
Abstract
The unsatisfactory outcomes for osteosarcoma necessitate novel therapeutic strategies. This study evaluated the effect of the telomerase inhibitor imetelstat in pre-clinical models of human osteosarcoma. Because the chaperone molecule HSP90 facilitates the assembly of telomerase protein, the ability of the HSP90 inhibitor alvespimycin to potentiate the effect of the telomerase inhibitor was assessed. The effect of single or combined treatment with imetelstat and alvespimycin on long-term growth was assessed in osteosarcoma cell lines (143B, HOS and MG-63) and xenografts derived from 143B cells. Results indicated that imetelstat as a single agent inhibited telomerase activity, induced telomere shortening, and inhibited growth in all 3 osteosarcoma cell lines, though the bulk cell cultures did not undergo growth arrest. Combined treatment with imetelstat and alvespimycin resulted in diminished telomerase activity and shorter telomeres compared to either agent alone as well as higher levels of γH2AX and cleaved caspase-3, indicative of increased DNA damage and apoptosis. With dual telomerase and HSP90 inhibition, complete growth arrest of bulk cell cultures was achieved. In xenograft models, all 3 treatment groups significantly inhibited tumor growth compared with the placebo-treated control group, with the greatest effect seen in the combined treatment group (imetelstat, p = 0.045, alvespimycin, p = 0.034; combined treatment, p = 0.004). In conclusion, HSP90 inhibition enhanced the effect of telomerase inhibition in pre-clinical models of osteosarcoma. Dual targeting of telomerase and HSP90 warrants further investigation as a therapeutic strategy.
Abbreviations
| COG | = | the Children's Oncology Group |
| CTEP | = | the National Cancer Institute Cancer Therapy Evaluation Program |
| ALT | = | alternative lengthening of telomeres |
| 17-DMAG | = | 17-NN-Dimethyl Ethylene Diamine-Geldanamycin |
| 7-AAD | = | 7-Amino-Actinomycin D |
| γH2AX | = | phosphorylation of H2AX, TERC, the RNA component of telomerase |
| TERT | = | the catalytic component of telomerase |
| TRAP | = | telomeric repeat amplification protocol |
Introduction
Osteosarcoma is the most common primary bone cancer in children and adolescents, accounting for approximately 3% of malignancies in individuals under 20 years old.Citation1 With modern treatment approaches, the survival estimates for patients with localized and metastatic osteosarcoma are 70% and 20%, respectively. Improvements in survival for osteosarcoma have stalled during the past 2 decades and it has been recognized by the NIH Office of Rare Diseases, the Children's Oncology Group (COG), and the National Cancer Institute Cancer Therapy Evaluation Program (CTEP) that novel biological targets will be critical for making further strides in osteosarcoma treatment.Citation2
Telomeres are specialized structures at chromosome ends that are composed of several thousand nucleotide repeats (TTAGGG in vertebrates) and associated telomere binding proteins.Citation3 Evidence has accumulated that telomere length is a biological clock that regulates the proliferative capacity of a cell.Citation4 As a normal somatic cell divides, its telomeres erode until one or more telomeres reaches a critical length, at which time the telomere becomes “uncapped,” the ATM-p53 pathway is activated, and the cell undergoes senescence or apoptosis.Citation5,6 The vast majority of cancer cells circumvent the problem of telomere attrition by activating the enzyme telomerase, which adds nucleotide repeats to telomeres, permitting unlimited proliferation.Citation7 Osteosarcoma is distinct from most human cancers in that 50% of tumors lack telomerase expression and use the alternative lengthening of telomeres (ALT) mechanism for telomere maintenance.Citation8,9 Studies have suggested that the telomere maintenance mechanism may have prognostic significance for osteosarcoma and that the presence of telomerase expression may correlate with unfavorable prognosis.Citation8,9
