
Abstract
RAD51-mediated recombinational repair is elevated in multiple myeloma (MM) and predicts poor prognosis. RAD51 has been targeted to selectively sensitize and/or kill tumor cells. Here, we employed a peptide nucleic acid (PNA) to inhibit RAD51 expression in MM cells. We constructed a PNA complementary to a unique segment of the RAD51 gene promoter, spanning the transcription start site, and conjugated it to a nuclear localization signal (PKKKRKV) to enhance cellular uptake and nuclear delivery without transfection reagents. This synthetic construct, (PNArad51_nls), significantly reduced RAD51 transcripts in MM cells, and markedly reduced the number and intensity of de novo and melphalan-induced nuclear RAD51 foci, while increasing the level of melphalan-induced γH2AX foci. Melphalan alone markedly induced the expression of 5 other genes involved in homologous-recombination repair, yet suppression of RAD51 by PNArad51_nls was sufficient to synergize with melphalan, producing significant synthetic lethality of MM cells in vitro. In a SCID-rab mouse model mimicking the MM bone marrow microenvironment, treatment with PNArad51_nls ± melphalan significantly suppressed tumor growth after 2 weeks, whereas melphalan plus control PNArad4µ_nls was ineffectual. This study highlights the importance of RAD51 in myeloma growth and is the first to demonstrate that anti-RAD51 PNA can potentiate conventional MM chemotherapy.
Abbreviations
| PNA | = | peptide nucleic acid |
Introduction
Genomic instability is a key feature of multiple myeloma (MM) cells.Citation1 We previously reported high-level expression of RAD51 and/or its paralogs and elevated homologous recombination (HR) rates in MM cell lines and primary bone marrow aspirates from MM patients, relative to normal plasma cells. These increased HR events contribute to karyotypic instability, disease progression and the development of chemotolerance.Citation2,3 RAD51 appears to be the key protein that drives HR, a mechanism that repairs DNA interstrand cross-links (ICLs), stalled/damaged replication forks and double strand breaks (DSBs), with high fidelity.Citation4 Thus, enhanced DNA repair resulting from RAD51 overexpression may protect cells from radiation- or chemotherapy-induced DNA damage.Citation5,6 High-level RAD51 expression has been shown to predict poor event-free and overall survival of MM patients,Citation7 and inhibition of HR by a RAD51 small-molecule inhibitor sensitizes MM cells to doxorubicin relative to normal human B cells.Citation3
Melphalan, an alkylating agent widely used in MM chemotherapy, produces bulky DNA adducts as well as ICLs, considered to be the most critical cytotoxic lesion.Citation8 During replication of genomic DNA, replication forks stall/collapse at these adducts and ICLs, resulting in the formation of DSBs that are lethal unless repaired—primarily via RAD51-mediated HR.Citation9 The ability of cells to repair ICLs is thus a critical determinant of response to melphalan, and this has been implicated in both inherent sensitivity and acquired resistance to the drug.Citation10,11 MM cells display elevated activity of the Fanconi Anemia (FA) pathway, which contributes to melphalan resistanceCitation11 by promoting RAD51-mediated HR repair of ICLs.Citation9,12 Moreover, melphalan treatment of MM cells induces HR repair,Citation13 which appears to be their principal line of defense since drug resistance correlates linearly with the levels of drug-induced nuclear RAD51 foci.Citation14
RAD51 is generally overexpressed in immortalized cellsCitation15 and in the majority of cancer cells, and predicts poor prognosis for diverse tumors.Citation16,17 Considerable effort has therefore been directed toward discovery and development of small-molecule inhibitors of RAD51.Citation18,19 However, such compounds are often associated with off-target toxicity. In contrast, nucleic acid-based inhibitors of transcription and/or translation are synthetic macromolecules designed to be specific to one target sequence, and thus avoid off-target toxicity. Such inhibitors have great potential for target validation and even as therapeutic agents. Previous studies have targeted RAD51 for cancer therapy using antisense oligonucleotides including siRNA or shRNACitation17,20,21 or mRNA-targeted ribozymes.Citation22 However, no previous study has used peptide nucleic acid (PNA), a DNA/RNA mimic in which the nucleic acid sugar phosphate backbone has been replaced with a synthetic polymer of 2-aminoethyl-glycine units,Citation23 to target RAD51 for cancer therapy. PNA binds to DNA or RNA via conventional Watson-Crick base pairing.Citation24 Relative to other types of oligonucleotides or oligonucleotide mimics, PNAs are remarkably stable to nucleases and proteasesCitation25 and their neutrality reduces electrostatic repulsion from nucleic acids, conferring exceptional specificity and stability of binding to DNA or RNA.Citation26 When bound to the non-template DNA strand, PNA can interfere with transcription via a steric-blockage mechanism.Citation27
The high stability of PNA:DNA hybrids enables PNAs to invade double-stranded DNA, binding to a complementary sequence while displacing the homologous strand.Citation23 For all oligonucleotides including PNAs, binding to chromosomal DNA is greatly enhanced if the target is single-stranded.Citation28 During transcription by polymerase II, an open complex containing single-stranded DNA forms at the transcription start site, predicted to extend from nucleotides −9 to +2 (29). In cancers, RAD51 is upregulated chiefly at the transcriptional level; using a transgenic reporter in which the RAD51 promoter drives luciferase, this construct was found to be 850-fold more active in cancer cells than in normal cells.Citation30 This implies that RAD51 is highly transcribed in cancer cells, and its transcription start site is likely to have an open configuration. Our strategy to downregulate RAD51 expression was to target the promoter's antisense strand with a PNA covering its transcription start site, conjugated to the nuclear localization signal (NLS) of SV40 large-T antigen. Basic leader peptides enable PNAs to efficiently enter cells without transfection reagents; the SV40 large-T NLS (PKKKRKVR) contains 6 basic residues and also directs the PNA to nuclei.Citation31,32 We then tested whether this construct, alone or combined with melphalan, inhibits survival and growth of myeloma cells.
