
Abstract
Renal cell carcinoma is an aggressive disease often asymptomatic and weakly chemo-radiosensitive. Currently, new biologic drugs are used among which everolimus, an mTOR inhibitor, that has been approved for second-line therapy. Since mTOR is involved in the control of autophagy, its antitumor capacity is often limited. In this view, chloroquine, a 4-alkylamino substituted quinoline family member, is an autophagy inhibitor that blocks the fusion of autophagosomes and lysosomes. In the present study, we evaluated the effects of everolimus alone or in combination with chloroquine on renal cancer cell viability and verified possible synergism. Our results demonstrate that renal cancer cells are differently sensitive to everolimus and chloroquine and the pharmacological combination everolimus/chloroquine was strongly synergistic inducing cell viability inhibition. In details, the pharmacological synergism occurs when chloroquine is administered before everolimus. In addition, we found a flow autophagic block and shift of death mechanisms to apoptosis. This event was associated with decrease of Beclin-1/Bcl-2 complex and parallel reduction of anti-apoptotic protein Bcl-2 in combined treatment. At last, we found that the enhancement of apoptosis induced by drug combination occurs through the intrinsic mitochondrial apoptotic pathway activation, while the extrinsic pathway is involved only partly following its activation by chloroquine. These results provide the basis for new therapeutic strategies for the treatment of renal cell carcinoma after appropriate clinical trial.
Abbreviations
| FITC | = | fluorescein isothiocyanate |
| PI | = | propidium iodide |
| VHL | = | Von Hippel-Lindau |
| CLC | = | chloroquine |
| RAD | = | everolimus |
| MFI | = | mean fluorescence intensity |
| MDC | = | monodansyl-cadaverine |
| AVOS | = | autophagic vacuoles |
Introduction
Renal cancer accounts for 2–3% of all adult malignancies. The annual incidence is around 100,000 new cases.Citation1,2 The kidneys may be affected by tumors of different nature: the best known is the renal cell carcinoma (RCC), which has a very high incidence. The highest peak is in the age group between 60 and 70 y and affects mostly men. Generally, renal cancer is asymptomatic and often when the first symptoms appear the stage is too advanced to be able to hope for a cure. The main risk factors are smoking, obesity and hypertension.Citation3,4 Today, 2 histotypes of renal cancer are known: a sporadic form, more frequently, usually unilateral and aggressive and a hereditary form, rarer, often bilateral, but with a less aggressive course.Citation5 In the last decades, thanks to the hereditary-familial forms studies, an enormous amount of information regarding the molecular pathogenesis of these tumors were acquired, with important implications also on therapies. Renal cancer is generally a poorly chemo-radiosensitive tumor and until a few years ago the only treatment for metastatic disease was made by cytokines (IL-2 and INF). The median survival with these treatments did not exceed 10 months,Citation6 with a small percentage of subjects alive at 5 y (<10%).Citation7 In addition, the tolerability was often unacceptable and frequent dose reduction or treatment interruption was necessary. This condition has been revolutionized by the recent molecular biology and genetics discoveries, in particular by some growth molecular mechanisms and by tumor angiogenesis concept. New targeted drugs have been developed, which can substantially change the natural history of this disease. The molecular pathogenesis model observed in both hereditary and sporadic forms have focused researcher attention on the Von Hippel-Lindau tumor suppressor gene (VHL). In physiological conditions, the VHL gene codes for a VHL protein, which forms a complex with other proteins and has as target the hypoxia inducible factor HIF1 sub-unit 1-α. In normal oxygen conditions, the VHL protein promotes HIF1-α degradation and destruction inside of proteasomes. In the presence of hypoxia, the HIF1-α is not degraded and migrates to the nucleus where binding to the β subunit, form the HIF1 heterodimer, which activates the proteins synthesis by hypoxia inducible genes. In this way, the cell activates the adaptation mechanisms to unfavorable environmental conditions, through the synthesis of proteins involved in the angiogenesis, proliferation and survival processes: vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), plated-derived growth factor (PDGF), transforming growth factor (TGF). In the presence of a VHL gene mutation, an abnormal VHL protein is encoded that is unable to degrade the HIF1 and the cell, in response to a presumed hypoxia condition, synthesizes proangiogenic growth factors such as VEGF and PDGF. The final results of the VEGF activation are: tumor angiogenesis, vascular permeability increased, lymphangiogenesis and metastasis. New biologic drugs, mainly directed against VEGF or its receptors, are currently into clinical practice and give excellent results in terms of therapeutic efficacy. These new drugs can be divided into 2 groups on the basis of their specific targets: multi-target kinase inhibitors (sunitinib, sorafenib, axitinib, pazopanib) and monoclonal antibodies against VEGF (bevacizumab).Citation8,9 Unfortunately, the patients after initial responses develop resistance to these agents and novel targets are required in order to control the disease. Currently, mTOR inhibitors are the pharmacological class most used in the renal cancer clinical treatment. Everolimus has been approved for second-line therapy of patients with renal cell carcinoma after failure of treatment with sunitinib and for the treatment of papillary renal carcinoma, pancreatic neuroendocrine tumor, some types of breast cancer, and subependymal giant cell astrocytoma associated with tuberous sclerosis.Citation10–14 Everolimus, a rapamycin analog, is an oral mammalian target of rapamycin (mTOR) inhibitor which belongs to the PI3K related family of protein kinases and is activated by phosphorylation at serine 2448 (S2448).Citation15,16 mTOR is a key effector in the PI3K/Akt/mTOR pathway and plays a critical role in regulating cell proliferation, survival, and angiogenesis.Citation17,18 mTOR works as part of 2 distinct multimeric complexes, known as mTORC1 and mTORC2. mTORC1 phosphorylates S6K1 and eIF-4E-binding protein 1 (4E-BP1) leading to protein synthesis. mTORC2 phosphorylates AKT in response to growth factors. As negative feedback, mTORC1 inhibits AKT by suppressing upstream pathways. The complexity of mTOR system implies contradictory functions both as tumor promoting and suppressor factors.Citation19,20 Furthermore, the PI3K/Akt/mTOR pathway is involved in the control of autophagy, an ubiquitous and evolutionarily conserved process that degrades cytosolic components via the lysosomes and allows cells to survive to various forms of stress.Citation21 There is increasing evidence that PI3K/Akt/ mTOR inhibitors initiate autophagy as a survival program that may interfere with their antitumor activity. Consequently, inhibition of autophagy was used as a strategy to enhance the efficacy of PI3K/Akt/mTOR inhibitors in different cancers.Citation22-24 Increasing experimental and clinical evidence may establish mTOR inhibition as a promising rationale for targeting human malignancy. Autophagy is known to limit tumor cell growth, reduce mutagenesis or other damage caused by reactive oxygen species.Citation25-27 Alternatively, autophagy may kill developing tumor cells. Stimulation of autophagy suppresses cancer development but may promote tumor growth and survival under conditions of nutrient deprivation in poorly angiogenic tumors. Autophagy may also protect tumor cells from undergoing apoptosis in response to treatment with anticancer agents. These evidences suggest that, depending on the context, autophagy can both stimulate and prevent cancer. To date, there are several autophagy inhibitors that are used to study the role of autophagy such as chloroquine, a 4-alkylamino substituted quinoline family member. It is lysosomotropic and inhibits the fusion of autophagosomes and lysosomes. Chloroquine is widely used as an anti-malarial, and also as an anti-inflammatory in the treatment of rheumatoid arthritis and Lupus Erythematosus. Recently, attention has focused on its potential anti-cancer action and chemotherapy sensitizer.Citation28,29 Although the precise basis of the anti-cancer effects of chloroquine is still under investigation, inhibition of autophagy most probably plays a part in anti-tumoral effect.
