin-4(3H)-one 1 emerged as the best compound (wt RT RNase H IC50 = 0.26 μM and C280A RT RNase H IC50 = 0.32 μM)Citation12.
Since bioisosterism is a well-known strategy for rational lead optimisation in drug discoveryCitation23 particularly to improve pharmacological activity, we applied it in an effort to enhance the thienopyrimidinone 1 biological effects. The initial candidate, 2–(3,4-dihydroxyphenyl)-6-methylquinazolin-4(3H)-one 2, showed promising activities, inhibiting HIV-1 RNase H in a submicromolar range (IC50 = 0.41 µM) ( and ). From a structural perspective, our studies indicated that 3,4-dihydroxyphenyl moiety was crucial for activity of every inhibitor, while the bioisosteric substitution of the thiophene core with a 6-methyl phenyl unit as in compound 2 slightly affected the activityCitation12 (). In fact, since the thiophene ring is a potential toxicophoreCitation24, with a strict tropism for liver, kidneys, and immune systemCitation25, its isosteric replacement in biologically active molecules is desirable.
Figure 1. Bioisosteric core replacement.
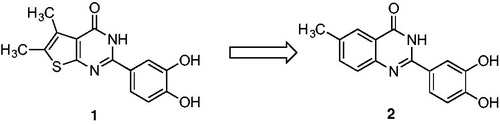
Table 1. Inhibition of wild type and p66/p51C280A mutant HIV-1 RNase H activity by compounds 2–17. 
Similar to thienopyrimidinones, we speculated that decorations of the benzene ring might positively affect inhibitory activity since the recently reportedCitation26 2–(3,4-dihydroxyphenyl)-quinazolin-4(3H)-one (17) is an 8-fold less potent HIV-1 RNase H inhibitor. Based on this evidence, we elected to investigate the introduction of different groups on the phenyl ring in order to establish their inhibitory role. A total of 22 new compounds were synthesised and tested against wt and C280A RT, also investigating their mode of action by means of magnesium-complexation and differential scanning fluorimetry experiments. Furthermore, since many compounds are reported to be dual inhibitors of different HIV-1 functionsCitation27,Citation28, to determine target specificity, we tested the effect of the entire set of quinazolinone-based derivatives on RDDP and IN activities. We also tested the compounds in a cell-based antiviral system.
Material and methods
Chemistry
All reagents were purchased from commercial sources and used without further purification. TLC performed on Aldrich silica gel 60 F254 (0.25 mm) with detection by UV was used for reaction monitoring. 1H and 13 C NMR spectra were recorded on a Varian Unity Inova 500 MHz spectrometer. High-resolution mass spectra were recorded using an Agilent 6520 LC-QTOF-MS system.
General procedure for arylpyrimidinone core formation
A mixture of the appropriate 2-amino arylamide (1.14 mmol), molecular iodine (1.48 mmol), and the appropriate aromatic aldehyde (1.37 mmol) in the presence of acetonitrile as solvent (20 ml) was stirred at room temperature for 6 h (TLC). The reaction was quenched with 40 ml of 5% Na2S2O3 solution, and the resulting precipitate was filtered off. The crude product was washed with n-hexane/ethyl acetate (1:1) and recrystallised from ethanol to give the final arylpyrimidinone as pure product.
For compounds 4 and 5, the corresponding 2-amino arylamide was prepared as follow.
To a magnetically stirred suspension of stannous chloride (24.68 mmol) in concentrated HCl (37%) (9.7 ml), the appropriate 2-nitrobenzamide (8.52 mmol) was added in about 1 h, maintaining the temperature of the slurry below 5 °C. After addition was complete, the mixture was stirred for 30 h at room temperature. A white slurry was obtained which was diluted with cold water (130 ml) and then treated with aqueous NaOH 40% (40 ml) to dissolve the tin salt. The mixture was extracted with ethyl acetate (4 × 100 ml), and the extracts dried on Na2SO4 and concentrated under vacuum to obtain the pure 2-amino arylamide.
2–(3,4-dihydroxyphenyl)-6-methylquinazolin-4(3H)-one (2). Yield (%) = 78. 1H NMR (500 MHz, DMSO): δ 12.12 (s, 1H), 9.59 (bs, 1H), 9.29 (bs, 1H), 7.91 (s, 1H), 7.67(s, 1H), 7.61 (d, J = 8.0 Hz, 1H), 7.57–7.52 (dd, J = 7.1 Hz, 2H), 6.83 (d, J = 8.0 Hz, 1H), 2.44 (s, 3H) ppm. 13 C NMR (500 MHz, DMSO): δ 161.17, 155.29, 148.12, 147.01, 146.76, 145.18, 137.56, 133.71, 128.17, 122.43,120.11, 117.17, 116.15, 21.01 ppm. HRMS calculated for C15H12N2O3 268.08, found 268.07.
2–(3,4-dihydroxyphenyl)-7-methylquinazolin-4(3H)-one (3). Yield (%) = 68. 1H NMR (500 MHz, DMSO): δ 12.06 (s, 1H), 9.11 (bs, 1H), 8.48 (bs, 1H), 7.98 (d, J = 7.5 Hz, 1H), 7.66 (s, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.46 (s, 1H), 7.27 (d, J = 8.0 Hz, 1H), 6.83 (d, J = 7.5 Hz, 1H), 2.45 (s, 3H) ppm. 13 C NMR (500 MHz, DMSO): δ 161.23, 155.45 148.67, 147.11, 146.56, 145.01, 137.58, 133.88, 128.16, 122.42,120.01, 117.00, 116.15, 20.56 ppm. HRMS calculated for C15H12N2O3 268.08, found 268.09.
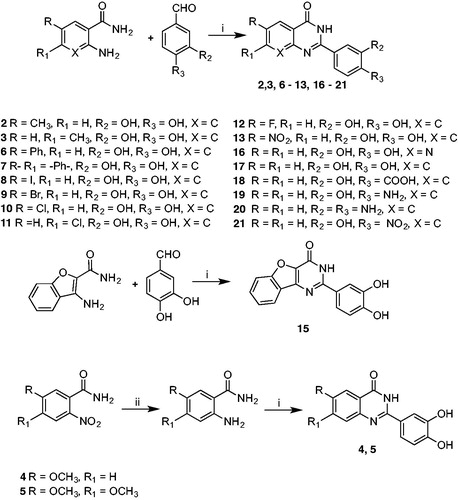
2–(3,4-dihydroxyphenyl)-6-methoxyquinazolin-4(3H)-one (4). Yield (%) = 73. 1H NMR (500 MHz, DMSO): δ 12.16 (s, 1H), 9.56 (bs, 1H), 9.23 (bs, 1H), 7.65 (s, 1H), 7.61 (d, J = 8.5 Hz, 1H), 7.51 (d, J = 6.0 Hz, 1H), 7.40 (d, J = 8.5 Hz, 1H), 6.82 (d, J = 6.0 Hz, 1H), 3.88 (s, 3H) ppm. 13C NMR (500 MHz, DMSO): δ 162.11, 153.99, 151.14, 148.78, 135.88, 129.00, 122.33, 119.10, 118.36, 116.11, 115.57, 114.38, 57.04 HRMS calculated for C15H12N2O4 284.08, found 284. 10.
2–(3,4-dihydroxyphenyl)-6,7-dimethoxyquinazolin-4(3H)-one (5). Yield (%) = 74. 1H NMR (500 MHz, DMSO): δ 12.07 (s, 1H), 9.60 (bs, 1H), 9.19 (bs, 1H), 7.63 (s, 1H), 7.50 (d, J = 7.0 Hz, 1H), 7.43 (s, 1H), 7.10 (s, 1H), 6.81 (d, J = 7.0 Hz, 1H), 3.90 (s, 3H), 3.85 (s, 3H) ppm. 13C NMR (500 MHz, DMSO): δ 162.21, 154.45, 151.16, 148.12, 135.89, 129.09, 122.23, 119.00, 118.34, 116.78, 115.57, 114.38, 57.08 ppm. HRMS calculated for C16H14N2O5 314.09, found 314.10.