Telomerase is an attractive therapeutic target for cancer because the enzyme plays a key role in conferring cellular immortality, is expressed in most cancers, and is relatively specific for cancer cells.Citation10 Imetelstat (GRN163L) is a lipidated thio-phosphoramidate-oligonucleotide with sequence complementarity to TERC, the RNA component of telomerase. By hybridizing to TERC, imetelstat inhibits the assembly of the telomerase enzyme complex, thereby inhibiting activity.Citation11 Studies in various adult and pediatric cancer cell lines have demonstrated that imetelstat, but not a mismatch oligonucleotide control, inhibited cell proliferation, colony formation, and invasiveness.Citation11-18 Phase 1 and 2 clinical trials demonstrated a favorable pharmacokinetic and side effect profile for imetelstat, though with mixed clinical results.Citation14,19-23
Although telomerase activity is regulated largely at the level of transcription of TERT, the gene that encodes the catalytic component of the telomerase enzyme complex, it has become clear that telomerase regulation is complex and also involves post-translational modifications, alternative splicing, and binding to chaperone proteins.Citation24,25 HSP90 plays a key role in the folding and function of TERT and more than 200 other oncogenic proteins including C-RAF, CDK4, AKT/PKB, mutant p53, and HIF-1α.Citation25-27 Depletion of functional HSP90 caused dramatic telomere shortening followed by apoptosis.Citation28,29 We hypothesized that HSP90 inhibition would potentiate the effect of telomerase inhibition therapy. In the present study, we evaluated the effect of telomerase inhibition therapy with imetelstat alone and in combination with the HSP90 inhibitor alvespimycin (17-NN-Dimethyl Ethylene Diamine-Geldanamycin, 17-DMAG) on human osteosarcoma cell lines and xenografts.
Results
Imetelstat inhibited telomerase activity and cell growth in osteosarcoma cell lines
To examine the effect of the telomerase inhibitor imetelstat on osteosarcoma cells, 3 telomerase-positive osteosarcoma cell lines (MG-63, HOS, and 143B) were treated with imetelstat or a mismatched negative control, GRN140S, for 3 days. As indicated in , imetelstat inhibited telomerase activity in all 3 cell lines in a dose-dependent manner. More than 50% telomerase inhibition occurred at imetelstat concentrations of 1 μM in MG-63 cells () and 2 μM in at HOS and 143B cells (). Maximal levels of telomerase inhibition were observed at higher concentrations (8 and 16 μM), but at these concentrations the cells exhibited an immediate cytotoxic effect most consistent with a telomere-independent effect because insufficient time elapsed to allow for telomere shortening. The mismatched control had no effect on telomerase activity in any of the cell lines.
Figure 1. Effect of imetelstat on telomerase activity and cell growth in osteosarcoma cell lines. (A–C). Three osteosarcoma cell lines (MG-63, HOS and 143B) were grown in media containing different concentrations of telomerase inhibitor, imetelstat or the mismatched negative control for 72 h. More than 50% telomerase inhibition occurred in all 3 cell lines when imetelstat concentrations reached 1 μM in MG-63 cells (A) and 2 μM in HOS and 143B cells (B, C). The mismatched control had no effect on telomerase activity. 0/H:heat-treated cell lysate samples to inactivate telomerase activity as a negative control, Buf: cell lysate buffer as a negative control, Intern: 36 bp internal PCR standard control. (D–F) Long-term treatment with imetelstat inhibited osteosarcoma cell growth. Cells were cultured in the presence of 1 μM imetelstat for MG-63 and 2 μM for HOS and 143B cells. Treatment duration is indicated as numbers of passages on the X- axis. Accumulated cell doubling numbers are indicated on the Y axis.