The aim of this study was to evaluate the effects of everolimus (RAD) alone or in combination with chloroquine (CLC) on renal cancer cells viability and to verify the possible synergism. Moreover, we have investigated cell death mechanisms and biochemical pathways involved in the cell proliferation and survival processes induced by drug combination.
Results
Everolimus and chloroquine effects on human renal cancer cells viability
We analyzed the antiproliferative effects of everolimus and chloroquine on human renal cancer cells lines viability (A498, RXF393, SN12C and 769P) by MTT assay. The antiproliferative effect induced by drugs, after 72 h of treatment, was dose-dependent in all cell lines examined (Figure1 A-B). The concentrations of everolimus and chloroquine able to induce 50% growth inhibition (IC50) in the different cell lines are shown in . The data obtained suggest that renal cancer cell lines are differently sensitive to everolimus and chloroquine and that RXF393 and SN12C cell lines seem to be the most resistant to both pharmacological treatments.
Table 1. Everolimus and chloroquine IC50 values (μM) in renal cancer cell lines
Evaluation of the synergistic effect induced by the drug combination everolimus/chloroquine in human renal cancer cell lines
We studied the drug combination effects on human renal cancer cell lines proliferation by MTT assay. The data obtained were processed with the dedicated program, CalcuSyn, which measures the drugs synergism, as described in “Materials and Methods.” In order to search the drugs combination index calculated for the 50% of surviving cells (CI50). The CI50 values of < 0.5, between 0.5 and 1, equal to 1 and > 1, indicate strong synergism, synergism, additivity and antagonism, respectively. We used equitoxic concentrations of 2 drugs in different sequences and in simultaneous administration. In all cases proliferative inhibition evaluation was performed after 72 h from the beginning of treatment. In particular, we observed that the drugs combination produced a strong synergism in renal cancer cell lines after 72 h of treatment, in both concomitant and sequential treatment, as shown in . The combination index for A498 cell line was 0.4 for the cells exposed to the simultaneous treatment, 0.85 for the cells exposed to RAD→CLC treatment and 0.33 for cells exposed to CLC→RAD treatment. The combination index for 769P cell line was 0.44 for cells exposed to simultaneous treatment, 1 for RAD→CLC treatment and 0.45 for CLC→RAD treatment. For RXF393 cell line we observed antagonism with the simultaneous combinations treatment, while synergism was observed when cells were treated with both sequences of administration. Finally for SN12C cell line we observed an antagonism in both sequences of administration and a strong synergism in the simultaneous combination, with a CI50 of 0.5. These data suggest that the sequence CLC→RAD is strongly synergistic in inducing cell growth inhibition for all renal cancer cell lines, except that in SN12C cell line, which was very sensitive to RAD/CLC in simultaneous treatment, while antagonism was observed by treating cells with the 2 different sequences of administration; this cell line responds differently to drug treatment compared to the other cell lines, probably due to molecular characteristics not yet known and that deserve additional investigations. Dose reduction index 50 (DRI50) represents the magnitude of dose reduction obtained for the 50% growth inhibitory effect in combination setting as compared to each drug alone. In our experimental conditions, these values were significant for all cell lines (), since DRI >1 values allow a decrease in the dosage, which leads to a considerable reduction of toxicity induced by the drugs.
Table 2. Combination Index (CI) and Dose Reduction Index (DRI) Values for everolimus and chloroquine (RAD/CLC) combinations
The synergistic effect of the RAD/CLC combination on growth inhibition is mediated by the induction of apoptosis
After treatment with the drug combinations, we found that A498 and RXF393 cell lines were more sensitive to drugs combination, therefore we evaluated in these cell lines death mechanisms involved. This study was conducted by FACS analysis after staining with AnnexinV/PtdIns, as described in Materials and Methods section. As shown in , we found a significant increase of mean fluorescence intensity (MFI) in both cell lines treated with the drug combination, compared to untreated or treated with single drugs. In particular, in RXF393 cell line we found an increased of late apoptosis in the cells treated with the drug combination, compared to untreated or treated with single drugs, while in the A498 cells line we found an increased also of early apoptosis in all type of drug administration, compared to untreated or treated with single drugs.
RAD/CLC combination induces an increase of autophagy markers in human renal cancer cell lines
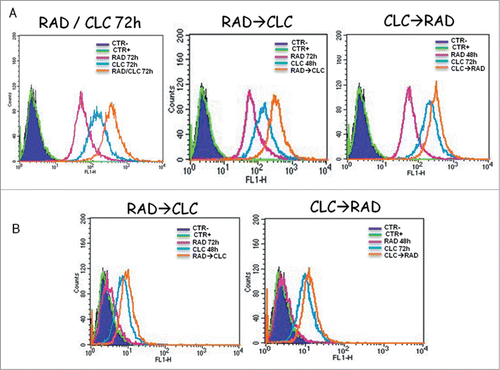
RAD/CLC combination induces an increase in markers of autophagy in the A498 and RXF393 cell lines. As shown in , the flow cytometric analysis, it was found an increase in the Mean Fluorescence Intensity (MFI) in the cells treated with the combination of drugs, compared to cells treated with drugs alone or not treated cells. This effect could be caused by late autophagy block by chloroquine which would, therefore, an accumulation of autofago-lysosomal vesicles with consequent paradox increase of MDC staining. So, autophagy may be a mechanism of protection against proliferative inhibition induced by everolimus.
Figure 1. Evaluation of everolimus and chloroquine effect on renal cancer cells growth. The curves show the percentage of renal cancer cells growth following everolimus (A) and chloroquine (B) dose-dependent exposure for 72 h. Each point is the average of at least 3 repeated experiments (Bars, SEs).