2–(3,4-dihydroxyphenyl)-6-phenylquinazolin-4(3H)-one (6). Yield (%) = 77. 1H NMR (500 MHz, DMSO): δ 12.35 (s, 1H), 9.69 (bs, 1H), 9.29 (bs, 1H), 8.37 (s, 1H), 8.07 (d, J = 7.0 Hz, 1H), 7.67–7.56 (m, 3H), 7.45 (s, 1H), 7.37–7.28 (m, 3H), 6.83 (dd, J = 6.7 Hz, 2H) ppm. 13C NMR (500 MHz, DMSO): δ 165.00, 155.61, 148.23, 141.09,138.12, 135.64, 135.20, 134.88, 133.56, 129.76,128.23, 127.22, 126.88, 124.67, 122.34, 120.54, 118.22, 117.11 ppm. HRMS calculated for C20H14N2O3 330.10, found 330.07.
2–(3,4-dihydroxyphenyl)benzo[g]quinazolin-4(3H)-one (7). Yield (%) = 80. 1H NMR (500 MHz, DMSO): δ 12.02 (s, 1H), 9.52 (bs, 1H), 9.23 (bs, 1H), 8.43 (d, J = 7.2 Hz, 2H), 8.23 (d, J = 7.5 Hz, 1H), 8.11 (d, J = 7.5 Hz, 1H), 7.32 (s, 1H), 7.03 (t, J = 7.5 Hz, 1H), 6.98 (t, J = 7.5 Hz, 1H), 6.84 (d, J = 8.1 Hz, 1H), 6.73 (d, J = 8.1 Hz, 1H) ppm. 13C NMR (500 MHz, DMSO): δ 160.00, 155.66, 153.09, 150.12, 147.89, 145.43, 140.06, 138.24, 133.56, 132.16, 130.87, 126.11, 122.34, 120.56, 114.98, 111.22 ppm. HRMS calculated for C18H12N2O3 304.08, found 304.07.
2–(3,4-dihydroxyphenyl)-6-iodoquinazolin-4(3H)-one (8). Yield (%) = 87. 1H NMR (500 MHz, DMSO): δ 11.56 (s, 1H), 9.45 (bs, 1H), 9.27 (bs, 1H), 8.35 (s, 1H), 8.03 (d, J = 8.0 Hz, 1H), 7.67(d, J = 8.0 Hz, 1H), 7.13 (s, 1H), 6.83 (d, J = 7.0 Hz, 1H), 6.78 (d, J = 7.0 Hz, 1H) ppm. 13 C NMR (500 MHz, DMSO): δ 162.00, 160.54,154.27, 147.12, 146.04, 145.76,138.59, 134.73, 127.17, 122.22, 119.19, 116.15, 110.90 ppm. HRMS calculated for C14H9IN2O3 379.97, found 379.97.
2–(3,4-dihydroxyphenyl)-6-bromoquinazolin-4(3H)-one (9). Yield (%) = 79. 1H NMR (500 MHz, DMSO): δ 12.11 (s, 1H), 9.56 (bs, 1H), 9.33 (bs, 1H), 8.15 (s, 1H), 7.98 (d, J = 8.2 Hz, 1H), 7.87 (d, J = 8.2 Hz, 1H), 7.22 (s, 1H), 6.95 (d, J = 7.5 Hz, 1H), 6.76 (d, J = 7.5 Hz, 1H) ppm. 13 C NMR (500 MHz, DMSO): δ 162.00, 155.21, 148.22, 147.04, 145.15, 144.17, 137.09, 134.07, 125.13, 121.22, 118.00, 116.00, 100.10 ppm. HRMS calculated for C14H9BrN2O3 331.98 found 331.97.
2–(3,4-dihydroxyphenyl)-6-chloroquinazolin-4(3H)-one (10). Yield (%) = 90. 1H NMR (500 MHz, DMSO): δ 12.34 (s, 1H), 9.47 (bs, 1H), 9.38 (bs, 1H), 8.02(s, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.67 (s, 1H), 7.54 (d, J = 7.5 Hz, 1H), 6.83 (d, J = 8.0 Hz, 1H), 6.79 (d, J = 7.5 Hz, 1H) ppm. 13 C NMR (500 MHz, DMSO): δ 162.34, 156.27, 151.01, 149.04, 147.66, 145.89, 137.12, 135.77, 123.00, 121.45, 118.50, 111.76 ppm. HRMS calculated for C14H9ClN2O3 288.03 found 288.02.
2–(3,4-dihydroxyphenyl)-7-chloroquinazolin-4(3H)-one (11). Yield (%) = 87. 1H NMR (500 MHz, DMSO): δ 12.45 (s, 1H), 9.82 (bs, 1H), 9.42 (bs, 1H), 8.20 (d, J = 10 Hz, 1H), 7.79 (d, J = 8.9 Hz, 1H), 7.77 (s, 1H), 7.67 (d, J = 10 Hz, 1H), 7.60 (d, J = 8.9 Hz, 1H), 6.95 (d, J = 8.9 Hz, 1H) ppm. 13 C NMR (500 MHz, DMSO): δ 162.31, 156.12, 150.99, 149.54, 147.16, 145.87, 137.22, 135.58, 123.16, 121.75, 118.50, 110.60 ppm. HRMS calculated for C14H9ClN2O3 288.03 found 288.03.
2–(3,4-dihydroxyphenyl)-6-fluoroquinazolin-4(3H)-one (12). Yield (%) = 93. 1H NMR (500 MHz, DMSO): δ 12.46 (s, 1H), 9.76 (bs, 1H), 9.39 (bs, 1H), 7.88 (d, J = 9.0 Hz, 1H), 7.85 (d, J = 8.0 Hz, 1H), 7.79 (s, 1H), 7.77 (d, J = 9.0 Hz, 1H), 7.66 (d, J = 8.0 Hz, 1H), 6.94 (d, J = 8.0 Hz, 1H) ppm. 13 C NMR (500 MHz, DMSO): δ 160.11, 157.22, 151.39, 149.67, 147.28, 145.88, 137.02, 136.56, 123.12, 121.79, 119.00, 111.00 ppm. HRMS calculated for C14H9FN2O3 272.06 found 272.05.
2–(3,4-dihydroxyphenyl)-6-nitroquinazolin-4(3H)-one (13). Yield (%) = 84. 1H NMR (500 MHz, DMSO): δ 12.66 (s, 1H), 9.49 (bs, 1H), 8.78 (bs, 1H), 8.42 (d, J = 7.5 Hz, 1H), 8.09 (d, J = 8.0 Hz, 1H), 7.80 (s, 1H), 7.78 (d, J = 7.5 Hz, 1H), 7.64 (d, J = 8.0 Hz, 1H), 6.86 (d, J = 7.5 Hz, 1H) ppm. 13 C NMR (500 MHz, DMSO): δ 162.01, 155.12, 151.67, 150.53, 148.12, 146.88, 139.14, 136.58, 123.00, 121.00, 118.10, 115.00 ppm. HRMS calculated for C14H9N3O5 299.05 found 299.05.