Results
Melphalan treatment induces expression of RAD51, its paralogs and BRCA1, and arrests Multiple Myeloma cells in S phase
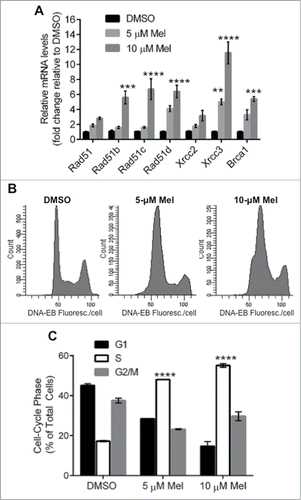
We previously showed that HR rates, and mRNA expression of RAD51 and/or its paralogs (RAD51B, RAD51C, RAD51D, XRCC2 and XRCC3), are elevated in MM cells.Citation2 Other studies showed that melphalan treatment increases HR in MM cells.Citation13 The FA pathway is highly expressed and mediates melphalan resistance in MM cells,Citation11 interacting directly with BRCA1 and RAD51 to promote HR repair of ICLsCitation9,12,37 during the S phase of the cell cycle.Citation37 We therefore wanted to know the effect of melphalan on the expression of RAD51, its paralogs and BRCA1, and whether the cell-cycle distribution was altered. Toward this end, we determined the mRNA levels of these genes in MM cells after 24 h exposure to increasing doses of melphalan (0, 5 or 10 µM). A portion of each cell culture was stained with propidium iodide (PI) for cell-cycle analysis. As shown in , melphalan induced a dose-dependent increase in the expression of all HR-associated genes analyzed, which was significant at the higher dose for all genes except RAD51 and XRCC2. RAD51 appeared to be induced roughly 2- and 3-fold by 5 and 10 µM melphalan, respectively. Five genes (RAD51B, C and D, XRCC3 and BRCA1) were at least 5-fold induced at the higher melphalan dose. The most upregulated gene (˜5- and 12-fold by 5 and 10 µM melphalan, respectively) was XRCC3, shown previously to support melphalan resistance by inducing RAD51 recombinational repair and S-phase checkpoint activation, while reducing apoptosis.Citation38 XRCC3 overexpression doubles the incidence of melphalan-induced RAD51 foci.Citation38 Perhaps as a byproduct of XRCC3 induction, melphalan treatment evoked a significant dose-dependent accumulation of MM cells in S phase (). These findings, in addition to previous observations that melphalan treatment triggers HR repair,Citation13 strongly implicate RAD51-mediated HR repair in the MM cell response to melphalan.
Figure 1. Effects of melphalan on MM cells in vitro. (A) Real-time quantitative RT-PCR analysis of RAD51, its paralogs and BRCA1 transcripts in H929 cells, was performed 24 h after indicated treatments. GAPDH was used as the internal control and the mRNA levels normalized to control (DMSO) samples. (B) Flow cytometric analysis of propidium iodide stained cells treated as indicated in (A) was performed. **, *** and ****P < 0.01, 0.001 and 0.0001, respectively, relative to DMSO treated group.
PNA targeting RAD51 reduces its transcript levels
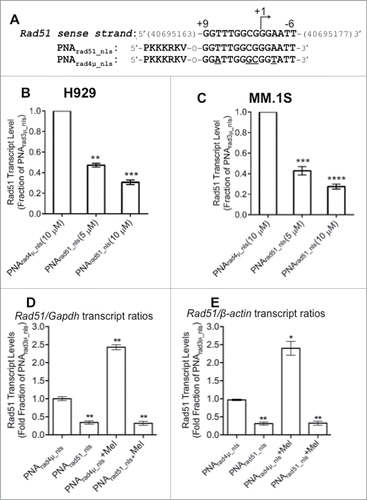
To test whether a PNA targeting RAD51 can effectively downregulate its expression, RAD51 transcript levels were monitored by RT-qPCR in cells that had been treated with active or control PNAs. We designed a PNA complementary to the template DNA strand of the RAD51 gene, encompassing its transcription start site, and compared the effect of this construct, PNArad51_nls, to that of a quadruple-mismatch control PNA with the same base composition, PNArad4µ_nls () in 2 established MM cell lines. We infer that PNA exposure is not toxic to these cells, since results for the control PNA were identical to those for untreated cells. Relative to the mismatched control PNA, cells exposed for 48 h to PNArad51_nls showed dose-dependent declines in RAD51 mRNA levels, increasing from ≤40% suppression at 0.2–1.3 µM PNArad51_nls (data not shown), to 53–58% at 5 µM and 70–73% at 10 µM PNArad51_nls (). In contrast, PNArad4µ_nls had no significant effect on RAD51 transcript levels relative to vehicle alone (not shown). We next performed RT-qPCR on cells treated with PNA in the absence or presence of melphalan. Results are expressed relative to 2 control genes (GAPDH and β-ACTIN); their similarity implies that the effects of PNA on RAD51 transcription are not specific to either control, nor can they be explained as global effects on gene transcript levels. We exposed H929 cells to PNA for 24 h, and then added melphalan (10 µM) or vehicle for an additional 24h; total RNA was then extracted for RT-qPCR. As shown in , pre-treatment with 10 µM PNArad51_nls reduced RAD51 mRNA levels by 70–90% in cells ±10-µM melphalan, relative to either GAPDH or β-ACTIN as the reference control gene.
Figure 2. RAD51 antigene PNA downregulates RAD51 mRNA expression in vitro. (A) PNA targeting RAD51 transcription. The top line shows the sense strand of the RAD51 gene, which is homologous to the active PNA; “+1” indicates the transcriptional start site. Thus, PNArad51_nls (second line) is complementary to the antisense strand of the RAD51 promoter, spanning bases −9 to +6. PNArad4μ_nls (third line) is a quadruple-mismatch control PNA with the same base composition as PNArad51_nls. (B) H929 and (C) MM.1S cells were treated with 10-μM PNArad4μ_nls or 5-μ and 10-μM PNArad51_nls for 48 h prior to harvest. RAD51 gene expression was determined by RT-qPCR, for 6 biological replicates per group. Results depict relative change normalized to control-PNA-treated samples, after standardization against GAPDH transcripts as internal control. **, ***, **** indicate P < 0.01, 0.001 or 0.0001, respectively, relative to control. (D, E) H929 cells were exposed to 10-μM PNArad4μ_nls or 10-μM PNArad51_nls for 24 h; then vehicle (H2O) or 10 μM Melphalan (Mel) was added for a further 24 h. Cells were harvested and RAD51 gene expression was determined as in (B) and (C) using either GAPDH (D) or β-actin (E) as the internal control (reference point). ** indicates P < 0.01, relative to control.