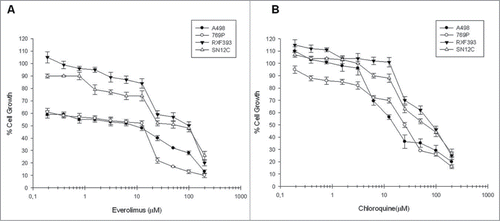
Figure 2. FACS analysis after double labeling A498 cell line with PI and Annexin V. The cells were treated with CLC and RAD alone and in sequence, compared to the control. Insets show the percentage of cells in the different quadrants. UL = Upper Left (necrosis); UR = Upper Right (late apoptosis); LL = Lower Left (viable); LR = Lower Right (early apoptosis). Untreated cells, CTR; CLC added for 72 h and RAD for the last 48 h, CLC→RAD; RAD added for 72 h and CLC added for the last 48 h, RAD→CLC; RAD and CLC added simultaneously for 72 h, RAD/CLC 72h. The figure is representative of 3 different experiments that always gave similar results.
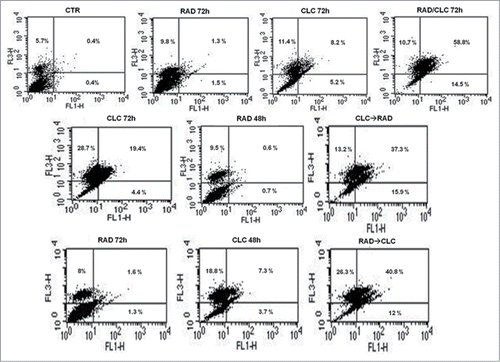
Figure 3. FACS analysis after double labeling RXF393 cell line with PI and Annexin V. The experimental conditions are identical to those shown in .
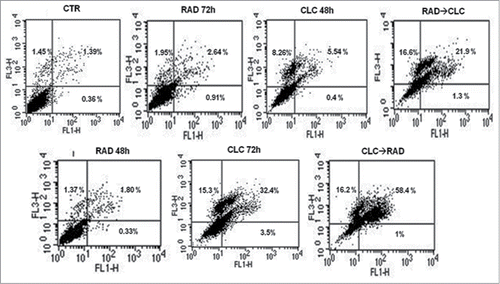
Figure 4. Autophagy evaluation after treatment with CLC and/or RAD alone or in sequence. A498 (A) and RXF393 (B) cells were incubated with MDC and analyzed by flow cytometry as described in “Materials and Methods” in order to evaluate the autophagy onset. Untreated cells unexposed to MDC, CTR; untreated cells exposed to MDC, CTR+; CLC added for 48 h and RAD 72 h, RAD→CLC; CLC added for 72 h and RAD for the last 48 h, CLC→RAD; CLC and RAD added for 72 h, RAD/CLC 72h. The experiments were repeated at least 3 times and always gave similar results (Bars, SDs).
Effects of CLC and RAD on autophagic molecular mechanisms
Next, we investigated the molecular mechanisms of cell death processes, in particular, we have focused our attention on autophagy by studying the interaction between 2 molecules involved in this process: Beclin1 and Bcl-2. It is known that Bcl-2, interacting with Beclin-1, inhibits Beclin-1-dependent autophagy.Citation30 Beclin1 is involved in the early autophagosome formation, therefore its seizure by Bcl-2 inhibits the autophagic process at that level switching the death process to apoptosis. These studies were conducted on cell lines that are more sensitive to drug combination (RXF393 and A498). When these proteins were co-immunoprecipitated, we found that the combined treatment of drugs decreases Beclin-1/Bcl-2 complex formation if compared to the 2 drugs alone, in both cell lines (). It is also noted that CLC alone reduces the Bcl-2/Beclin-1 complex formation, which is in agreement with late action of CLC on autophagic inhibition. In fact, in this case, Beclin-1 has the opportunity to participate in early autophagosome formation, but autophagy is blocked downstream by CLC. The reduction of Beclin-1/Bcl-2 complex formation induced by combined treatment takes place in parallel to the reduction of the Bcl-2 anti-apoptotic protein expression, in both cell lines, and especially in the sequence CLC→RAD, which is also able to determine that a greater proliferative inhibition synergism. Therefore, the reduction of the 2 proteins interaction is paralleled by a reduction of total Bcl2 expression with consequent lack of its anti-apoptotic effect. These results suggest a block of late autophagic flux with a concomitant increase of apoptosis activation through the mitochondrial pathway.
Evaluation of pathways involved in the proliferation and survival regulation
Another key component that regulates the balance between cell growth and autophagy in response to cellular physiological conditions and environmental stress is mTOR, the mammalian target of rapamycin, target of everolimus. The mTOR activation leads to the phosphorylation of its 2 downstream main components p70S6K and eIF4E-binding protein 1 (4EBP1). 4EBP1 is necessary mainly to control the cap-dependent translation binding and inactivating eIF4E, an initiation factor, which binds the cap structure of mRNA, mediating the start of translation. The phosphorylation of 4EBP1 causes the release of eIF4E and subsequent activation of the eIF4G binding protein. Both PI3kinase/AKT and mTOR regulate the activity of 4EBP1.Citation29 As shown in , in both cell lines, the combination RAD→CLC decreases 4EBP1 phosphorylation if compared to untreated cells. It should be noted, however, that in the simultaneous treatment with RAD and CLC in A498 cell line, 4EBP1 phosphorylation increases if compared to that of untreated cells. Furthermore, this drug combination determines a lower interaction than the other 2 sequences of administration. It 's interesting to note that, in the same cell line, RAD alone, in both times of treatment, causes an increase of 4EBP1 phosphorylation which is antagonized by the combined treatment, especially in the 2 sequences of drug administration.
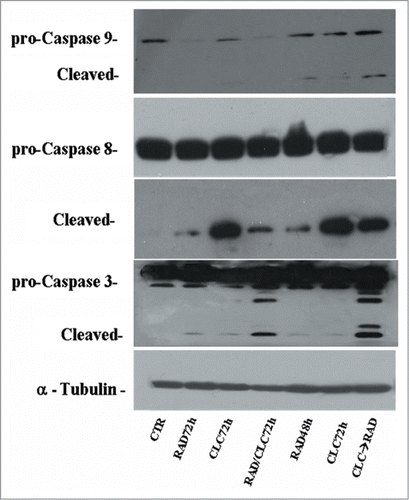
We also studied the regulation of a molecule involved in cell proliferation and in survival pathways, Erk1/2, whose activity is decreased in both cell lines, especially in the sequence CLC→RAD, compared with drugs alone. Also in this case the reduction of the activity of anti-autophagic and anti-apoptotic mTOR-dependent pathways occurs together with the reduction of an Erk-dependent signaling survival moving cell death to apoptosis. In A498 cell line, we have also evaluated which apoptotic pathway is activated, and as shown in , we found that the apoptosis enhancement induced by the drug combination is mediated by the initiator caspase-9 and the effector caspase-3. In fact, an evident increase of the cleaved fragments of these caspases is observed in cells treated with drug combination compared to the cells treated with drug alone. On the other hand, there isn't a clear enhancement of the initiator caspase-8 fragmentation induced by drug combination, while CLC at 72 h induces a caspase-8 activation, as demonstrated by the evident caspase-8 fragments formation. These data suggest that the apoptosis enhancement induced by the drug combination occurs through the activation of the intrinsic mitochondrial apoptosis pathway, while the extrinsic pathway is involved only partly, following its activation by CLC.