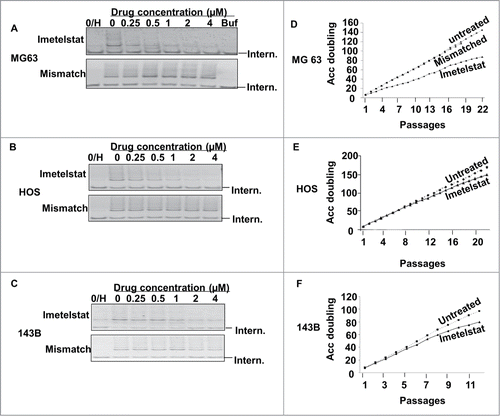
To assess the effect of protracted exposure to imetelstat on osteosarcoma cells, cells were treated with imetelstat applied twice a week for 14 to 36 weeks. Cell growth inhibition in imetelstat-treated MG-63 cells appeared at early passages after drug exposure and persisted into late passages, though the cells did not undergo complete arrest of proliferation after 36 weeks of treatment (). In HOS and 143B cells, modest cell growth inhibition in imetelstat-treated cells was not evident until passage 12 and 6, respectively (). As seen with the MG-63 cells, the bulk cultures never ceased proliferating for the duration of the experiment. Cell growth was not affected in 3 cell lines treated with mismatched control ( for MG63, data not shown for HOS and 143B). Telomerase activity was reassessed at late passages to ascertain whether reactivation of telomerase activity explained the continued growth of the bulk cultures. Telomerase activity was effectively inhibited by imetelstat even at late passages (data not shown).
Addition of the HSP90 inhibitor alvespimycin enhanced telomerase inhibition and accelerated telomere shortening
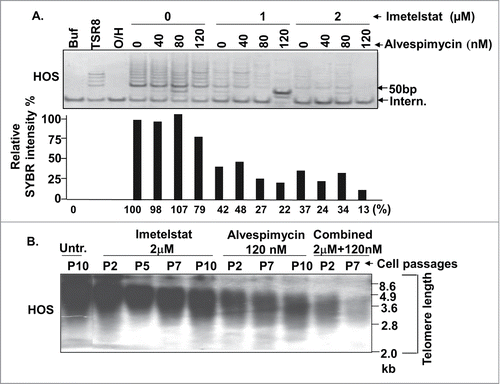
To overcome the limitations of imetelstat as a single agent, we assessed the effect of combined inhibition of telomerase and HSP90, a chaperone molecule of telomerase protein (TERT) that promotes its stability and the assembly of the telomerase enzyme complex. In HOS cells, the HSP90 inhibitor alvespimycin displayed a modest inhibitory effect on telomerase activity (79% activity compared to the untreated control) at a concentration of 120 nM (), with no inhibitory effect observed at lower concentrations. The combination of alvespimycin and imetelstat displayed stronger telomerase inhibition than with either agent alone, with maximum telomerase activity inhibition of 13% activity compared to the untreated control seen at imetelstat and alvespimycin concentrations of 2 μM and 120 nM, respectively (). Consistent with the effect on telomerase activity inhibition, combined treatment with imetelstat and alvespimycin resulted in greater telomere shortening () compared to either agent alone. Interestingly, telomere shortening was seen as soon as passage 2 after exposure to alvespimycin as a single agent (, 6th lane) even though the effect of alvespimycin as a single agent on telomerase activity was modest (, 7th lane). This suggests that inhibition of HSP90 may promote telomere shortening through mechanisms other than telomerase inhibition.
Figure 2. Combined effect of the HSP90 inhibitor on HOS cells. (A) Telomerase activity assay. HOS cells were grown in media containing the indicated concentrations of imetelstat and alvespimycin. The lower panel represents relative SYBR green fluorescence intensity corresponding to the lanes of the gel electrophoresis picture in the upper panel. Buf: cell lysate buffer as a negative control. TSR8: telomerase quantitation template control. 0/H:heat-treated cell lysate samples to inactivate telomerase activity as a negative control. Intern: 36 bp internal PCR standard control. (B) Telomere length Southern blot. Digested DNA from indicated passage numbers (P) of HOS cells was subjected to Southern blot using a digoxigenin-labeled telomere repeat (TTAGGG)6 probe. DNA marker size (kb) is indicated along the right side of the blot. Untr.: Untreated control, Combined: imetelstat plus alvespimycin treatment.