RAD51 antigene PNA inhibits de novo and melphalan-induced RAD51 foci and increases the extent of melphalan-induced DNA damage
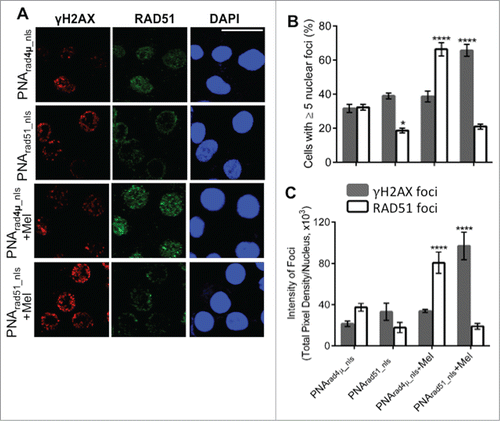
In response to DSBs, histone H2AX becomes phosphorylated on serine 139 (forming γH2AX) to initiate DNA-damage repair. Consequently, γH2AX is often used as a marker of DSBsCitation39 while nuclear RAD51 foci represent nucleoprotein filaments engaged in HR repair.Citation40 MM cells exhibit γH2AX foci constitutively,Citation41 and overexpression of RAD51 in cancer cells can produce nuclear RAD51 foci even in the absence of exogenous DNA damage.Citation42 We assessed γH2AX and RAD51 foci by immunostaining and confocal microscopy, to determine the effects of RAD51 antigene PNA, melphalan treatment, and both combined. As shown in , about 32% of nuclei had ≥5 RAD51 foci after exposure to control PNArad4µ_nls without melphalan. This fraction fell to ∼19% (p<0.05) for cells exposed only to PNArad51_nls, whereas γH2AX foci increased slightly and insignificantly (). Melphalan exposure nearly doubled the fraction of cells with ≥5 RAD51 foci in the absence of RAD51 knockdown, with little perceptible induction of γH2AX foci. PNArad51_nls had a profound effect on melphalan-treated cells, reducing the fraction with RAD51 foci by >3-fold while boosting the fraction of γH2AX-positive cells by ∼70% (each p<0.0001), relative to PNArad4µ_nls control (). The same trends were apparent when comparing the total pixel intensity of either RAD51 or γH2AX foci, integrated density per nucleus (). Thus the effects of PNArad51_nls on RAD51 focus formation were consistent with the qPCR data, where RAD51 is silenced both in the presence and absence of melphalan. The implications are as follows: in the absence of exogenously induced DNA damage, a PNA targeting RAD51 reduces the number and intensity of RAD51 foci but has little effect on γH2AX foci mediating their repair. However, when cells are challenged with melphalan, the same suppression of RAD51 foci has more serious consequences in that the level of unrepaired DNA damage (as indicated by γH2AX foci) rises substantially.
Figure 3. PNA targeting RAD51 reduces de novo nuclear RAD51 foci and those induced by melphalan, and potentiates melphalan-induced DNA damage. (A, B) H929 cells were treated as indicated in “Materials and Methods.” Cytospins were made and stained for RAD51 and γH2AX. (A) Immunofluorescence pictures of cells after staining for RAD51, γH2AX and DAPI. (B) Quantitation of cells with ≥5 discrete nuclear foci of γH2AX or RAD51. At least 100 nuclei were analyzed for each treatment and nuclei with ≥5 foci were scored as positive. (C) Mean fluorescence (integrated pixel intensity per nucleus) ±SEM, of γH2AX and RAD51 foci after the indicated drug exposures. Data are shown as mean ± SEM of 3 experiments. * P < 0.05 ****P< 0.0001, for comparisons to controls. Prad4μ_nls, Prad51_nls and Mel indicate PNArad4μ_nls, PNArad51_nls and Melphalan, respectively. The scale bar (—) is 20 μm.
RAD51 antigene PNA potentiates the sensitivity to melphalan of MM cells in vitro
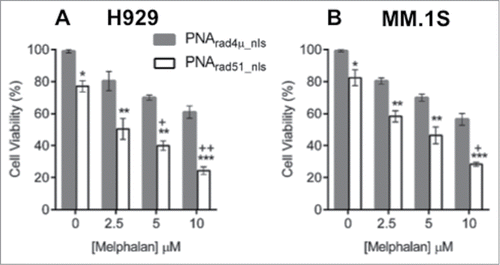
A single DSB, if unrepaired, is a lethal event for a cell.Citation43 Having observed that PNArad51_nls potentiates melphalan-induced DNA damage, we next asked whether this translates into enhanced cytotoxicity upon melphalan treatment. We assessed cell proliferation for 2 human MM cell lines, H929 and MM.1S. Treatment with PNArad51_nls in the absence of melphalan reduced the survival of H929 and MM.1S cell lines, by ∼23% and ∼18% respectively, relative to PNArad4µ_nls treatment (each P < 0.05, zero-dose points in ). MM cells were then exposed 24 h to PNArad51_nls or PNArad4µ_nls, followed by 48-h treatment with melphalan (at 2.5–10 µM) with the PNAs still present. Relative to PNArad4µ_nls control treatment, PNArad51_nls significantly potentiated melphalan cytotoxicity for both cell lines, at all doses tested (each p ≤ 0.01, ). In the presence of PNArad51_nls, the IC50 for melphalan was 2.7 µM for H929 and 3.8 µM for MM.1S. In contrast, melphalan in the presence of PNArad4µ_nls did not reach 50% toxicity for the dose range tested (i.e. each IC50 was >10 µM). We evaluated whether there is significant synergy or synthetic lethality between PNArad51_nls and melphalan treatments by comparing the observed responses for the PNArad51_nls plus melphalan combinations with the product of their individual effects (i.e., the predicted responses). For three of the 6 combinations, myeloma cell viability was substantially less than the survival predicted if they acted independently (i.e., the product of their individual survival fractions), with nominal significances ranging from P < 0.05 to <0.01 (“+” symbols in ).
Figure 4. PNA targeting RAD51 inhibits myeloma cell growth and sensitizes MM cells to melphalan in vitro. (A, B) Cell viability of H929 (A) and MM.1S (B) cell lines was measured by WST-1 assay after cells were treated as described in “Materials and Methods.” The data represent mean ± SEM of triplicate samples in each of 3 independent experiments, each considered as a single point (i.e. considering only the biological N of 3). Viability of PNArad4μ_nls was set to 100%, and other treatments expressed relative to that value. *, ** and *** indicate P < 0.05, 0.01 and 0.001, respectively, for the indicated PNArad51_nls bar relative to the corresponding PNArad4μ_nls control, at each melphalan exposure (0, 2.5, 5 or 10 µM) indicated on the x-axis. Significance of synergy was calculated by a 1-tailed, heteroscedastic t test comparing the normalized cell viability obtained with PNArad51_nls plus melphalan (a%) with the product of the survival fractions for each agent administered separately (i.e., comparing a% to d% = b% × c%). + and ++ indicate P < 0.05 and P < 0.01, respectively, for significance of synergy between the observed viability inhibition with the PNArad51_nls plus melphalan combination, relative to the loss of viability expected from the product of their individual survival fractions.
PNA targeting RAD51 inhibits myeloma tumor growth in vivo
We next examined whether RAD51 antigene PNA would inhibit MM tumor growth and/or enhance the anti-myeloma effect of melphalan in our SCID-rab model, designed to mimic the normal bone-marrow microenvironment in which myelomas arise.Citation34 We injected H929-Luc cells directly into the marrow of implanted rabbit bones in mice and tumor growth was monitored via bioluminescence imaging of the intensity of constitutive luciferase expression. We monitored the change in tumor growth 2 and 4 weeks after a single injection of 100 µl of 100 µM PNA (either PNArad4µ_nls or PNArad51_nls) directly into the implanted bones, with or without twice-weekly intraperitoneal injections of 2.0 mg/kg melphalan. Treatment with PNArad51_nls, either alone or in combination with melphalan, reduced total tumor bioluminescence at 2 weeks to roughly half of that seen after PNArad4µ_nls treatment (p = 0.01 and <0.05, respectively; ). Treatment with PNArad4µ_nls + melphalan also inhibited tumor growth, although not significantly (p = 0.07; ). After 4 weeks, however, tumor inhibition by anti-RAD51 PNA ± melphalan was less pronounced and no longer statistically significant. Based on the doses used and without regard to the route of administration, these in vivo data indicate that PNArad51_nls has anti-myeloma activity comparable to melphalan, and suggest a slight further improvement when they are combined.