2–(3,4-dihydroxyphenyl)benzofuro[3,2-d]pyrimidin-4(3H)-one (15). Yield (%) = 91. 1H NMR (500 MHz, DMSO): δ 12.73 (s, 1H), 9.65 (bs, 1H), 9.31 (bs, 1H), 8.08 (d, J = 8.2 Hz, 1H), 7.82 (d, J = 7.5 Hz, 1H), 7.69–7.65 (m, 2H), 7.52 (s, 1H), 6.96 (d, J = 7.5 Hz, 1H), 6.85 (d, J = 8.2 Hz, 1H) ppm. 13 C NMR (500 MHz, DMSO): δ 162.26, 149.89, 146.12, 135.86, 133.22, 123.36, 122.12, 121.09, 120.14, 118.90, 118.21, 113.45 ppm. HRMS calculated for C16H10N2O4 294.06 found 294.05.
2–(3,4-dihydroxyphenyl)pyrido[2,3-d]pyrimidin-4(3H)-one (16). Yield (%) = 81. 1H NMR (500 MHz, DMSO): δ 12.45 (s, 1H), 9.57 (bs, 1H), 9.01 (bs, 1H), 8.77 (d, J = 7.8 Hz, 1H), 8.32 (d, J = 7.8 Hz, 1H), 7.70 (s, 1H), 7.58 (t, J = 7.8 Hz, 1H), 7.12 (d, J = 8.0 Hz, 1H), 6.86 (d, J = 8.0 Hz, 1H) ppm. 13 C NMR (500 MHz, DMSO): δ 160.91, 159.14, 155.87, 146.22, 139.88, 138.15, 135.76, 133.33, 122.10, 120.00, 117.45, 114.10 ppm. HRMS calculated for C13H9N3O3 255.06 found 255.06.
2–(3,4-dihydroxyphenyl)quinazolin-4(3H)-one (17). Yield (%) = 93. 1H NMR (500 MHz, DMSO): δ 12.20 (s, 1H), 9.63 (bs, 1H), 9.28 (bs, 1H), 8.11 (d, J = 9.0 Hz, 1H), 7.79 (d, J = 9.0 Hz, 1H), 7.67(s, 1H), 7.65 (t, J = 9.0 Hz, 2H), 7.43 (d, J = 8.0 Hz, 1H), 6.85 (d, J = 9.0 Hz, 1H), ppm. 13 C NMR (500 MHz, DMSO): δ 161.00, 159.17, 157.12, 149.72, 145.43, 138.12, 134.96, 133.31, 122.33, 120.00, 116.42, 114.15 ppm. HRMS calculated for C14H10N2O3 254.07 found 254.06.
2-hydroxy-4-(4-oxo-3,4-dihydroquinazolin-2-yl)benzoic acid (18). Yield (%) = 89. 1H NMR (500 MHz, DMSO): δ 13.12 (bs, 1H), 12.15 (s, 1H), 11.06 (bs, 1H), 8.22 (d, J = 8.0 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7. 71 (s, 1H), 7.54 (t, J = 8.0 Hz, 2H), 7.33 (d, J = 7.5 Hz, 1H), 6.98 (d, J = 7.5 Hz, 1H), ppm. 13 C NMR (500 MHz, DMSO): δ 172.00, 160.17, 159.24, 151.15, 148.63, 145.12, 139.64, 133.96, 133.11, 121.33, 119.00, 116.22, 111.78 ppm. HRMS calculated for C15H10N2O4 282.06 found 282.04.
2-(4-amino-3-hydroxyphenyl)quinazolin-4(3H)-one (19). Yield (%) = 83. 1H NMR (500 MHz, DMSO): δ 12.34 (s, 1H), 10.03 (bs, 1H), 8.03 (d, J = 7.5 Hz, 1H), 7.67 (d, J = 7.5 Hz, 1H), 7.54 (s, 1H), 7.11 (t, J = 7.5 Hz, 1H), 7.02 (t, J = 7.5 Hz, 1H), 6.81 (d, J = 8.0 Hz, 1H), 6.76 (d, J = 8.0 Hz, 1H), 5.90 (bs, 2H) ppm. 13 C NMR (500 MHz, DMSO): δ 159.20, 157.19, 155.32, 147.79, 144.67, 141.12, 135.00, 122.33, 121.14, 118.65, 116.22, 113.17 ppm. HRMS calculated for C14H11N3O2 253.09 found 253.10.
2–(3,4-diaminophenyl)quinazolin-4(3H)-one (20). Yield (%) = 82. 1H NMR (500 MHz, DMSO): δ 11.98 (s, 1H) 8.25 (d, J = 8.0 Hz, 1H), 8.09 (d, J = 8.0 Hz, 1H), 7.68 (s, 1H), 7.43 (t, J = 8.0 Hz, 2H), 7.04 (d, J = 7.4 Hz, 1H), 6.76 (d, J = 7.4 Hz, 1H), 5.45 (bs, 2H), 5.34 (bs, 2H) ppm. 13 C NMR (500 MHz, DMSO): δ 163.09, 160.00, 159.12, 152.07, 148.41, 139.12, 136.00, 135.31, 123.13, 121.00, 117.33, 113.18 ppm. HRMS calculated for C14H12N4O 252.10 found 252.09.
2–(3-hydroxy-4-nitrophenyl)quinazolin-4(3H)-one (21). Yield (%) = 92. 1H NMR (500 MHz, DMSO): δ 12.21 (s, 1H), 8.13 (d, J = 7.5 Hz, 1H), 7.97 (d, J = 7.5 Hz, 1H), 7.34 (s, 1H), 7.15 (t, J = 7.5 Hz, 1H), 7.00 (t, J = 7.5 Hz, 1H), 6.91 (d, J = 8.0 Hz, 1H), 6.80 (d, J = 8.0 Hz, 1H), 6.25 (bs, 2H) ppm. 13 C NMR (500 MHz, DMSO): δ 159.70, 156.13, 154.32, 147.43, 145.68, 142.24, 135.10, 126.37, 122.56 117.85, 115.00, 111.17, 110.67 ppm. HRMS calculated for C14H10N4O3 282.08 found 282.08.
Synthetic protocols for the preparation of compounds (14), (22), and (23)
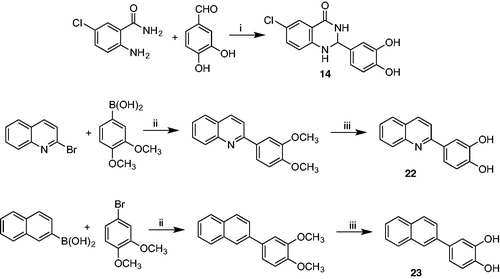
Synthesis of 6-chloro-2–(3,4-dihydroxyphenyl)-2,3-dihydroquinazolin-4(1H)-one (14). 2-amino-5-chlorobenzamide (0.7 mmol) and 3,4-dihydroxybenzaldehyde (0.91 mmol) were dissolved in a mixture of dichloromethane (10 ml) and acetonitrile (7 ml) and refluxed for 40 h. After completion, the solvent was removed under vacuum. The solid residue was treated with water, filtered and then washed again with water to give compound 14 as a pale yellow crystal. Yield (%) = 82. 1H NMR (500 MHz, DMSO): δ 9.42 (s, 1H), 8.47 (bs, 1H), 8.38 (bs, 1H), 7.99 (s, 1H), 7.85 (d, J = 7.0 Hz, 1H), 7.54 (d, J = 7.5 Hz, 1H), 6.93 (s, 1H), 6.77 (d, J = 7.5 Hz, 1H), 6.44 (d, J = 7.5 Hz, 1H),6.29 (bs, 1H), 6.13 (s, 1H) ppm. 13 C NMR (500 MHz, DMSO): δ 159.78 149.27, 147.11, 145.64, 137.76, 135.99, 132.00, 124.75, 122.10, 121.00, 116.70, 110.75, 80.05 ppm. HRMS calculated for C14H11ClN2O3 290.05 found 290.05.