Figure 5. Evaluation of Beclin-1 and Bcl-2 interaction. (A) RXF393 were treated with CLC and RAD alone or in sequence. Then we performed western blotting assay (WB) for the expression of the total Bcl-2 and Beclin-1 proteins and immunoprecipitation (IP) for the evaluation of Bcl-2/Beclin-1 and Beclin-1/Bcl-2 complex formation. (B) Representation of the complexes expressed as the ratio between the relative intensities of the bands associated with the Bcl-2/Beclin-1 and Beclin-1/Bcl-2 complexes vs. the bands associated with total Bcl-2 and Beclin-1, respectively. The intensities of the bands were expressed as arbitrary units when compared to those of the untreated cells (CTR) The figure is representative of 3 different experiments that always gave similar results.
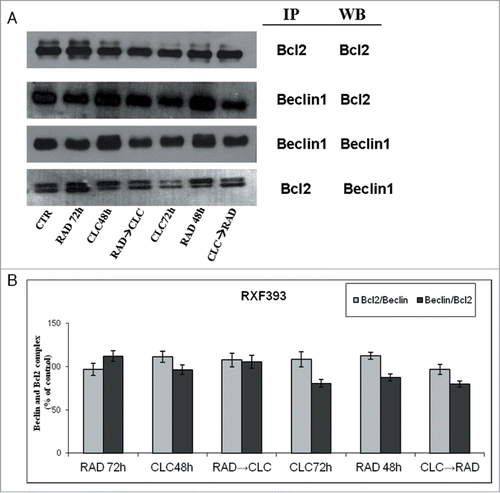
Figure 6. Evaluation of pathways involved in the regulation of proliferation and survival. (A) RXF393 were treated with RAD and/or CLC alone or in combination. Thereafter, both the activity and the expression of the different proteins were evaluated. Expression of the house-keeping protein α-tubulin was used as loading control. (B) Representation of the intensities of the bands associated to the different proteins normalized for the expression of the total proteins. The intensities of the bands were expressed as arbitrary units when compared to those of the untreated cells. The figure is representative of 3 different experiments that always gave similar results.
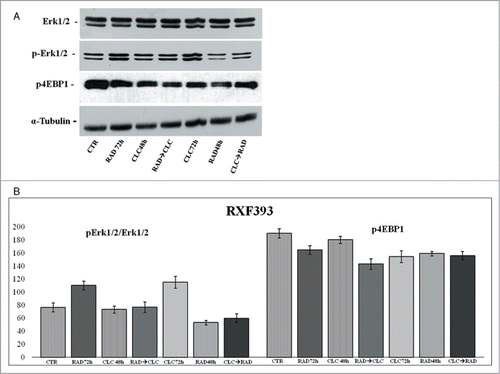
Figure 7. Evaluation of Beclin-1 and Bcl-2 interaction. (A) A498 were treated with CLC and RAD alone or in sequence. Thereafter we performed protein gel blotting assay (WB) for the expression of the total Bcl-2 protein and immunoprecipitation (IP) for the evaluation of Bcl-2/Beclin-1 complex formation. (B) Representation of the Bcl-2/Beclin-1 complexes expressed as the ratio between the relative intensities of the bands of the complex versus the bands associated with total Bcl-2. The intensities of the bands were expressed as arbitrary units when compared to those of the untreated cells. The figure is representative of 3 different experiments that always gave similar results.
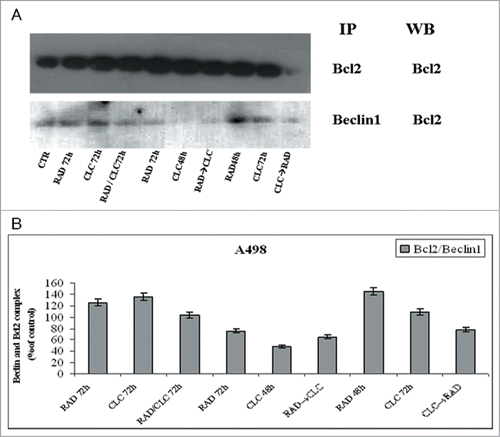
Figure 8. Evaluation of pathways involved in the regulation of proliferation and survival. (A) A498 were treated with RAD and/or CLC alone or in combination. Thereafter, both the activity and the expression of the different proteins were evaluated after western blotting assay with specific antibodies, as described in “Materials and Methods.” Expression of the house-keeping protein α-tubulin was used as loading control. (B) Representation of the intensities of the bands associated to the different proteins normalized for the expression of the total proteins. The intensities of the bands were expressed as arbitrary units when compared to those of the untreated cells. The figure is representative of 3 different experiments that always gave similar results.
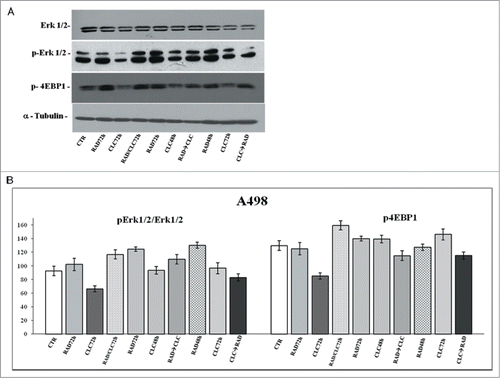
Figure 9. Evaluation of caspases involved in apoptotic process. A498 were treated with RAD and/or CLC alone or in combination. Thereafter, both the activity and the expression of the different proteins were evaluated after protein gel blotting assay with specific antibodies, as described in “Materials and Methods.” Expression of the house-keeping protein α-tubulin was used as loading control.