Figure 5. PNA targeting RAD51 in vivo inhibits MM tumor growth, sensitizes MM cells to melphalan, and inhibits expression of RAD51, its paralogs and BRCA1. The effects of RAD51 antigene PNA ± melphalan on MM tumor growth in vivo. (A, B) Luciferase-expressing H929 myeloma cells were engrafted into rabbit bone implanted in SCID mice, and bioluminescence intensity (RLU) of luciferase expression was determined after 2 and 4 weeks. (A) Representative live-animal bioluminescence images before the start of treatment (week 0), and after 2 or 4 weeks of treatment. (B) Changes in tumor bioluminescence (log10 of fold change) after 2 or 4 weeks of treatment. (C) RT-qPCR analysis of transcripts of RAD51, its paralogs and BRCA1, in cells recovered from myeloma-seeded bone implants, was performed as described in “Materials and Methods.” GAPDH transcripts were used as an internal control to which other mRNA levels were normalized and expressed as a ratio to control (PNArad4μ_nls) samples. Data represent the mean ± SEM of at least 5 samples (from 5 mice) per treatment group. Nominal significance levels, indicated in the figure, have not been corrected for multiple comparisons. Based on a conservative Bonferroni correction, P < 0.05/7 = 0.007 (true for **, ***, **** and *****) would ensure a total false-positive rate α < 0.05.
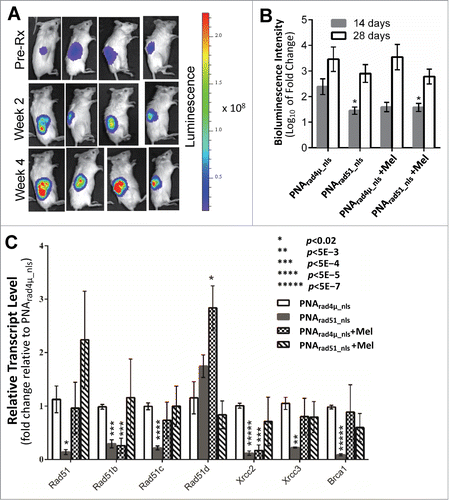
Melphalan is a widely used and effective agent in MM chemotherapy, but did not show potent activity in the SCID-rab model. We performed RT-qPCR on total RNA extracted from bone marrow cells recovered from mice (≥5 mice per group) implanted with myeloma cells seeded inside rabbit bone segments. In these cells, expression of RAD51, its paralogs and BRCA1, were all induced 1.6- to >11-fold by melphalan treatment in vitro (). In vivo, however, only RAD51D transcripts were significantly induced by melphalan in the presence of control PNA (). Due to large variance, melphalan in the presence of anti-RAD51 PNA did not cause a significant alteration in the expression of any gene; transcripts for RAD51, RAD51B, and RAD51C increased slightly, whereas those for RAD51D, XRCC2, XRCC3 and BRCA1 decreased marginally. Treatment with PNArad51_nls alone consistently lowered the expression of RAD51, all of its paralogs (except RAD51D), and BRCA1, relative to PNArad4µ_nls treatment (; nominal significances of P < 0.02 to P < 5E-7).
Discussion
Melphalan is one of the key drugs used in MM chemotherapy. However, dose-dependent adverse effects limit its efficacy, so that at tolerated doses the eradication of tumor cells is typically incomplete, myeloma recurs, and drug resistance may be acquired. These shortcomings could be eliminated if tumor cells can be sensitized to the drug. Enhanced ICL repair in myeloma cells has been suggested as a route to melphalan resistance,Citation11,14 which can be mediated by repair proteins in the FA pathwayCitation11,44 and by RAD51-mediated HR repair.Citation10,14 We previously reported high-level expression of genes encoding RAD51 and its paralogs (RAD51B, RAD51C, RAD51D, XRCC2 and XRCC3), accompanying elevated HR rates in MM cells.Citation2 The FA pathway acts upstream of RAD51 to promote its repair of ICLs.Citation9,12 RAD51 is overexpressed in the majority of cancer cellsCitation16 and predicts therapy resistance; it has therefore been designated as a cancer-therapeutic target (see review in refCitation45.). Oncogenic transformation induces RAD51 overexpressionCitation46 mainly at the transcriptional level; the RAD51 promoter is hyperactive in cancer cells compared with normal cells, often strikingly so (e.g. >800-fold), and RAD51 promoter-driven toxin constructs selectively kill cancer cells both in vitro and in vivo.Citation30,47 Thus the RAD51 promoter represents an appropriate target for silencing RAD51 in cancer cells.
In the present study, we employed a PNA targeting the RAD51 gene's transcription start site, and conjugated an NLS to its N-terminus to facilitate cellular and nuclear delivery without transfection reagents. PNArad51_nls significantly suppressed RAD51 expression, and reduced both de novo and melphalan-induced nuclear RAD51 protein foci. The current study is the first to use PNA to inhibit RAD51, although previous studies have evaluated the role of RAD51 in cancer therapy by using either small-molecule drugs that have off-target toxicitiesCitation48 or siRNA/shRNA,Citation17,20,21 or mRNA-targeted ribozymesCitation22 that are less stable to biological degradation than PNAs,Citation25 and for which appropriate delivery may be problematic. Our results demonstrate an effective strategy to downregulate RAD51, which appears to have oncogenic roles in many cancer cells.Citation2,17
Melphalan treatment increased the in vitro expression of RAD51, its paralogs and BRCA1, and induced nuclear RAD51 protein foci. Also, the drug induced cell-cycle arrest during S phase, a key interval during which the FA pathway interacts with BRCA1 and RAD51 to activate RAD51-mediated repair of ICLs.Citation9,37 BRCA1 and paralogs of RAD51 play critical roles in the recruitment of RAD51 protein to damaged DNA sites. Previous studies have shown that XRCC3 activates melphalan-induced S phase checkpoint arrest, stimulates RAD51-mediated HR repair of melphalan-induced ICLs, and reduces apoptosis.Citation38 Sensitivity to melphalan correlates linearly with levels of XRCC3 and RAD51 foci.Citation14 In the present study, XRCC3 was the gene most highly induced by melphalan, followed by RAD51, B-D and BRCA1. Induction of these genes during chemotherapy, and the ensuing enhanced repair, can contribute to the acquisition by myeloma cells of clinical drug resistance, in addition to melphalan resistance arising by de novo mutations. Previous studies have shown that melphalan treatment increases the rate of functional HR repair in MM cells,Citation13 findings fully consistent with our current results.