Synthesis of 4-(quinolin-2-yl)benzene-1,2-diol (22). To a mixture of (3,4-dimethoxyphenyl)boronic acid (3.29 mmol), K2CO3 (3.29 mmol), and Pd(OAc)2 (0.027 mmol) in ethanol (9 ml) and water (3 ml) was slowly added 2-bromoquinoline (2.74 mmol). The mixture was stirred at room temperature for 18 h. To the resulting mixture was added water (50 ml), and then extracted with dichloromethane. The organic phase was separated, dried over Mg2SO4 and concentrated under reduced pressure to give a solid residue which was purified by flash chromatography (silica gel and hexane/acetone 3/1). The pure 2–(3,4-dimethoxyphenyl)quinoline was obtained as light grey solid and used for the next reaction. Yield (%) = 92. 1H NMR (500 MHz, DMSO): δ 8.76 (d, J = 8.0 Hz, 1H), 8.11 (d, J = 7.0 Hz, 1H), 7.93 (d, J = 7.0 Hz, 1H), 7.77 (t, J = 7.0 Hz, 1H), 7.68 (s, 1H), 7.59 (d, J = 7.5 Hz, 1H), 7.55 (t, J = 7.0 Hz, 1H),7.29 (d, J = 8.0 Hz, 1H), 7.02 (d, J = 7.0 Hz, 1H), 3.85 (s, 6H) ppm. 13 C NMR (500 MHz, DMSO): δ 156.00, 153.12, 145.12, 135.82, 130.27, 128.00, 125.26, 122.30, 119.33, 117.01, 115.40, 113.13, 57.89 ppm. HRMS calculated for C17H15NO2 265.11 found 265.10.
To a stirred solution of 2–(3,4-dimethoxyphenyl)quinoline (0.604 mmol) in dichloromethane (4 ml) at 0 °C was added boron tribromide 1 M solution in dichloromethane (9.66 mmol). After 1.5 h, the resulting mixture was stirred for 4 more hours at room temperature. The resulting mixture was the poured into ice water and the yellow residue obtained was dissolved in ethanol. The mixture was then extracted with dichloromethane and the organic phase, previously dried over anhydrous MgSO4, was evaporated to give compound 22 as a yellow solid. Yield (%) = 91. 1H NMR (500 MHz, DMSO): δ 9.09 (bs, 1H), 8.87 (bs, 1H), 8.73 (d, J = 8.0 Hz, 1H), 8.09 (d, J = 7.0 Hz, 1H), 7.89 (d, J = 7.0 Hz, 1H), 7.80 (t, J = 7.0 Hz, 1H), 7.64 (s, 1H), 7.53 (d, J = 7.5 Hz, 1H), 7.51 (t, J = 7.0 Hz, 1H),7.29 (d, J = 8.0 Hz, 1H), 7.12 (d, J = 7.0 Hz, 1H) ppm. 13 C NMR (500 MHz, DMSO): δ 155.20, 153.44, 144.23, 136.81, 133.17, 127.06, 125.97, 122.32, 118.56, 117.15, 116.40, 112.99, 110.78 ppm. HRMS calculated for C15H11NO2 237.08 found 237.08.
Synthesis of 4-(naphthalen-2-yl)benzene-1,2-diol (23). To a mixture of 2-naphtylboronic acid (5.81 mmol), K2CO3 (5.81 mmol), and Pd(OAc)2 (0.048 mmol) in ethanol (15 ml) and water (5 ml) was slowly added 4-bromo-1,2-dimethoxybenzene (4.84 mmol). The mixture was stirred at room temperature for 18 h. To the resulting mixture was added water (80 ml), and then extracted with dichloromethane. The organic phase was separated, dried over Mg2SO4 and concentrated under reduced pressure to give a solid residue which was purified by flash chromatography (silica gel, hexane/ethyl acetate 5/1). The pure 2–(3,4-dimethoxyphenyl)naphthalene was obtained as white solid and used for the next reaction. Yield (%) = 93. 1H NMR (500 MHz, DMSO): δ 8.29 (d, J = 8.0 Hz, 1H), 8.12 (d, J = 8.0 Hz, 1H), 7.98 (d, J = 7.4 Hz, 1H), 7.82 (t, J = 8.0 Hz, 1H), 7.75 (t, J = 8.0 Hz, 1H), 7.61 (s, 1H), 7.45 (d, J = 8.2 Hz, 1H), 7.39 (d, J = 7.4 Hz, 1H), 7.12 (s, 1H), 7.02 (d, J = 8.2 Hz, 1H), 3.85 (s, 3H), 3.78 (s, 3H) ppm. 13 C NMR (500 MHz, DMSO): δ 151.20, 148.19, 139.45, 135.45, 132.13, 127.00, 124.15, 119.32, 117.56, 116.03, 113.40, 112.26, 110.67, 108.65, 57.23 ppm. HRMS calculated for C18H16O2 264.12 found 264.12.
To a stirred solution of 2–(3,4-dimethoxyphenyl)naphthalene (0.36 mmol) in dichloromethane (3 ml) at 0 °C was added boron tribromide 1 M solution in dichloromethane (6.00 mmol). After 1 h, the resulting mixture was stirred for 3 more hours at room temperature. The resulting mixture was the poured into ice water and treated with methanol (20 ml). The pale pink mixture was then extracted with dichloromethane and the organic phase, previously dried over anhydrous MgSO4, was evaporated to give compound 23 as a grey solid. Yield (%) = 89. 1H NMR (500 MHz, DMSO): δ 9.56 (bs, 1H), 9.38 (bs, 1H), 8.31 (d, J = 8.1 Hz, 1H), 8.15 (d, J = 8.1 Hz, 1H), 7.88 (d, J = 7.5 Hz, 1H), 7.83 (t, J = 8.1 Hz, 1H), 7.75 (t, J = 8.1 Hz, 1H), 7.62 (s, 1H), 7.47 (d, J = 8.0 Hz, 1H), 7.39 (d, J = 7.5 Hz, 1H), 7.22 (s, 1H), 7.00 (d, J = 8.0 Hz, 1H) ppm. 13 C NMR (500 MHz, DMSO): 149.80, 148.00, 140.46, 136.41, 133.16, 128.00, 125.16, 118.82, 117.76, 116.27, 112.47, 112.16, 111.57, 108.65 ppm. HRMS calculated for C18H12O2 236.08 found 236.08.
Biology
Expression and purification of recombinant HIV-1 RT
Wt and mutant heterodimeric RT were expressed essentially as previously describedCitation29,Citation30.
RNase H inhibitor analysis
IC50 values were determined as previously reportedCitation31 using an 18-nt 3′-fluorescein-labeled RNA annealed to a complementary 18-nt 5′-dabcyl-labeled DNA. To a 96-well plate was added 1 μL of each inhibitor (in DMSO), followed by 10 μL of the appropriate RT (15−80 ng/mL) in reaction buffer. Hydrolysis was initiated by adding 10 μL of RNA/DNA hybrid (2.5 μM). Final assay conditions were 50 mM Tris·HCl, pH 8.0, 60 mM KCl, 10 mM MgCl2, 1% DMSO, 150−800 ng RT, 250 nM substrate, and increasing concentrations of inhibitor. Wells containing only DMSO was used as negative control. Plates were incubated at 37 °C in a Spectramax Gemini EM fluorescence spectrometer for 10 min, and fluorescence (lex= 485 nm; lem = 520 nm) was measured at 1-min intervals such that linear initial rates could be measured in the presence (vi) and absence (vo) of inhibitor. Percent inhibition was calculated as 100 × (vo–vi)/vo and plotted against log[I]. IC50 values were determined using Prism5 (GraphPad Software, San Diego, CA). All assays were performed in triplicate.