Combined or sequential treatment of imetelstat and alvespimycin induced cell death
To explore the impact of combined telomerase and HSP90 inhibition on cell growth, we compared long-term cell growth in the presence of 2 uM imetelstat, 120 nM alvespimycin, and the combination of 2 uM imetelstat and 120 nM alvespimycin. As shown in , HOS cells treated with both agents underwent cell growth arrest after passage 9, whereas cells treated with either agent alone continued to grow. To assess the nature of the proliferative arrest in cells treated with combination therapy, annexin V staining was performed to measure apoptosis. Treated cells showed significantly increased apoptosis compared to untreated cells, with 4.7% in early apoptosis (P = 0.0091), 28.3% in late apoptosis (P < 0.0001) and 10.9% dead cells (P = 0.0009) after exposure to imetelstat and alvespimycin for10 passages (). Similarly, combined treatment with 2 uM imetelstat and 50 nM alvespimycin caused cell death in 143B cells after passage 12, however, the combination of 1uM imetelstat and 120 nM alvespimycin did not cause cell death in MG-63 cells (data not shown).
Figure 3. Effect of combined treatment with imetelstat and alvespimycin on cell growth and apoptosis. (A) HOS cells were cultured in alvespimycin (120 nM), imetelstat (2 μM), or both agents (120 nM alvespimycin +2 μM imetelstat, Alv+Imet) for the indicated number of passages on the X-axis. (B) Flow cytometric assay of annexin V staining versus 7-Amino-Actinomycin D (7-AAD) provided a measure of viable cells (lower left quadrant), early apoptosis (lower right quadrant), late apoptosis (upper right quadrant), or dead cells (upper left quadrant). The lower panel shows a histogram depicting the quantitation of cells in the upper panel. The asterisk (*) denotes a statistically significant difference. (C) Effect of different concentrations of alvespimycin on cell viability in untreated HOS cells vs. cells pre-treated with imetelstat for 9 passages. The y-axis represents the percent cell viability compared to untreated cells.
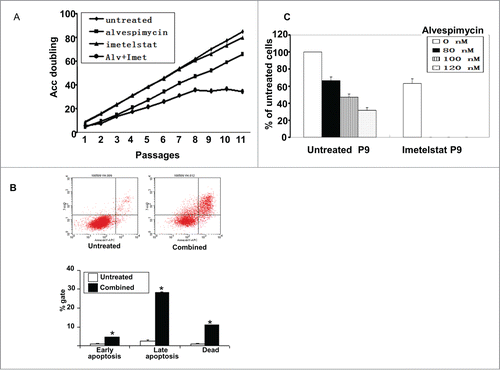
In a complementary experiment, cell viability was assessed by adding alvespimycin to cells pre-treated with imetelstat. HOS cells pretreated with 2 μM imetelstat for 9 passages had markedly decreased viability after treatment with 80–120 nM alvespimycin for one week compared to cells that were not pre-treated (). Similar results were observed for MG-63 cells and 143B cells (data not shown).
Effect of telomerase and HSP90 inhibition on colony formation
Colony formation is a marker of growth potential of cancer cells. To evaluate the impact of imetelstat treatment on colony formation, plating efficiency was assessed in 3 cell lines exposed to imetelstat for various durations of treatment. As shown in , the colony forming capacity of all 3 cell lines decreased with increasing duration of imetelstat treatment and progressive telomere shortening. In MG-63 cells, colony forming ability was completely eliminated by passage 18 in imetelstat ().
Figure 4. Colony formation assays. (A–C) Imetelstat reduced colony formation of osteosarcoma cells. Cells were grown in the presence of imetelstat 1 μM (MG-63) or 2 μM (HOS and 143B), the mismatched oligonucleotide control 2 μM or no treatment for the indicated number of passages. The y-axis indicates plating efficiency relative to untreated cells. Untreated: white bars, imetelstat: hashed bars, mismatched control: black bars. (D) Clonogenic survival assay showing enhanced sensitivity to alvespimycin with increasing duration of exposure to imetelstat. Untreated cells or cells treated with imetelstat for different passage numbers (Imet P # number) were checked for colony formation while grown in the presence of differing concentrations of alvespimycin (X-axis). % colony formation is indicated relative to untreated controls (Y-axis).