PNA-mediated disruption of RAD51 expression significantly potentiated melphalan's ability to inhibit myeloma tumor growth in vitro. Thus, RAD51 knockdown is sufficient to inhibit ICL repair, as indicated by the reduced number and intensity of RAD51 foci and increased γH2AX foci with PNArad51_nls plus melphalan treatment (relative to PNArad4µ_nls ± melphalan). PNArad51_nls alone caused significant MM cell toxicity in the absence of melphalan. These results reflect the strong dependence of MM cells on RAD51 for HR repair of both routine and chemotherapy-induced DNA damage. Myeloma cells were shown to be impaired in nonhomologous end-joining (NHEJ), the other major pathway for repairing DSBs.Citation49 We recently reported that RAD51 disruption by a small-molecule inhibitor, or RAD51 knockdown via RNA interference, both led to marked inhibition of myeloma cell survival in the absence of DNA damage, and also sensitized tumor cells to agents that cause double-strand DNA breaks.Citation3 In a similar study, RAD51 was found to be overexpressed in triple-negative breast cancer cells and shRNA-mediated knockdown of RAD51 was shown to inhibit their growth and metastasis in vivo,Citation17 supporting a key role of RAD51 in tumor progression.
In order to recapitulate the bone marrow microenvironment where MM cells flourish, we employed our SCID-rab model to examine the in vivo effects of PNArad51_nls ± melphalan. Short-term treatment with PNArad51_nls alone caused significant tumor growth inhibition. As noted above, MM cells rely on HR for survival of double-strand DNA breaks, so that HR inhibition should reduce their survival of clastogenic (break-inducing) drugs. A similar finding was made in triple-negative breast cancer (TNBC), which also overexpresses RAD51: shRNA-mediated knockdown of RAD51 resulted in significant inhibition of tumor growth and metastasis in vivo.Citation17
The bone-marrow microenvironment plays an important role in proliferation, viability and drug resistance of MM cells, which may help to explain the reduced activity of conventional anti-MM drugs (e.g., alkylating agents) against myeloma cells co-cultured in vitro with bone marrow stromal cells, relative to their potency against MM cells cultured alone.Citation50,51 Such micro-environment effects, along with other host defenses that are only at play in vivo, may have contributed to the lower efficacy of melphalan in vivo, with respect to either inhibition of tumor growth or induction of RAD51-complex genes, than was evident against the same cells grown in vitro. Those differences may suggest insufficient ICL generation by melphalan, at the dose used in vivo, to seriously challenge myeloma cells in the absence of RAD51 knockdown. Under those conditions, normal myeloma-cell levels of the RAD51-HR complex may be sufficient to repair most DNA damage without further induction of the genes involved; PNA knockdown of RAD51, however, could exacerbate the response to melphalan so as to induce somewhat higher expression of HR-related genes, and may have moderately reduced tumor growth.
In summary, our study is the first to demonstrate that antigene PNA can be used to inhibit RAD51 transcription, reducing a pivotal HR protein that is overexpressed in most cancers and that predicts therapy resistance. We propose that RAD51-mediated repair represents a drug target that could improve cancer treatment in combination with DNA-damaging agents, serving to block tumor progression and the acquisition of drug resistance.
Materials and Methods
Cell culture and reagents
The human MM cell line NCI-H929 was provided by Dr. Shmuel Yaccoby (Myeloma Institute for Research and Therapy, University of Arkansas for Medical Sciences) and MM.1S cells were obtained from the ATCC (catalog # ATCC CRL-2974). The cells were maintained in RPMI 1640 medium with L-glutamine and NaHCO3 (ATCC, catalog # 30-2001) containing 10% FBS (ATCC, catalog # 30-2020), 100 U/mL of penicillin and 100 µg/mL of streptomycin (Sigma-Aldrich, cat # P4458). All cultures were maintained at 37°C, 5% CO2, and 70% relative humidity. Melphalan (Santa Cruz Biotechnology, catalog # sc-204799) was dissolved in DMSO, and diluted in cell culture medium for cell treatment.
Peptide nucleic acid (PNA)
We designed 2 types of PNA: one targeting the antisense strand of the RAD51 transcription start site, called PNArad51_nls; and as a control, a quadruple-mismatch sequence, PNArad4µ_nls, targeting no counterpart in the human genome. The nuclear localization signal of the SV40 large T antigen (PKKKRKV), which is rich in basic residues, was covalently conjugated via an O-linker at the N terminus of both PNAs. The PNAs (synthesized by PNA Bio Inc.., Thousand Oaks, CA, USA) have the following sequences:
PNArad51_nls: PKKKRKV-O-GGTTTGGCGGGAATT
PNArad4µ_nls: PKKKRKV-O-GGATTGGGCGGTATT
Cell-cycle analysis
After incubation under the indicated conditions for 24 h, cells were harvested and counted to ensure equal numbers of cells were processed (1 × 10Citation6) per sample. Cells were then washed with ice-cold PBS, transferred to 70% ethanol at −20°C, and stored for 2–12 h. Subsequently, cells were washed in ice-cold PBS and stained with propidium iodide in a solution containing RNase A (FxCycle PtdIns/RNase staining solution, Invitrogen Life Technologies, catalog # F10797) and analyzed for DNA content on a fluorescence activated cell sorter (LSRFortessa, BD Biosciences) and data processed with FACS-Diva software (BD Biosciences).
Real time quantitative polymerase chain reaction (RT-qPCR)
For in vitro assays, total RNA was extracted from cells after the indicated treatments using RNAeasy Mini Kit (Qiagen, catalog # 74104). For in vivo analysis, PureLink® RNA Mini Kits (Ambion, catalog # 12183020) were used to extract total RNA from recovered samples. Total RNA (1µg) was used to reverse transcribe cDNA using SuperScript First Strand cDNA synthesis kit (Invitrogen, catalog # 18080-051). The cDNA was amplified by RT-qPCR with an ABI PRISM 7900HT Sequence Detection system (Applied Biosystems, Foster City, CA, USA). Amplified PCR products were detected using SYBR Green PCR Master Mix (Roche, catalog # 04673484001). RT-PCRs for the endogenous GAPDH and/or β-ACTIN gene served as internal controls. Results of RT-qPCR were analyzed using the 2−ΔΔCT method.Citation33 The mRNA levels in treated samples were standardized against samples exposed to vehicle or the control, PNArad4µ_nls.