Expression and purification of recombinant HIV-1 in and LEDGF
HIV IN was expressed as previously describedCitation32–34.
HTRF in LEDGF-dependent assay
The IN LEDGF/p75-dependent assay measures inhibition of IN activity in the presence of LEDGF/p75 proteinCitation35–37. Briefly, 50 nM IN was pre-incubated with increasing concentration of compounds for 1 h at room temperature in reaction buffer containing 20 mM HEPES pH 7.5, 1 mM DTT, 1% Glycerol, 20 mM MgCl2, 0.05% Brij-35 and 0.1 mg/ml BSA. To this mixture, 9 nM DNA donor substrate (5′-ACAGGCCTAGCACGCGTCG-Biotin-3′ annealed with 5′-CGACGCGTGGTAGGCCTGT-Biotin3’) and 50 nM DNA acceptor substrate (5′-Cy5-ATGTGGAAAATCTCTAGCAGT-3′ annealed with 5′-Cy5- TGAGCTCGAGATTTTCCACAT-3′) and 50 nM LEDGF/p75 were added and incubated at 37 °C for 90 min. After incubation, 4 nM of Europium–Streptavidin was added to the reaction mixture and the HTRF signal was recorded using a Perkin Elmer Victor 3 plate reader using a 314 nm for excitation wavelength and 668 and 620 nm for the wavelength of the acceptor and the donor substrates emission, respectively.
HIV-1 RNA-dependent DNA polymerase activity determination
HIV-1 RT-associated RDDP activity was measured as describedCitation17,Citation38. Briefly, in 25 μL volume containing 60 mM Tris-HCl pH 8.1, 8 mM MgCl2, 60 mM KCl, 13 mM DTT, poly(A)-oligo(dT), 100 μM dTTP, and 6 ng wt RT. After enzyme addition, the reaction mixture was incubated for 30 min at 37 °C and stopped by addition of EDTA. Reaction products were detected by PicoGreen addition and measured with a Victor 3 (Perkin) at 502/523 nm. IC50 values were determined using GraphPad Prism version 6.01 software (GraphPad Software, Inc.; San Diego, CA). Figures were prepared with GraphPad Prism 6 version 6.01. All assays were performed in triplicate.
Evaluation of MgCl2 coordination
The coordination properties for the compounds were determined as reported previouslyCitation39–41. Briefly, compounds were solubilised in 1 ml of 96% ethanol at a final concentration of 100 μM. The UV–Vis spectrum was recorded from 200 to 600 nm before and after titration with increasing final concentrations of MgCl2, from 100 to 10 mM. UV–Vis spectra were recorded from 200 to 600 nm using an Ultrospec 2100 pro (Amersham Biosciences, Little Chalfont, UK) and spectra were plotted using SigmaPlot for Windows version 11.0 (Systat Software Inc., San Jose, CA). Colour legend indicates the final Mg2+ micromolar concentration within the sample.
Differential scanning fluorimetry (ThermoFluor)
Thermal stability assays were performed according to Nettleship et al.Citation42 To a LightCycler®480 96-well plate (Roche, Basel, Switzerland) was added 1 μL of 500 μM inhibitor in DMSO, followed by 49 μL of 300 nM HIV-1 RT in reaction buffer containing 20 mM HEPES, pH 7.5, 10 mM MgCl2, 100 mM NaCl, and a 1:1000 dilution of Sypro® Orange dye (Invitrogen, Carlsbad, CA). The mixture was heated from 30 to 90 °C in increments of 0.2 °C. Fluorescence intensity was measured using excitation/emission wavelengths of 483 and 568 nm, respectively. Changes in protein thermal stability (ΔTm) upon inhibitor binding were analysed by using LightCycler® 480 Software. All assays were performed in triplicate.
Yonetani–Teorell analysis
Yonetani–Theorell analysisCitation43 was performed as reportedCitation44. Briefly, HIV RT-associated RNase H activity was measured in 100 µL reaction volume containing 50 mM Tris–HCl buffer pH 7.8, 6 mM MgCl2, 1 mM dithiothreitol (DTT), 80 mM KCl, 0.25 µM hybrid RNA/DNA 5′-GAUCUGAGCCUGGGAGCU-Fluorescin-3′ (HPLC, dry, QC: Mass Check) (available from Metabion) 5′-Dabcyl-AGCTCCCAGGCTCAGATC-3′(HPLC, dry, QC: Mass Check), and different amount of enzymes according to a linear range of dose-response curve: 20 ng of WT RT, and increasing concentrations of inhibitor (diluted) in water and incubated for 1 h at 37 °C, at 200 rpm. Products were measured with a multilabel counter plate reader Victor 3 (Perkin Elmer model 1420-051) equipped with filters for 490/528 nm (excitation/emission wavelength). A calibration curve for the fluorescein signal was generated in parallel, calculating pmols of product generated by the reaction. The reciprocal of the reaction product was plotted vs. the compound concentration a different condition using the Sigma-Plot software.
Antiviral activity
Antiviral activity of selected thienopyrimidinones was determined via the 2,3-bis[2-methoxy-4-nitro-5-sulfophenyl]-5-[(phenylamino)carbonyl]-2H tetrazolium hydroxide (XTT)-based cell viability assay of Weislow et al.Citation45, using the HIV-1RF isolate and human T-cell line CEM-SS.
Results and discussion
Chemistry
As reported in Scheme 1, compounds 2, 3, 6 − 13, 15 − 21 were synthesised by a one-pot direct oxidative condensation of the appropriate commercially available 2-amino arylamide with the 3,4-disubstituted benzaldehyde in the presence of molecular iodineCitation46. After 6 h, the reaction, carried out in acetonitrile at room temperature, afforded the final aryl pyrimidinone derivative in good yields. Synthesis of quinazolinones 4 and 5 required synthesising the 2-amino-5-methoxybenzamide and 2-amino-4,5-dimethoxybenzamide. Starting from the corresponding 2-nitrobenzamide, we carried out a reduction of the nitro groupCitation47 in concentrated HCl in the presence of SnCl2, obtaining the corresponding mono or dimethoxy −2-amino arylamide as pure product.
Scheme 1. Synthetic protocol for compounds 2–13 and 15–21. Reagents and conditions: i) I2/CH3CN, rt, 6 h. ii) SnCl2/HCl 37%,−5 °C (1 h), rt (30 h).
In contrast, cyclisationCitation48 (Scheme 2) of 2-amino-6-chlorobenzamide with 3,4-dihydroxybenzaldehyde carried out at reflux in a mixture of dichloromethane/acetonitrile gave the racemic 6-chloro-2–(3,4-dimethoxyphenyl)-2,3-dihydroquinazolin-4(1H)-one 14, which is more flexible than the corresponding derivative 10. Moreover, it adds a stereocenter on C-2, making the molecule chiral. Synthesis of compounds 22 and 23 started with a Suzuki C-C coupling reaction between the appropriate arylboronic acid and the proper aryl bromide under basic conditions and in the presence of Pd(OAc)2 as catalystCitation49. Subsequent demethylation was performed using BBr3 as demethylating agent in dichloromethaneCitation50.
Scheme 2. Synthetic protocol for compounds 14, 22, and 23. Reagents and conditions: i) CH2Cl2/CH3CN, reflux, 40 h. ii) Pd(OAc)2/K2CO3, EtOH/H2O 3/1, rt (18 h). iii) BBr3 1 M in CH2Cl2, CH2Cl2, 0 °C (1 h), rt (3 h).
Biology
All compounds were tested for inhibitory activity against both wt and p66/p51 C280A RT. In fact, p51 residue Cys280 is crucial for binding of thienopirimidinonesCitation51. provides IC50 values for the first panel of quinazolinone derivatives. It is immediately evident that every substitution gave rise to compounds with submicromolar activity, equally effective on both RT wild type and p66/p51 C280A mutant.