Discussion
Renal cancer usually occurs without symptoms if not in an advanced stage of the disease.Citation3,4 The recent molecular biology and genetic knowledge has revealed new targeted agents that modify substantially the natural history of this disease. In particular, sunitinib, a pan inhibitor of multiple membrane tyrosine kinases receptors (e.g., VEGFR, PDGFR, FGFR and the c-Kit) has radically changed the prognosis of this disease resulting in clinical responses and disease-free survival in a large number of patients (approximately 45%). Furthermore, it has been demonstrated that chloroquine enhanced sunitinib cytotoxicity in a synergistic manner via inducing apoptosis while switching off autophagic and angiogenic machineries.Citation32 Another drug used is sorafenib that inhibits Raf kinase and VEGF; it has also been used successfully in the second-line of renal cancer treatment. However patients develop resistance to sunitinib and sorafenib so second-line therapies are required. In this context, we include the everolimus which is an analog of rapamycin, which mammals target is mTOR, that is a serine threonine kinase, key effector in the PI3K\Akt\mTOR pathway and plays a critical role in the regulation of proliferation, cell survival and angiogenesis. Everolimus binds to the intracellular protein FKBP-12 to form a complex that inhibits the activity of mTORC1. This inhibition interferes the translation and proteins synthesis reducing the S6K1 and 4E-BP1 proteins activity, which regulate the proteins involved in cell-cycle, angiogenesis and glycolysis. In addition, everolimus reduces the levels of VEGF, which potentiates tumor angiogenic processes and it has been demonstrated to induce anti-tumor effects, such as cell cycle arrest and modulation of senescence, as described in Faggiano et al.Citation33 The PI3K\Akt\mTOR pathway is also involved in the control of autophagy, programmed cell death characterized by the cellular cytoplasmatic autophagic vacuoles formation that incorporate and degrade cellular components. Autophagy is stimulated in response to stress and normally represents a cell survival mechanism which is opposed to other types of programmed cell death, such as apoptosis. Therefore, the switch from apoptosis to autophagy is widely considered as a mechanism of cell resistance to cell death itself. Even for anticancer drugs, autophagy appears as a resistance mechanism to cell death, unless there is a protracted exposure of drug which leads to cell death. Among autophagy inhibitors we remember the chloroquine that is widely used as an antimalarial and works blocking DNA synthesis and the activities of some important enzymes of the parasites. Recently, the attention has focused on its potential as anticancer agent, indeed it induces the suppression of the autophagy process, which in turn is inhibited in cells that have active mTOR.
In this study we evaluated the effects of everolimus alone or in combination with chloroquine on the renal cancer cells viability and verified the possible synergism. Moreover, we have investigated cell death mechanisms and biochemical pathways involved in the cell proliferation and survival processes induced by drug combination. First, we studied the drug interaction, in vitro, on renal cancer cell growth inhibition (A498, 769P, RXF393, SN12C) and we found a synergistic effect induced by pharmacological combination. In particular, the pharmacological synergism occurs when chloroquine is administered before everolimus. Flow cytometric studies showed an induction of apoptotic cell death caused by the drug combination and a paradoxical increase of some late autophagic markers (AVOS formation); these data suggested a block of autophagic flow and a shift of death mechanisms to apoptosis. The antiproliferative effect induced by everolimus and chloroquine was dose-dependent in all examined cell lines. Subsequently, we studied the drug combination effects treating the cells with different drug concentrations, in different sequences of administration CLC→RAD, RAD→CLC, RAD/CLC. The data obtained were processed by the dedicated program, CalcuSyn which measures the synergism degree between the drugs, in order to search the combination index calculated for 50% of surviving cells (IC50). In particular, for A498 cell line, the drugs are strongly synergistic in both sequences of administration and in simultaneous administration; for RXF393 cell line the drugs are synergistic in CLC→RAD sequence, while for 769P and SN12C cell lines a synergistic effect was detected in RAD/CLC and CLC→RAD treatments. The apoptosis induced by drug combination was evaluated on A498 and RXF393 cell lines. This study was conducted by FACS analysis, after staining with Annexin V/PI. A significant increase of' apoptosis was found in cells treated with 2 drugs in combination or in sequence, compared to untreated cells or cells treated with drugs alone. On the same cell lines was assessed the ability of the 2 drugs to induce autophagy, the alternative mechanism of cell death that can develop as a phenomenon of protection of tumor cells from everolimus. Autophagy was evaluated by FACS analysis after staining with the monodansylcadaverine (MDC). We found a strong increase of Mean Fluorescence Intensity (MFI) in cells treated with the pharmacological combination, compared to cells treated with drugs alone and to untreated cells. These data suggest that chloroquine, blocking autophagy at a late stage, inhibits recycling autofagolysosomal vesicles. This induces an increase in these vesicles, producing an inhibition of autophagic flux and a paradox increase of MDC staining (autophagic later vesicles marker). Similarly, it was recently reported in the literature that clomipramine blocks autophagy in Hela cells, producing an paradox increase of late autophagic vesicles, blocking their recycling.Citation34 Based on these results, we have focused our attention on the modulation of the biochemical pathways involved in the proliferation processes, survival and cell death. These studies were conducted on A498 and RXF393 cell lines that are more sensitive to everolimus/chloroquine combination. In particular, we studied autophagy and the interaction between 2 molcules involved in this process: Beclin-1 and Bcl-2. It well known that Bcl-2 interacting with Beclin-1, inhibits Beclin-1-dependent autophagy. Co-immunoprecipitating the 2 proteins, we found that the combined treatment decreases the formation of Beclin-1/Bcl-2 complex respect to 2 drugs used in a single dose, on both cell lines. However, this effect occurs in parallel with a significant reduction of the total expression of anti-apoptotic protein Bcl-2, while Beclin-1 levels remain almost unchanged. These results suggest that the pro-apoptotic effect of pharmacological combination occurs through the modulation of the anti-apoptotic mitochondrial proteins expression, such as Bcl-2. Another key component that regulates the balance between cell growth and autophagy in response to physiological cellular conditions and to stress is mTOR, the mammalian target of rapamycin, target of everolimus. The mTOR activation leads to the phosphorylation of its 2 downstream main components p70S6K and eIF4E-binding protein 1 (4EBP1). 4EBP1 is necessary mainly to control the cap-dependent translation binding and inactivating eIF4E, an initiation factor, which binds the cap structure of mRNA, mediating the start of translation. The phosphorylation of 4EBP1 causes the release of eIF4E and subsequent activation of the eIF4G binding protein. Both PI3kinase/AKT and mTOR regulate the activity of 4EBP1.Citation31
The RAD→CLC combination decreases 4EBP1 phosphorylation if compared to untreated cells. It should be noted, however, that in the simultaneous treatment for A498 cell line 4EBP1 phosphorylation increases if compared to untreated cells. It should be noted that a simultaneous drug combination determines a lower pharmacological interaction than the other 2 sequences of administration. It 's interesting to note that, in the same cell line, everolimus alone at 48 and 72 hours of treatment, causes an increase of 4EBP1 phosphorylation, which is antagonized by the combined treatment, especially in the 2 sequences of administration. These data suggest that in pharmacological conditions in which there is a greater synergism, there is a 4EBP1 phosphorylation reduction, which is a surrogate marker for mTOR activity. Therefore, the drug combination has a synergistic effect to inhibit the Akt-mTOR-dependent pathway. This effect occurs in parallel to a decrease of Erk1/2 activity in both cell lines and in both sequences of administration. These effects are in agreement with the enhancement of a pro-apoptotic pathway, as mTOR is involved in the apoptosis and autophagy regulation (), and Erk one of the most important cellular survival enzymes. In order to clarify the caspases involvement and apoptotic pathway activated by the drug combination, we studied the expression of full and cleaved forms of the initiator caspases-8 and -9 and effector caspase-3, on A498 cell line. We found that apoptosis is mediated by initiator caspase- 9 and the effector caspase-3. In fact, a clear increase of cleaved caspase-3 fragments is detected in cells treated with the pharmacological combination compared to cells treated with drug alone. On the other hand there isn't a clear enhancement of the initiator caspase-8 fragmentation induced by the drug combination, while CLC 72 h induces a caspase-8 activation, as demonstrated by the evident caspase-8 fragment formation. These data suggest that the enhancement of apoptosis induced by drug combination occurs through the intrinsic mitochondrial apoptotic pathway activation, while the extrinsic pathway is involved only partly following its activation by chloroquine. However, it is reported that the use of chloroquine to treat cancer could induce side effects on normal cells and in particular in kidney, since autophagy plays also a protective role by the damage induced by pharmacological agents.Citation35 On these bases, we intend to evaluate the possible toxic effects in in vivo experiments before to translate this results in a clinical study. Nonetheless, taken together, these preliminary data provide the rationale for the design of new therapeutic strategies for the treatment of renal cancer.