The following are the forward (F) and reverse (R) primer sequences used
RAD51: 5′-CAATGCAGATGCAGCTTGAA-3′ (F);
5′-CCTTGGCTTCACTAATTCCCT-3′ (R)
RAD51B: 5′-TTTCCCCACTGGAGCTTATG-3′ (F);
5′-CTTCGTCCAAAGCAGAAAGG-3′ (R)
RAD51C: 5′-AGACGTTCCGCTTTGAAATG-3′ (F);
5′-GGAGTTCCTCAGCAGTCTGG-3′ (R)
RAD51D: 5′-AGTGGTGGACCTGGTTTCTG-3′ (F);
5′-CCAAGTCCTGCCTTCTTCAG-3′ (R)
XRCC2: 5′-TAGTGCCTTCCATAGGGCTGA-3′ (F);
5′-TGGGAAGTATACATCGTGGTG-3′ (R)
XRCC3: 5′-AAGAAGGTCCCCGTACTGCT-3′ (F);
5′-CTGTCACCTGGTTGATGCAC-3′ (R)
BRCA1: 5′-TAGGGCTGGAAGCACAGAGT-3′ (F);
5′-AATTTCCTCCCCAATGTTCC-3′ (R)
GAPDH: 5′-GTCCACTGGCGTCTTCACCA-3′ (F);
5′-GTGGCAGTGATGGCATGGAC-3′ (R)
ACTIN: 5′-CTCGACACCAGGGCGTTATG-3′ (F);
5′-TCTCCCACGTAGCAGTCCTTC-3′ (R)
Cell immunofluorescence
Cells were treated with PNArad51_nls or PNArad4µ_nls for 48 h, and then treated with melphalan (10 µM) or DMSO (vehicle) for an additional 6 h. Cells were harvested, washed with PBS and resuspended in PBS. Aliquots (10Citation4 cells in 0.12 mL PBS) were centrifuged onto clean glass slides using a Shandon Cytospin. Immediately after centrifugation, preparations were fixed in 4% paraformaldehyde in PBS for 12 min at 22°C followed by 3 5-minute washes in PBS. Cells were permeabilized by incubation with 0.1% Triton X-100 in PBS for 10 min, followed by 3 5-minute washes in PBS. Cells were then incubated with blocking solution (1.5% BSA in PBS) for 1 h at 22°C and then incubated at 4°C overnight with primary antibodies diluted in blocking solution (1:1000 goat polyclonal anti-RAD51 IgG [Santa Cruz Biotechnology, catalog # sc-6862]; 1:1000 mouse monoclonal anti-γH2AX [ser139] clone JBW301 [EMD Millipore, catalog # 05-636]). After three 5-minute washes in PBS, cells were incubated at 22°C in the dark, with appropriate secondary antibodies diluted in blocking buffer (donkey anti-goat IgG Alexa Fluor 488 for RAD51, and goat anti-mouse IgG Alexa Fluor 594 for γH2AX [Jackson Immuno-Research, catalog # 705-546-147 and 115-586-003, respectively]). Cells were washed 3 times in PBS and mounted under coverslips in Prolong® antifade reagent with DAPI (Invitrogen, catalog # P-36931). Images were acquired with an LSM 510 Zeiss confocal laser-scanning microscope with a 63x oil objective. For quantitative analysis, ≥100 cells from each treatment group were chosen at random and nuclear foci were counted manually to determine the percent positive for RAD51 and/or γH2AX (i.e., having ≥5 discrete foci per nucleus). Focus intensities (summarized as the integral of pixel density over all foci per nucleus) were measured using ImageJ software, with subtraction of background peripheral to each nucleus. Results were averaged from at least 3 biological replicates per group.
Cell proliferation and viability assay
Cell viability was measured by the WST-1 (Clontech, catalog # MK400) colorimetric survival assay. MM cell lines were seeded in 96-well plates at ∼8000 cells/well. The cells were treated with10-µM PNArad51_nls or PNArad4µ_nls for 24 h, with addition of melphalan (at 0–10 µM) for a further 48 h. WST-1 reagent was then added to each well at a 1:10 ratio, and incubation continued 4 h at 37°C. Viability was estimated from absorbance at 450 nm using a spectrophotometer (Molecular Devices Corp., Sunnyvale, CA, USA). The percent viability for PNArad4µ_nls was taken as 100% and the results of other treatments calculated relative to that. PNArad4µ_nls had no effect on cell viability relative to cell culture medium alone.
SCID-rab myeloma xenograft model
To evaluate the in vivo effects of antigene PNA ± melphalan, we used our SCID-rab mouse model of MM as previously described.Citation34 The Institutional Animal Care and Use Committee, University of Arkansas for Medical Sciences, approved all experimental procedures and protocols. We constructed luciferase-expressing H929 cells (H929-Luc) as described.Citation35 H929-Luc MM cells (1×10Citation6 cells in 50-µL PBS) were injected directly into the marrow cavity of rabbit-bone segments implanted in SCID mice. In situ growth of the inoculated cells as primary tumors was estimated by bioluminescence imaging, taking the total intensity (integrated over tumor area) of constitutive luciferase expression.Citation35,36 At 4 weeks post-injection, mice were randomized into 4 groups (each n = 10) and treated as follows:
50 µL of 100-µM PNArad4µ_nls injected directly into implanted bones bi-weekly;
50 µL of 100-µM PNArad51_nls injected directly into the implanted bones bi-weekly;
2.0 mg/kg melphalan injected intraperitoneally (i.p) twice per week in combination with treatment (a).
2.0 mg/kg melphalan injected intraperitoneally (i.p) twice per week in combination with treatment (b).
After 4 weeks of treatments a–d, mice were sacrificed and cells recovered from bone for total RNA extraction and RT-qPCR analysis of the mRNA levels of RAD51, its paralogs and BRCA1.