Interestingly, steric hindrance of the chosen substituents seemed not to affect RNase H activity. In fact, compounds 6 (IC50 = 0.37 μM) and 2 (IC50 = 0.41 μM) share the same potency. Conversely, compound 7 (IC50 = 1.10 μM) demonstrated that adding rigidity to the molecule by introducing another aromatic ring ortho condensed to the quinazolinone core impaired the activity while replacing naphthalene with benzofuran as in 15 (IC50 = 0.64 μM) was only slightly detrimental to inhibitory potency. Noteworthy, electron withdrawing groups, and in particular halogens in position 6, provided the most active compounds, with 8 being the best compound (IC50 = 0.15 μM) of the entire series of derivatives. Finally, the 2–(3,4-dihydroxyphenyl) unit was demonstrated to be crucial for the inhibitory activity similarly to thienopyrimidinones. In fact, as reported in , replacing one or both OH groups with –COOH, NH2, or NO2 led to a complete loss of activity.
Table 2. Inhibition of HIV-1 RNase H activity by compounds 18–21. 
We also explored the role of the pyrimidinone moiety. First, in order to determine the importance of the rigidity of the quinazolinone core, we decided to synthesise compound 14, in which removal of the double bond between N-1 and C-2 gave more flexibility to the pyrimidinone ring and introduced a stereocenter on C-2. Since the synthetic pathway was not stereoselective, we obtained and subsequently tested, compound 14 as a racemic mixture. As shown in , this structural modification did not affect RNase H inhibitory activity on both wt and p66/p51 C280A mutant RTs. Encouragingly, this modification rendered quinazoline 14 more selective for RNase H activity than 9 () and prompted us to explore a stereoselective synthetic approach that will be the subject of a future paper.
As shown in , removing the amide unit gave rise to 2–(3,4-dihydroxyphenyl) quinoline 22 with almost the same inhibitory potency of 2–(3,4-dihydroxyphenyl)- quinazolin-4(3H)-one 17 against wt RT (IC50 = 2.9 uM)Citation26. Noteworthy, 22 inhibited p66/p51 C280A mutant RT RNase H activity 4-fold more potently than wt RT (IC50 = 0.61 μM). Substituting the pyridine core with its simplest bioisoster, a benzene ring, generated compound 23, 5-fold more potent than the reported compound 17. However, 23 inhibited p66/p51 C280A mutant RT RNase H activity with the same potency of the wt enzyme ().
Table 3. Inhibition of wild type and p66/p51C280A mutant HIV-1 RNase H activity by compounds 22 and 23.
Inhibitory activity against RNA-dependent DNA polymerase (RDDP) and integrase (IN)
In order to determine the specificity of our quinazolinone-based derivatives, we tested their effect on RDDP and IN activities. With respect to RDDP function, all compounds were assayed using efavirenz, a non-nucleoside RT inhibitor, as positive control. Data of indicate that all the quinazolinone derivates, albeit with a different degree, were poorly active against RDDP with IC50 values ranging from 23 to more than 100 µM. Thus, they are highly selective RNase H inhibitors.
Table 4. Effect of compounds 2–17 on HIV-1 RDDP and IN functions.
Since the catechol moiety has been identified in a variety of HIV-1 IN inhibitorsCitation52,Citation53, we also tested our compounds against this target, using the strand-transfer inhibitor raltegravir, as positive control. Almost all quinazolinones were moderately active on HIV-1 IN, displaying, and IC50 range of 2.3–89 µM, showing high selectivity for RNase H. Remarkably, the pyridine core was subtly deleterious for RT activity but powerful in IN function suppression. In fact, compounds 16 and 17 showed a different trend. In particular, these compounds inhibited IN activities in the presence of LEDGF/p75 protein with IC50 values of 0.69 and 1.44 µΜ, respectively. Moreover, these derivatives exhibited a higher selectivity against IN.
Investigation of mode of action
Typically, RNase H active site inhibitors chelate the catalytically crucial Mg2+ ions. Since catechol-containing compounds usually display metal-chelating properties; we performed UV spectrometric analysis to determine the possible involvement of Mg2+ cofactors in the inhibition mode of our quinazolinones. In this respect, a magnesium effect was examined at increasing concentrations of the divalent metal (Supplementary Material). Results showed that the presence of the Mg2+ failed to shift the peak of absorbance of the compounds spectra, excluding the involvement of Mg2+ cofactor coordination on the mode-of-action of compounds. Only compound 2 showed a hyperchromic effect at the highest concentration tested.
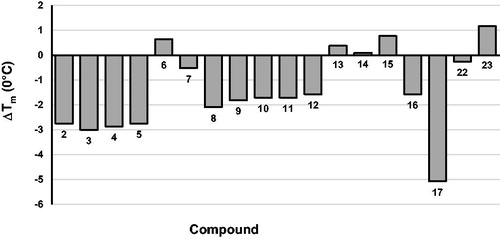
To gain additional evidence, thermal denaturation experiments were carried out. Differential scanning fluorimetry was the technique of choice, as can determine the effect of an inhibitor on RT stability by measuring changes in its melting temperature (Tm)Citation54. Results graphically reported in , indicate that almost all tested inhibitors destabilise the RT heterodimer stability, i.e. they decrease the Tm. Since it had been demonstrated that compounds that bind in the proximity of the p66/p51 interface usually reduce RT TmCitation11, we can assume that present quinazolinones, similarly to thienopyrimidinones, might act as allosteric inhibitors.
Figure 2. Effect of compounds 2–17, 22, and 23 on the thermal stability of p66/p51 HIV-1 RT. Tm values are the average of triplicate analysis.
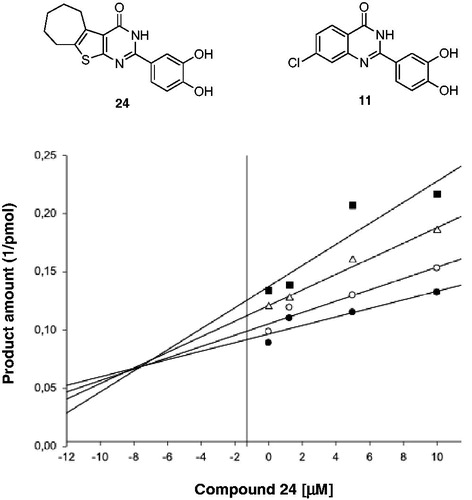
To confirm this hypothesis, we performed a Yonetani-Theorell analysis of simultaneous action of compound 11 vs. the 2–(3,4-dihydroxyphenyl)-3,5,6,7,8,9-hexahydro-4H-cyclohepta[4,5]thieno[2,3-d]pyrimidin-4-one 2412 on HIV-1 RT-associated RNase H activity. Such an analysis reveals whether simultaneous binding (or inhibition) of two compounds is possible or not. Results showed that compound 11 and compound 24 inhibition are not kinetically mutually exclusive (), suggesting that they may bind simultaneously to RT. However, it is worth noting that the calculated interaction constant α had a value of greater than 1, suggesting a negative interference between the two compounds (i.e. compound binding 11 has a negative influence on compound 24 binding, and vice versa).
Figure 3. Yonetani–Theorell analysis. Combination of compound 11 and 24 on HIV-1 RNase H activity. HIV-RT was incubated in the presence of 24 alone (●) or combined with increasing concentrations of compound 11: 2,5 µM (O); 5 µM(Δ) and 10 µM (■).