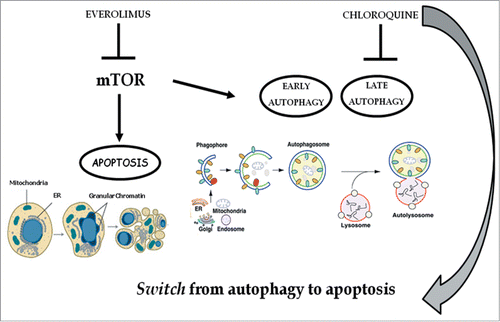
Figure 10. Chloroquine blocks autophagy induced by everolimus and consequently strengthens the apoptosis induced by the latter. mTOR is involved in the apoptosis and autophagy regulation: everolimus inhibits the mTOR pathway triggering both apoptosis and early autophagy; the addition of chloroquine inhibits late autophagy and consequently blocks autophagic flux thus inducing a switch from autophagy to apoptosis. These effects mediate a potentiation of the growth inhibition of renal cancer cells due to the abrogation of a protective anti-apoptotic and autophagic pathway.
Materials and Methods
Materials
Dulbecco's modified medium (DMEM), RPMI 1640 medium, Fetal Bovine Serum (FBS), BSA were purchased from GIBCO (Life Technologies, Milan, Italy). Tissue culture plastic ware was from Becton Dickinson (Lincoln Park, NJ). Anti-Caspase-3, -8, -9, anti-Beclin1, anti-p-4EBP1, anti-MAPK p42/44 and anti-p-MAPK p42/44 were purchased from Cell Signaling Technology Inc. (Beverly, MA, USA). Anti-Bcl2 was from Santa Cruz Biotechnology. Annexin-V-FITC was from Med Systems Diagnostics (Vienna, Austria). Everolimus (RAD) (Afinitor, Novartis), chloroquine (CLC), protein A-agarose, PtdIns and Monodansyl-cadaverine were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
The human renal adenocarcinoma A498, RXF393, SN12C and 769P cells, obtained from the National Institute for cancer treatment and study, "Giovanni Pascale" foundation, (Naples, Italy), were grown in DMEM (A498, RXF393, SN12C) and RPMI (769P) supplemented with heat inactivated 10% FBS, 20 mM HEPES, 100 U/ml penicillin, 100mg/ml streptomycin, 1% L-glutamine, and 1% sodium pyruvate. The cells were grown in a humidified atmosphere of 95% air/5% CO2 at 37°C.
Cell viability assay
Cell viability was analyzed by the MTT [3-(4,5-dimethylthiazolyl)-2,5-diphenyl tetrazolium bromide] assay. Cells were seeded in 96-well plates at the density of 1.5 × 103 cells/well for A498, RXF393, SN12C and at the density of 3 × 103 cells/well for 769P, in a final volume of 100 ml. Cells were then incubated at 37°C in a humidified atmosphere to allow exponential growth. The following day, cells were treated with RAD and CLC at a range of concentrations between 0.19 and 200 μM. At 72 h from the treatments, cells were exposed to a solution of 10% MTT for 4 h at 37°C to form formazan crystals by reacting with metabolically active cells. The formazan crystals were solubilized in a 1 N isopropanol/HCl 10% solution at 37°C, on a shaking table for 20 min. The absorbance values of the solution in each well were measured at 570 nm using a microplate reader. Cell viability was determined by the formula: cell viability (%): absorbance of the treated wells -absorbance of the blank control wells / absorbance of the negative control wells - absorbance of the blank control wells × 100%. All MTT experiments were performed in triplicate and repeated at least 3 times.
Drug combination studies
This study allowed the evaluation of the synergistic inhibition of cell growth produced by RAD/CLC combination in human renal adenocarcinoma cells. Cells were seeded in 96-well plates at the previously indicated density. After 24 h of incubation at 37°C, cells were treated at the drugs range of concentrations described above. To explore the relative contribution of each agent to the synergism equiactive doses of RAD/CLC were tested (IC50). Particularly we have tested equiactive molar ratios between 2 drugs in different sequences of administration (RAD for 48 h and CLC for 72 h (CLC→RAD) or RAD for 72 h and CLC for 48 h (RAD→CLC) and in co-administration for 72 h. The proliferative response was estimated by MTT test.
Drug combination studies were based on concentration–effect curves generated as a plot of the fraction of unaffected (surviving) cells versus drug concentration after 72 h of treatment. Assessment of synergy was performed quantitating drug interaction by CalcuSyn computer program (Biosoft, Ferguson, MO, USA). Combination index (CI) values of <1, 1, and >1 indicate synergy, additivity and antagonism, respectively. Dose reduction index (DRI) representing the measure of how much the dose of each drug in a combination may be reduced at a given effect level compared with the doses of each drug alone.
Apoptosis assay
Annexin V-FITC (fluorescein isothiocyanate) was used in conjunction with a vital dye, Propidium Iodide (PI), to distinguish apoptotic from necrotic cells. Briefly, the renal cancer cells were seeded in 6-multiwell plates at the density of 5 × 106 cells/ml medium and then incubated 24 h in humidified atmosphere at 37°C. The day after cells were treated with either the single agents or the synergistic sequence. After the treatment the cells were collected and centrifuged for 5 min at 1,300 rpm. Pellet was washed in PBS and incubated with Annexin-V-FITC and PtdIns in a binding buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2.5 mM CaCl2) for 10 min at room temperature, washed and resuspended in the same buffer as described by the manufacturer. Analysis of apoptotic cells was performed by flow cytometry (FACScan, Becton Dickinson). For each sample, 1 × 104 events were acquired. Analysis was carried out by triplicate determination on at least 3 separate experiments.