Statistical analysis
GraphPad Prism software (Prism ver. 6, San Diego, CA, USA) and Excel were used for statistical analyses. Significance of the difference between 2 groups was calculated by heteroscedastic t-tests, or among 3 or more groups by ANOVA with Tukey's post hoc test for multiple comparisons. Where indicated, differences with P < 0.05 were stated to have “nominal statistical significance” and were presented without correction, to reduce the frequency of type-I errors and to allow the reader to determine the appropriate correction for multiple endpoints.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Additional information
Funding
References
- Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet JP, Ahmann GJ, Adli M, et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011; 471:467-72; PMID:21430775; http://dx.doi.org/10.1038/nature09837
- Shammas MA, Shmookler Reis RJ, Koley H, Batchu RB, Li C, Munshi NC. Dysfunctional homologous recombination mediates genomic instability and progression in myeloma. Blood 2009; 113:2290-7; PMID:19050310; http://dx.doi.org/10.1182/blood-2007-05-089193
- Alagpulinsa DA, Ayyadevara S, Shmookler Reis RJ. A small molecule inhibitor of RAD51 reduces homologous recombination and sensitizes multiple myeloma cells to doxorubicin. Front Oncol 2014 ( In press); 4:289; PMID:25401086
- San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 2008; 77:229-57; PMID:18275380; http://dx.doi.org/10.1146/annurev.biochem.77.061306.125255
- Vispe S, Cazaux C, Lesca C, Defais M. Overexpression of RAD51 protein stimulates homologous recombination and increases resistance of mammalian cells to ionizing radiation. Nucleic Acids Res 1998; 26:2859-64; PMID:9611228; http://dx.doi.org/10.1093/nar/26.12.2859
- Yang Z, Waldman AS, Wyatt MD. Expression and regulation of RAD51 mediate cellular responses to chemotherapeutics. Biochem Pharmacol 2012; 83:741-6; PMID:22222428; http://dx.doi.org/10.1016/j.bcp.2011.12.022
- Kassambara A, Gourzones-Dmitriev C, Sahota S, Reme T, Moreaux J, Goldschmidt H, Constantinou A, Pasero P, Hose D, Klein B. A DNA repair pathway score predicts survival in human multiple myeloma: the potential for therapeutic strategy. Oncotarget 2014; 5(9):2487-98; PMID:24809299
- Lawley PD, Brookes P. Molecular mechanism of the cytotoxic action of difunctional alkylating agents and of resistance to this action. Nature 1965; 206:480-3; PMID:5319105; http://dx.doi.org/10.1038/206480a0
- Long DT, Raschle M, Joukov V, Walter JC. Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science 2011; 333:84-7; PMID:21719678; http://dx.doi.org/10.1126/science.1204258
- Spanswick VJ, Craddock C, Sekhar M, Mahendra P, Shankaranarayana P, Hughes RG, Hochhauser D, Hartley JA. Repair of DNA interstrand crosslinks as a mechanism of clinical resistance to melphalan in multiple myeloma. Blood 2002; 100:224-9; PMID:12070031; http://dx.doi.org/10.1182/blood.V100.1.224
- Chen Q, Van der Sluis PC, Boulware D, Hazlehurst LA, Dalton WS. The FA/BRCA pathway is involved in melphalan-induced DNA interstrand cross-link repair and accounts for melphalan resistance in multiple myeloma cells. Blood 2005; 106:698-705; PMID:15802532; http://dx.doi.org/10.1182/blood-2004-11-4286
- Nakanishi K, Yang YG, Pierce AJ, Taniguchi T, Digweed M, D'Andrea AD, Wang ZQ, Jasin M. Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proc Natl Acad Sci U S A 2005; 102:1110-5; PMID:15650050; http://dx.doi.org/10.1073/pnas.0407796102
- Neri P, Ren L, Gratton K, Stebner E, Johnson J, Klimowicz A, Duggan P, Tassone P, Mansoor A, Stewart DA, et al. Bortezomib-induced “BRCAness” sensitizes multiple myeloma cells to PARP inhibitors. Blood 2011; 118:6368-79; PMID:21917757; http://dx.doi.org/10.1182/blood-2011-06-363911
- Wang ZM, Chen ZP, Xu ZY, Christodoulopoulos G, Bello V, Mohr G, Aloyz R, Panasci LC. In vitro evidence for homologous recombinational repair in resistance to melphalan. J Natl Cancer Inst 2001; 93:1473-8; PMID:11584063; http://dx.doi.org/10.1093/jnci/93.19.1473
- Xia SJ, Shammas MA, Shmookler Reis RJ. Elevated recombination in immortal human cells is mediated by HsRAD51 recombinase. Mol Cell Biol 1997; 17:7151-8; PMID:9372947
- Raderschall E, Stout K, Freier S, Suckow V, Schweiger S, Haaf T. Elevated levels of RAD51 recombination protein in tumor cells. Cancer Res 2002; 62:219-25; PMID:11782381
- Wiegmans AP, Al-Ejeh F, Chee N, Yap PY, Gorski JJ, Da Silva L, Bolderson E, Chenevix-Trench G, Anderson R, Simpson PT, et al. RAD51 supports triple negative breast cancer metastasis. Oncotarget 2014; 5(10):3261-72; PMID:24811120
- Budke B, Logan HL, Kalin JH, Zelivianskaia AS, Cameron McGuire W, Miller LL, Stark JM, Kozikowski AP, Bishop DK, Connell PP. RI-1: a chemical inhibitor of RAD51 that disrupts homologous recombination in human cells. Nucleic Acids Res 2012; 40:7347-57; PMID:22573178; http://dx.doi.org/10.1093/nar/gks353
- Normand A, Riviere E, Renodon-Corniere A. Identification and characterization of human RAD51 inhibitors by screening of an existing drug library. Biochem Pharmacol 2014; 91:293-300; PMID:25124703; http://dx.doi.org/10.1016/j.bcp.2014.07.033
- Ito M, Yamamoto S, Nimura K, Hiraoka K, Tamai K, Kaneda Y. RAD51 siRNA delivered by HVJ envelope vector enhances the anti-cancer effect of cisplatin. J Gene Med 2005; 7:1044-52; PMID:15756713; http://dx.doi.org/10.1002/jgm.753
- Sak A, Stueben G, Groneberg M, Bocker W, Stuschke M. Targeting of RAD51-dependent homologous recombination: implications for the radiation sensitivity of human lung cancer cell lines. Br J Cancer 2005; 92:1089-97; PMID:15785736; http://dx.doi.org/10.1038/sj.bjc.6602457
- Collis SJ, Tighe A, Scott SD, Roberts SA, Hendry JH, Margison GP. Ribozyme minigene-mediated RAD51 down-regulation increases radiosensitivity of human prostate cancer cells. Nucleic Acids Res 2001; 29:1534-8; PMID:11266555; http://dx.doi.org/10.1093/nar/29.7.1534
- Nielsen PE, Egholm M, Berg RH, Buchardt O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991; 254:1497-500; PMID:1962210; http://dx.doi.org/10.1126/science.1962210