Antiviral activity
The ability of all compounds to inhibit HIV-1 virus replication was evaluated. Only compounds 3, 8, 10, 11, and 23, showed a modest antiviral activity within low micromolar range. Unfortunately, they displayed very low SI values, demonstrating to be highly cytotoxic. In fact, the most active compound in cell-based assays, compound 8, showed a very small selectivity index (SI) (EC50 = 8.3 µM, CC50 = 17 µM, SI = 2). Similar results were obtained for 3 (EC50 = 12 µM, CC50 = 25 µM, SI = 2) and 23 (EC50 = 14 µM, CC50 = 26 µM, SI = 2). Interestingly, the simple substitution of an iodine atom with a chlorine in 10 displayed a slightly better SI (EC50 = 5.4 µM, CC50 = 22 µM, SI = 4). Ring walking of the chlorine from position 6–7 as in 11, reduced the SI (EC50 = 1.8 µM, CC50 = 6 µM, SI = 3). All other compounds were inactive (data not shown).
Conclusions
The isosteric replacement approach allowed us to synthesise a series of 22 quinazolinone-based derivatives. All compounds were equally effective on both wt RT and p66/p51 C280A mutant, unable to chelate the Mg2+ ions, and in most cases destabilised the RT heterodimer by decrease its Tm. These combined findings prompted us to hypothesise that they are not active site, but most likely allosteric, and in particular RT heterodimer interface inhibitors. Yonetani–Theorell analysis showed that compound 11 and the reference thienopyrimidinone 24 are not kinetically mutually exclusive and the value of the calculated interaction constant α indicated a negative interference between the two compounds. Thus, they may bind simultaneously to different, but probably close, RT allosteric sites since compound 11 binding has a negative influence on compound 24 binding, and vice versa.
Moreover, all compounds were highly selective for RNase H function, since they were poorly active on RDDP activity and moderately active on HIV-1 IN. The exception to this was compounds 16 and 17, which exhibited a higher selectivity against IN.
Interestingly, compounds 3, 8, 10, 11, and 23 demonstrated to suppress HIV-1 replication in a cell-based assay albeit with a modest antiviral activity, within the low micromolar range, but unfortunately showed to be cytotoxic, too.
As previously reported for other allosteric RNase H inhibitorsCitation11,Citation55, the presence of the catechol moiety was essential for the inhibitory activity of our compounds. However, it cannot be discarded that, because of its redox reactivity, it might be in part responsible for the poor antiviral activity and cytotoxicity, too. In fact, toxicity of catechol-containing drugs is often related to the ability of semiquinone radical, the catechol oxidised form, to participate in production of a superoxide anion radical deleterious for proteins and cell membranesCitation56,Citation57.
In summary, bioisosteric replacement of the thiophene with a benzene ring proved a worthwhile strategy since all compounds reported here retained their inhibitory activity against both RT wild type and p66/p51 C280A mutant. Thus, the quinazolinone-based derivatives reported herein may be promising hit molecules for the development of a novel generation of selective RNase H allosteric inhibitors without the liability of the thiophene series.
Supplemental Material
Download PDF (793.4 KB)Disclosure statement
The authors have no other relevant affiliations or financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilised in the production of this manuscript.
Additional information
Funding
References
- UNAIDS/World Health Organization. United Nations report on 2020 Global AIDS update-seizing the moment-tackling entrenched inequalities to end epidemics. June 2020. Available from: https://www.unaids.org/sites/default/files/media_asset/2020_global-aids-report_en.pdf.
- Henderson LJ, Reoma LB, Kovacs JA, Nath A. Advances toward curing HIV-1 infection in tissue reservoirs. J Virol 2019;94:1–21.
- Cohen J. Combo of two HIV vaccines fails its big test. Science 2020;367:611–2.
- Deeks SG, Lewin SR, Havlir DV. The end of AIDS: HIV infection as a chronic disease. Lancet 2013;382:1525–33.
- FDA - Approved HIV Medicines Report. Available from: https://hivinfo.nih.gov/understanding-hiv/fact-sheets/fda-approved-hiv-medicines.
- Engelman A, Cherepanov P. The structural biology of HIV-1: mechanistic and therapeutic insights. Nat Rev Microbiol 2012;10: 279–90.
- Gupta RK, Gregson J, Parkin N, et al. HIV-1 drug resistance before initiation or re-initiation of first-line antiretroviral therapy in low-income and middle-income countries: a systematic review and meta-regression analysis. Lancet Infect Dis 2018;18:346–55.
- The TenoRes Study Group. Global epidemiology of drug resistance after failure of WHO recommended first-line regimens for adult HIV-1 infection: a multicentre retrospective cohort study. Lancet Infect. Dis 2016;16:565–75.
- Grandi N, Pisano MP, Scognamiglio S, et al. Comprehensive analysis of HERV transcriptome in HIV + cells: absence of HML2 activation and general downregulation of individual HERV loci. Viruses 2020;12:481.
- Schatz O, Cromme FV, Naas T. Lindemann D, Mous J, Le Grice SFJ Inactivation of the RNase H domain of HIV-1 reverse transcriptase blocks viral infectivity.In: Gene Regulation and AIDS. Houston, TX: Portfolio Publishing Company; 1990,p. 293–303
- Masaoka T, Chung S, Caboni P, et al. Exploiting drug-resistant enzymes as tools to identify thienopyrimidinone inhibitors of human immunodeficiency virus reverse transcriptase-associated ribonuclease H. J Med Chem 2013;56:5436–45.
- Corona A, Masaoka T, Tocco G, et al. Active site and allosteric inhibitors of the ribonuclease H activity of HIV reverse transcriptase. Future Med Chem 2013;5:2127–39.
- Tramontano E. HIV-1 RNase H: recent progress in an exciting, yet little explored, drug target. Mini-Rev Med Chem 2006;6:727–37.
- Distinto S, Maccioni E, Meleddu R, et al. Molecular aspects of the RT/drug interactions. Perspective of dual inhibitors. Curr Pharm Design 2013;19:1850–9.
- Corona A, Esposito F, Tramontano E. Can the ever-promising target HIV reverse transcriptase-associated RNase H become a success story for drug development? Future Virol 2014;9:445–8.
- Boyer PL, Smith SJ, Zhao XZ, et al. Developing and evaluating inhibitors against the RNase H active site of HIV-1 reverse transcriptase. J Virol 2018;92:1–26.
- Schneider A, Corona A, Spöring I, et al. Biochemical characterization of a multi-drug resistant HIV-1 subtype AG reverse transcriptase: antagonism of AZT discrimination and excision pathways and sensitivity to RNase H inhibitors. Nucleic Acids Res 2016;44:2310–22.
- Corona A, Ballana E, Distinto S, et al. Targeting HIV-1 RNase H: N'-(2-Hydroxy-benzylidene)-3,4,5-trihydroxybenzoylhydrazone as selective inhibitor active against NNRTIs-resistant variants. Viruses 2020;12:729.
- Messore A, Corona A, Madia VN, et al. Pyrrolyl pyrazoles as non-dike to acid inhibitors of the HIV-1 ribonuclease H function of reverse transcriptase. ACS Med Chem Lett 2020;11:798–805.
- Xi Z, Wang Z, Sarafianos SG, et al. Determinants of active-site inhibitor interaction with HIV-1 RNase H. ACS Infect Dis 2019;5:1963–74.
- Tramontano E, Corona A, Menéndez-Arias L. Ribonuclease H, an unexploited target for antiviral intervention against HIV and hepatitis B virus. Antiviral Res 2019;171:104613.
- Wang L, Sarafianos SG, Wang Z. Cutting into the substrate dominance: pharmacophore and structure-based approaches toward inhibiting human immunodeficiency virus reverse transcriptase-associated ribonuclease H. Acc Chem Res 2020;53:218–30.