Autophagy assay
To quantify the induction of the autophagic process in renal cancer cells treated with drugs (single agents or synergistic sequence), the staining with autofluorescent agent monodansyl-cadaverine (MDC), a selective marker for autophagic vacuoles (AVOS) and especially autolysosomes, was performed. Briefly, cells were incubated with 50 μM MDC in PBS at 37°C for 15 min. washed with PBS, trypsinized, and immediately analyzed by flow cytometry. All fluorescences were analyzed with a FACScalibur flow cytometer (Becton Dickinson). The fluorescent emissions were collected through a 530 nm band pass filter (FL-1 channel). At least 10,000 events were acquired in log mode. For the quantitative evaluation of MDC, CellQuest software (Becton Dickinson) was used to calculate mean fluorescence intensities (MFIs) by the formula (MFI-treated/MFI-control), where MFI-treated is the fluorescence intensity of cells treated with the various compounds and MFI-control is the fluorescence reported in the figures are the means ± SD from 3 independent experiments.
Western blot analysis
Renal cancer cells were grown for 72 h with or without RAD and/or CLC in the previously described experimental conditions. For cell extract preparation, cells were washed twice with ice-cold PBS/BSA, scraped, and lysed for 30 min at 4°C in lysis buffer (50 mM Tris, pH 7.4, 150 mM sodium chloride, 1% Nonidet P-40, 1 mM EDTA, 1 mM sodium orthovanate, 1 mM sodium fluoride, 1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 μg/ml pepstatin A, 1 mM phenylmethylsulfonyl fluoride (PMSF)). Lysates were spinned at 10000 × g for 10 min and supernatants were collected. Protein concentration was determined by Lowry method and compared with bovine serum albumin standard curve. Approximately, 80 μg of protein extract were separated by 12.5% SDS-PAGE as described. The gel was transblotted by Trans blot turbo (BIORAD) on a nitro-cellulose membrane and reacted with the different antibody. After incubation with secondary antibodies, the signal was detected using enhanced chemoluminescence detection reagents (Super Signal West Pico, Pierce) and exposed to X-ray film. The bands derived from western blotting were scanned with a laser scanner (Epson 1260) and their intensities were quantified with Image J software (NIH). Membranes were normalized with a polyclonal antibody against α-tubulin. The error bars shown in the histograms represent the standard deviation from the mean of different densitometric scanning in at least 3 different experiments.
Immunoprecipitation
Protein lysates (120 μg) were incubated with primary antibody (anti-Beclin1 and anti-Bcl2) overnight at 4°C, with gentle rocking. Immune complexes were collected with 20 μl of protein A-agarose and incubated for 16 h at 4°C. The protein A-agarose/immune complex was washed twice with cold PBS, resuspended in 20 μl of SDS-loading buffer, heated to 95°C for 5 min and used for protein gel blotting analysis using either anti-Bcl2 or anti-Beclin 1 antibodies.
Statistical analysis
All data are expressed as mean ± DS. Statistical analysis was performed by ANOVA with Neumann–Keul's multiple comparison test or Kolmogorov–Smirnov where appropriate.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
MC was supported by the Italian Ministry of Education, University and Research (MIUR) with a project (FIRB-ACCORDI DI PROGRAMMA 2011) entitled: “Application of high-throughput technology platforms for the characterization of new biomarkers and molecular targets in nanovectors for the diagnosis and treatment of human cancer.” MC was supported by Regione Campania with a project entitled “Laboratori Pubblici Progetto Hauteville."
References
- Vogelzang NJ, Stadler WM. Kidney cancer. Lancet 1998; 352 (9141): 1691-6; PMID:9853456; http://dx.doi.org/10.1016/S0140-6736(98)01041-1
- McLaughlin JK, Lipworth L. Epidemiologic aspects of renal cell cancer. Semin Oncol 2000; 27 (2): 115-23; PMID:10768591
- Lindblad P. Epidemiology of renal cell carcinoma. Scand J Surg 2004; 93 (2): 88-96; PMID:15285559
- Pischon T, Lahmann PH, Boeing H, Tjønneland A, Halkjaer J, Overvad K, Klipstein-Grobusch K, Linseisen J, Becker N, Trichopoulou A, et al. Body size and risk of renal cell carcinoma in the European Prospective Investigation into Cancer and Nutrition (EPIC). Int J Cancer 2006; 118 (3): 728-38; PMID:16094628; http://dx.doi.org/10.1002/ijc.21398
- Linehan WM, Bates SE, Yanhg JC. Cancers of the genitourinary system. In Cancer Principles & Practice of Oncology 7th Edition. De Vita TV, Hellman S, Rosenberg AS Eds. 2005, Philadelphia: Lippincott & Williams,Chapter 30, 113-40.