- Egholm M, Buchardt O, Christensen L, Behrens C, Freier SM, Driver DA, Berg RH, Kim SK, Norden B, Nielsen PE. PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogen-bonding rules. Nature 1993; 365:566-8; PMID:7692304; http://dx.doi.org/10.1038/365566a0
- Demidov VV, Potaman VN, Frank-Kamenetskii MD, Egholm M, Buchard O, Sonnichsen SH, Nielsen PE. Stability of peptide nucleic acids in human serum and cellular extracts. Biochem Pharmacol 1994; 48:1310-3; PMID:7945427; http://dx.doi.org/10.1016/0006-2952(94)90171-6
- Kaihatsu K, Janowski BA, Corey DR. Recognition of chromosomal DNA by PNAs. Chem Biol 2004; 11:749-58; PMID:15217608; http://dx.doi.org/10.1016/j.chembiol.2003.09.014
- Belotserkovskii BP, Hanawalt PC. PNA binding to the non-template DNA strand interferes with transcription, suggesting a blockage mechanism mediated by R-loop formation. Mol Carcinog 2014; 30:22209
- Janowski BA, Kaihatsu K, Huffman KE, Schwartz JC, Ram R, Hardy D, Mendelson CR, Corey DR. Inhibiting transcription of chromosomal DNA with antigene peptide nucleic acids. Nat Chem Biol 2005; 1:210-5; PMID:16408037; http://dx.doi.org/10.1038/nchembio724
- Holstege FC, Fiedler U, Timmers HT. Three transitions in the RNA polymerase II transcription complex during initiation. EMBO J 1997; 16:7468-80; PMID:9405375; http://dx.doi.org/10.1093/emboj/16.24.7468
- Hine CM, Seluanov A, Gorbunova V. Use of the RAD51 promoter for targeted anti-cancer therapy. Proc Natl Acad Sci U S A 2008; 105:20810-5; PMID:19106292; http://dx.doi.org/10.1073/pnas.0807990106
- Cutrona G, Carpaneto EM, Ulivi M, Roncella S, Landt O, Ferrarini M, Boffa LC. Effects in live cells of a c-myc anti-gene PNA linked to a nuclear localization signal. Nat Biotechnol 2000; 18:300-3; PMID:10700145; http://dx.doi.org/10.1038/73745
- Cogoi S, Codognotto A, Rapozzi V, Meeuwenoord N, van der Marel G, Xodo LE. Transcription inhibition of oncogenic KRAS by a mutation-selective peptide nucleic acid conjugated to the PKKKRKV nuclear localization signal peptide. Biochemistry 2005; 44:10510-9; PMID:16060660; http://dx.doi.org/10.1021/bi0505215
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25:402-8; http://dx.doi.org/10.1006/meth.2001.1262
- Yata K, Yaccoby S. The SCID-rab model: a novel in vivo system for primary human myeloma demonstrating growth of CD138-expressing malignant cells. Leukemia 2004; 18:1891-7; PMID:15385929; http://dx.doi.org/10.1038/sj.leu.2403513
- Li X, Pennisi A, Zhan F, Sawyer JR, Shaughnessy JD, Yaccoby S. Establishment and exploitation of hyperdiploid and non-hyperdiploid human myeloma cell lines. Br J Haematol 2007; 138:802-11; PMID:17760811; http://dx.doi.org/10.1111/j.1365-2141.2007.06742.x
- Pennisi A, Li X, Ling W, Khan S, Zangari M, Yaccoby S. The proteasome inhibitor, bortezomib suppresses primary myeloma and stimulates bone formation in myelomatous and nonmyelomatous bones in vivo. Am J Hematol 2009; 84:6-14; PMID:18980173; http://dx.doi.org/10.1002/ajh.21310
- Taniguchi T, Garcia-Higuera I, Andreassen PR, Gregory RC, Grompe M, D'Andrea AD. S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood 2002; 100:2414-20; PMID:12239151; http://dx.doi.org/10.1182/blood-2002-01-0278
- Xu ZY, Loignon M, Han FY, Panasci L, Aloyz R. XRCC3 induces cisplatin resistance by stimulation of RAD51-related recombinational repair, S-phase checkpoint activation, and reduced apoptosis. J Pharmacol Exp Ther 2005; 314:495-505; PMID:15843498; http://dx.doi.org/10.1124/jpet.105.084053
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 1998; 273:5858-68; PMID:9488723; http://dx.doi.org/10.1074/jbc.273.10.5858
- Raderschall E, Golub EI, Haaf T. Nuclear foci of mammalian recombination proteins are located at single-stranded DNA regions formed after DNA damage. Proc Natl Acad Sci U S A 1999; 96:1921-6; PMID:10051570; http://dx.doi.org/10.1073/pnas.96.5.1921
- Walters DK, Wu X, Tschumper RC, Arendt BK, Huddleston PM, Henderson KJ, Dispenzieri A, Jelinek DF. Evidence for ongoing DNA damage in multiple myeloma cells as revealed by constitutive phosphorylation of H2AX. Leukemia 2011; 25:1344-53; PMID:21566653; http://dx.doi.org/10.1038/leu.2011.94
- Raderschall E, Bazarov A, Cao J, Lurz R, Smith A, Mann W, Ropers HH, Sedivy JM, Golub EI, Fritz E, et al. Formation of higher-order nuclear RAD51 structures is functionally linked to p21 expression and protection from DNA damage-induced apoptosis. J Cell Sci 2002; 115:153-64; PMID:11801733
- Resnick MA, Martin P. The repair of double-strand breaks in the nuclear DNA of Saccharomyces cerevisiae and its genetic control. Mol Gen Genet 1976; 143:119-29; PMID:765749; http://dx.doi.org/10.1007/BF00266917
- Yarde DN, Oliveira V, Mathews L, Wang X, Villagra A, Boulware D, Shain KH, Hazlehurst LA, Alsina M, Chen DT, et al. Targeting the Fanconi anemia/BRCA pathway circumvents drug resistance in multiple myeloma. Cancer Res 2009; 69:9367-75; PMID:19934314; http://dx.doi.org/10.1158/0008-5472.CAN-09-2616
- Carvalho JF, Kanaar R. Targeting homologous recombination-mediated DNA repair in cancer. Expert Opin Ther Targets 2014; 18(4):427-58; PMID:24491188; http://dx.doi.org/10.1517/14728222.2014.882900
- Hine CM, Li H, Xie L, Mao Z, Seluanov A, Gorbunova V. Regulation of RAD51 promoter. Cell Cycle 2014; 13:2038-45; PMID:24781030; http://dx.doi.org/10.4161/cc.29016
- Hine CM, Seluanov A, Gorbunova V. RAD51 promoter-targeted gene therapy is effective for in vivo visualization and treatment of cancer. Mol Ther 2012; 20:347-55; PMID:22008909; http://dx.doi.org/10.1038/mt.2011.215
- Qiao B, Kerr M, Groselj B, Teo MT, Knowles MA, Bristow RG, Phillips RM, Kiltie AE. Imatinib radiosensitizes bladder cancer by targeting homologous recombination. Cancer Res 2013; 73:1611-20; PMID:23302228; http://dx.doi.org/10.1158/0008-5472.CAN-12-1170
- Yang C, Betti C, Singh S, Toor A, Vaughan A. Impaired NHEJ function in multiple myeloma. Mutat Res 2009; 660:66-73; PMID:19028508; http://dx.doi.org/10.1016/j.mrfmmm.2008.10.019
- Mitsiades CS, Mitsiades N, Munshi NC, Anderson KC. Focus on multiple myeloma. Cancer Cell 2004; 6:439-44; PMID:15542427; http://dx.doi.org/10.1016/j.ccr.2004.10.020
- Hideshima T, Bergsagel PL, Kuehl WM, Anderson KC. Advances in biology of multiple myeloma: clinical applications. Blood 2004; 104:607-18; PMID:15090448; http://dx.doi.org/10.1182/blood-2004-01-0037