- Badland M, Compère D, Courté K, et al. Thiophene and bioisostere derivatives as new MMP12 inhibitors. Bioorg Med Chem Lett 2011; 21:528–30.
- Rance DJ. In: Damani LA, ed. Sulfur-containing drugs and related organic compounds, chemistry, biochemistry and toxicology: metabolism of sulfur functional groups. Vol. 1, Part B. Chichester: Ellis Horwood;1989 p. 217–68.
- Dansette PM, Bonierbale E, Minoletti C, et al. Drug-induced immunotoxicity. Eur J Drug Metab Pharmacokinet 1998;23:443–51.
- Massari S, Corona A, Distinto S, et al. From cycloheptathiophene-3-carboxamide to oxazinone-based derivatives as allosteric HIV-1 ribonuclease H inhibitors. J Enzyme Inhib Med Chem 2019;34:55–74.
- Cuzzucoli Crucitti G, Métifiot M, Pescatori L, et al. Structure-activity relationship of pyrrolyl diketo acid derivatives as dual inhibitors of HIV-1 integrase and reverse transcriptase ribonuclease H domain. J Med Chem 2015;58:1915–28.
- Ngoutane Mfopa A, Corona A, Eloh K, et al. Uvaria angolensis as a promising source of inhibitors of HIV-1 RT-associated RNA-dependent DNA polymerase and RNase H functions. Nat Prod Res 2018;32:640–7.
- Tintori C, Corona A, Esposito F, et al. Inhibition of HIV-1 reverse transcriptase dimerization by small molecules. ChemBioChem 2016;17:683–8.
- Tocco G, Begala M, Esposito F, et al. ZnO-mediated regioselective C-arylsulfonylation of indoles: a facile solvent-free synthesis of 2- and 3-sulfonylindoles and preliminary evaluation of their activity against drug-resistant mutant HIV-1 reverse transcriptases (RTs). Tetrahedron Lett 2013;54:6237–41.
- Corona A, Di Leva FS, Thierry S, et al. Identification of highly conserved residues involved in inhibition of HIV-1 RNase H function by Diketo acid derivatives. Antimicrob Agents Chemother 2014;58:6101–10.
- Virgilio A, Amato T, Petraccone L, et al. Improvement of the activity of the anti-HIV-1 integrase aptamer T30175 by introducing a modified thymidine into the loops. Sci Rep 2018;8:7447.
- Tintori C, Esposito F, Morreale F, et al. Investigation on the sucrose binding pocket of HIV-1 integrase by molecular dynamics and synergy experiments. Bioorg Med Chem Lett 2015;25:3013–6.
- Esposito F, Tintori C, Martini R, et al. Kuwanon-L as a new allosteric HIV-1 integrase inhibitor: molecular modeling and biological evaluation. ChemBioChem 2015;16:2507–12.
- Esposito F, Sechi M, Pala N, et al. Discovery of dihydroxyindole-2-carboxylic acid derivatives as dual allosteric HIV-1 Integrase and Reverse Transcriptase associated Ribonuclease H inhibitors. Antiviral Research 2020;174:104671.
- Sala M, Spensiero A, Esposito F, et al. Development and identification of a novel anti-HIV-1 peptide derived by modification of the N-terminal domain of HIV-1 integrase. Front Microbiol 2016;845:1–9.
- Esposito V, Esposito F, Pepe A, et al. Probing the importance of the G-quadruplex grooves for the activity of the anti-HIV-integrase aptamer T30923. Int J Mol Sci 2020;21:5637.
- Pisano MB, Cosentino S, Viale S, et al. Biological activities of aerial parts extracts of euphorbia characias. Biomed Res Int 2016;2016:1538703.
- Corona A, Onnis V, Del Vecchio C, et al. 2-(Arylamino)-6-(trifluoromethyl)nicotinic acid derivatives: new HIV-1 RT dual inhibitors active on viral replication. Molecules 2020;25:1338.
- Esposito F, Sanna C, Del Vecchio C, et al. Hypericum hircinum L. components as new single-molecule inhibitors of both HIV-1 reverse transcriptase-associated DNA polymerase and ribonuclease H activities. Pathog Dis 2013;68:116–24.
- Desantis J, Massari S, Corona A, et al. 1,2,4-Triazolo[1,5-a]pyrimidines as a novel class of inhibitors of the HIV-1 reverse transcriptase-associated Ribonuclease H activity. Molecules 2020;25:1183.
- Nettleship JE, Brown J, Groves MR, Geerlof A. Methods for protein characterization by mass spectrometry, thermal shift (ThermoFluor) assay, and multiangle or static light scattering. Methods Mol Biol 2008;426:299–318.
- Yonetani T. The Yonetani-Theorell graphical method for examining overlapping subsites of enzyme active centers. Methods Enzymol 1982;87:500–9.
- Sonar VP, Corona A, Distinto S, et al. Natural product-inspired esters and amides of ferulic and caffeic acid as dual inhibitors of HIV-1 reverse transcriptase. Eur J Med Chem 2017;130:248–60.
- Weislow OS, Kiser R, Fine DL, et al. New soluble-formazan assay for HIV-1 cytopathic effects: application to high-flux screening of synthetic and natural products for AIDS-antiviral activity. J Natl Cancer Inst 1989; 81:577–86.
- Bakavoli M, Bagherzadeh G, Vaseghifar M, et al. Iodine catalysed synthesis and antibacterial evaluation of thieno[2,3-d]pyrimidine derivatives. J Chem Res 2009;2009:653–5.
- Raffa D, Maggio B, Plescia F, et al. Synthesis, antiproliferative activity, and mechanism of action of a series of 2-{[(2E)-3-phenylprop-2-enoyl]amino}benzamides. Eur J Med Chem 2011; 46:2786–96.
- Zheng Y, Bian M, Deng X, et al. Synthesis and anticonvulsant activity evaluation of 5-phenyl-[1,2,4]triazolo[4,3-c]quinazolin-3-amines. Arch Pharm Chem Life Sci 2013;346:119–26.
- Del Zotto A, Amoroso F, Baratta W, Rigo P. Very fast suzuki–miyaura reaction catalyzed by Pd(OAc)2 under aerobic conditions at room temperature in EGME/H2O. Eur J Org Chem 2008;2009:110–6.
- Roussaki M, Kontogiorgis CA, Hadjipavlou-Litina D, et al. A novel synthesis of 3-aryl coumarins and evaluation of their antioxidant and lipoxygenase inhibitory activity. Bioorg Med Chem Lett 2010;20:3889–92.
- Chung S, Miller JT, Johnson BC, et al. Mutagenesis of human immunodeficiency virus reverse transcriptase p51 subunit defines residues contributing to vinylogous urea inhibition of ribonuclease H activity. J Biol Chem 2012;287:4066∓75.
- Dubey S, Satyanarayana YD, Lavania H. Development of integrase inhibitors for treatment of AIDS: an overview. Eur J Med Chem 2007;42:1159–68.
- Esposito F, Tramontano E. Past and future. Current drugs targeting HIV-1 integrase and reverse transcriptase-associated ribonuclease H activity: single and dual active site inhibitors. Antiv Chem Chemother 2014;23:129–44.
- Cummings MD, Farnum M, Nelen MI. Universal screening methods and applications of ThermoFluor. J Biomol Screen 2006;11:854–63.
- Himmel DM, Sarafianos SG, Dharmasena S, et al. HIV-1 reverse transcriptase structure with RNase H inhibitor dihydroxy benzoyl naphthyl hydrazone bound at a novel site. ACS Chem Biol 2006;1:702–12.
- Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 2010;53:2719–40.
- Ingólfsson HI, Thakur P, Herold KF, et al. Phytochemicals perturb membranes and promiscuously alter protein function. ACS Chem Biol 2014;9:1788–9178.