- Coppin C, Porzsolt F, Awa A, Kumpf J, Coldman A, Wilt T. Immunotherapy for advanced renal cell cancer. Cochrane Database Syst Rev 2005; 1:CD001425; PMID:15674877
- Chown WH, Devesa SS, Warren JL, Fraumeni JF Jr. Rising incidence of renal cell cancer in the United States. JAMA 1999; 281:1628–31; PMID:10235157; http://dx.doi.org/10.1001/jama.281.17.1628
- Iacovelli R, Verzoni E, De Braud F, Procopio G. First line treatment of metastatic renal cell carcinoma: two standards with different toxicity profile. Cancer Biol Ther 2014; 15(1): 19-21; PMID:24253418; http://dx.doi.org/10.4161/cbt.27150
- Hamed HA, Das SK, Sokhi UK, Park MA, Cruickshanks N, Archer K, Ogretmen B, Grant S, Sarkar D, Fisher PB, et al. Combining histone deacetylase inhibitors with MDA-7/IL-24 enhances killing of renal carcinoma cells. Cancer Biol Ther 2013; 14(11): 1039-49; PMID:24025359; http://dx.doi.org/10.4161/cbt.26110
- Agarwala S S, Case S. Everolimus (RAD001) in the treatment of advanced renal cell carcinoma: a review. Oncologist 2010; 15:236-45; PMID:20215359; http://dx.doi.org/10.1634/theoncologist.2009-0141
- Yim KL. Everolimus and mTOR inhibition in pancreatic neuroendocrine tumors. Cancer Manage Res 2012; 4:207-14; PMID:22904642; http://dx.doi.org/10.2147/CMAR.S25979
- Baselga J, Campone M, Piccart M, Burris HA 3rd, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. New Engl J Med 2012; 366:520-9; PMID:22149876; http://dx.doi.org/10.1056/NEJMoa1109653
- Curran MP. Everolimus: in patients with subependymal giant cell astrocytoma associated with tuberous sclerosis complex. Pediat Drugs 2012; 14:51-60; PMID:22136276; http://dx.doi.org/10.2165/11207730-000000000-00000
- Mirzoeva OK, Hann B, Hom YK, Debnath J, Aftab D, Shokat K, Korn WM. Autophagy suppression promotes apoptotic cell death in response to inhibition of the PI3KmTOR pathway in pancreatic adenocarcinoma. J Mol Med 2011; 89:877-89; PMID:21678117; http://dx.doi.org/10.1007/s00109-011-0774-y
- Keith CT, Schreiber SL. PIK-related kinases: DNA repair, recombination, and cell cycle checkpoints. Science 1995; 270:50-1; PMID:7569949; http://dx.doi.org/10.1126/science.270.5233.50
- Schmelzle T, Hall MN. mTOR, a central controller of cell growth. Cell 2000; 103:253-62; PMID:11057898; http://dx.doi.org/10.1016/S0092-8674(00)00117-3
- Bjornsti MA, Houghton PJ. The mTOR pathway: a target for cancer therapy. Nat Rev Cancer 2004; 4:335-48; PMID:15122205; http://dx.doi.org/10.1038/nrc1362
- Chen W, Hu QD, Xia XF, Liang C, Liu H, Zhang Q, Ma T, Liang F, Liang TB.. Rapamycin enhances cetuximab cytotoxicity by inhibiting mTOR-mediated drug resistance in mesenchymal hepatoma cells. Cancer Biol Ther 2014; 15(8): 992-9; PMID:24800850; http://dx.doi.org/10.4161/cbt.29113
- Rossia M, Di Martino MT, Morellia E, Leotta M, Rizzo A, Grimaldi A, Misso G, Tassone P, Caraglia M. Molecular targets for the treatment of multiple myeloma. Curr Cancer Drug Targets 2012; 12:757-67; PMID:22671925; http://dx.doi.org/10.2174/156800912802429300
- Okabe S, Tauchi T, Tanaka Y, Kitahara T, Kimura S, Maekawa T, Ohyashiki K. Efficacy of the dual PI3K and mTOR inhibitor NVP-BEZ235 in combination with nilotinib against BCR-ABL-positive leukemia cells involves the ABL kinase domain mutation. Cancer Biol Ther 2014; 15(2): 207-15; PMID:24100660; http://dx.doi.org/10.4161/cbt.26725
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010; 22:124-31; PMID:20034776; http://dx.doi.org/10.1016/j.ceb.2009.11.014
- Degtyarev M, De Mazière A, Orr C, Lin J, Lee BB, Tien JY, Prior WW, van Dijk S, Wu H, Gray DC, et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J Cell Biol 2008; 183:101-16; PMID:18838554; http://dx.doi.org/10.1083/jcb.200801099
- Fan QW, Cheng C, Hackett C, Feldman M, ,Houseman BT, Nicolaides T, Haas-Kogan D, James CD, Oakes SA, Debnath J, et al. Akt and autophagy cooperate to promote survival of drug resistant glioma. Sci Signal 2010; 3 (147): ra81; PMID:21062993; http://dx.doi.org/10.1126/scisignal.2001017
- Xu CX, Zhao L, Yue P, Fang G, Tao H, Owonikoko TK, Ramalingam SS, Khuri FR, Sun SY. Augmentation of NVP-BEZ235's anticancer activity against human lung cancer cells by blockage of autophagy. Cancer Biol Ther 2011; 12:549-55; PMID:21738008; http://dx.doi.org/10.4161/cbt.12.6.16397
- Bergamini E, Cavallini G, Donati A, Gori Z. The anti-ageing effects of caloric restriction may involve stimulation of macroautophagy and lysosomal degradation, and can be intensified pharmacologically. Biomed Pharmacother 2003; 57:203-8; PMID:12888255; http://dx.doi.org/10.1016/S0753-3322(03)00048-9
- Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cells. Int J Biochem Cell Biol 2004; 36:2445-62; PMID:15325584; http://dx.doi.org/10.1016/j.biocel.2004.02.002
- Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol 2004; 36:2491-502; PMID:15325587; http://dx.doi.org/10.1016/j.biocel.2004.02.005
- Solomon VR, Lee H. Chloroquine and its analogs: a new promise of an old drug for effective and safe cancer therapies. Eur J Pharmaco 2009; 625:220-33; PMID:19836374; http://dx.doi.org/10.1016/j.ejphar.2009.06.063
- Sasaki K, Tsuno NH, Sunami E, Tsurita G, Kawai K, Okaji Y, Nishikawa T, Shuno Y, Hongo K, Hiyoshi M, et al. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer 2010; 10:370; PMID:20630104; http://dx.doi.org/10.1186/1471-2407-10-370
- Pattingre S, Levine B. Bcl-2 inhibition of autophagy: a new route to cancer? Cancer Res 2006; 66:2885-8; PMID:16540632; http://dx.doi.org/10.1158/0008-5472.CAN-05-4412
- Smolewski P. Recent developments in targeting the mammalian target of rapamycin (mTOR) kinase pathway. Anticancer Drugs 2006; 17:487-94; PMID:16702804; http://dx.doi.org/10.1097/00001813-200606000-00001
- Abdel-Aziz AK, Shouman S, El-Demerdash E, Elgendy M, Abdel-Naim AB. Chloroquine synergizes sunitinib cytotoxicity via modulating autophagic, apoptotic and angiogenic machineries. Chem Biol Interact 2014; 217:28-40; PMID:24751611; http://dx.doi.org/10.1016/j.cbi.2014.04.007
- Faggiano A, Ramundo V, Dicitore A, Castiglioni S, Borghi MO, Severino R, Ferolla P, Crinò L, Abbruzzese A, Sperlongano P, et al. Everolimus is an active agent in medullary thyroid cancer: a clinical and in vitro study. J Cell Mol Med 2012 Jul; 16(7):1563-72; PMID:21883896; http://dx.doi.org/10.1111/j.1582-4934.2011.01438.x
- Rossi M, Munarriz ER, Bartesaghi S, Milanese M, Dinsdale D, Guerra-Martin MA, Bampton ET, Glynn P, Bonanno G, Knight RA, et al. Desmethylclomipramine induces the accumulation of autophagy markers by blocking autophagic flux. J Cell Sci 2009; 122:3330-9; PMID:19706685; http://dx.doi.org/10.1242/jcs.048181
- Kimura T, Takabatake Y, Takahashi A, Isaka Y. Chloroquine in cancer therapy: a double-edged sword of autophagy. Cancer Res 2013; 73(1): 3-7; PMID:23288916; http://dx.doi.org/10.1158/0008-5472.CAN-12-2464