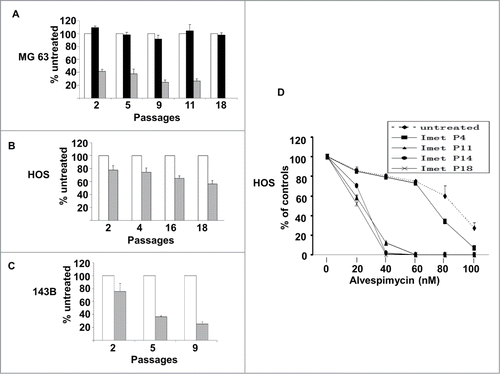
To explore whether telomere shortening sensitizes cells to alvespimycin, a clonogenic survival assay was performed with different passages of imetelstat pre-treated HOS cells. As telomere lengths decreased (imetelstat pre-treated P11-P18), cells became more sensitive to alvespimycin compared to untreated cells and cells with longer telomeres (imetelstat pre-treated P4) (P < 0.01, ). Similar results were observed in MG-63 and 143B cells (data not shown).
Effect of imetelstat and alvespimycin on the ATM-p53 DNA damage pathway
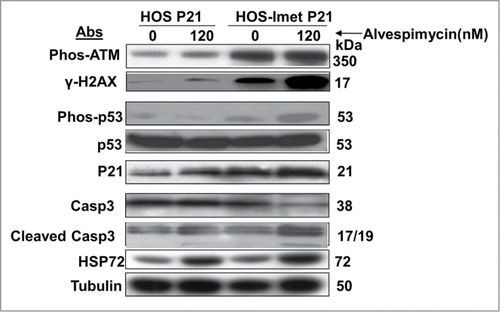
To further assess the mechanism through which HSP90 inhibition enhances the effect of telomerase inhibition, components of the DNA damage response pathway were evaluated. As expected with telomere shortening, treatment with imetelstat as a single agent resulted in phosphorylation of H2AX (γH2AX), a measure of DNA damage (, compare lanes 1 and 3). Treatment with imetelstat also led to activation of ATM (phosphorylation of ATM, Phos-ATM), but no appreciable change in phosphorylation of its downstream target p53. Treatment with alvespimycin alone did not result in a change in γH2AX level (, compare lanes 1 and 2) or any other components of the DNA damage response pathway, though HSP72 levels were increased, confirming that the drug was hitting its target.Citation30 Addition of alvespimycin to imetelstat pre-treated cells further increased the level γH2AX compared to imetelstat alone, indicating a greater degree of DNA damage, or alternatively, delayed DNA repair. This was accompanied by increased phosphorylated p53 and p21. Levels of cleaved caspase-3 were increased and levels of total caspase-3 were decreased only in cells treated with both imetelstat and alvespimycin, indicating that the apoptotic pathway was activated most significantly with combination therapy.
Figure 5. Activation of the DNA damage and apoptosis pathways in HOS cells treated with imetelstat and alvespimycin. Immunoblot analysis with the indicated antibodies (Abs) to examine the target proteins. Tubulin served as the protein loading control. The molecular weight markers are indicated on the right.
Inhibitory effect of imetelstat and alvespimycin on xenograft growth
To evaluate the effect of imetelstat and alvespimycin on osteosarcoma tumor growth in vivo, subcutaneous tumors were established in the flanks of immunodeficient mice using the 143B cell line, as this line generated xenografts at a higher rate than the other cell lines. After appearance of palpable tumors in one week, mice were injected intraperitoneally with PBS, imetelstat (30 mg/kg twice/week), alvespimycin (25mg/kg twice/week), or the combination of the 2 agents. After one month of treatment, 2 mice for each group were sacrificed and their tumors were collected to assess the pharmacodynamics of imetelstat and alvespimycin to confirm that the drugs were hitting their targets. As shown in , imetelstat-treated mice showed telomerase activity of 62% and 37% compared to the untreated controls. Likewise, administration of alvespimycin resulted in increased expression of HSP72, indicating that alvespimycin was hitting its target (data not shown).
Figure 6. Imetelstat, alvespimycin or combination treatment inhibited tumor growth of osteosarcoma xenografts. 106 143B cells were subcutaneously injected into flanks of CB17SC-SCID immunodeficient mice. Drug treatment was started once palpable tumors appeared. Mice were injected intraperitoneally with PBS, imetelstat (30 mg/kg twice/week), alvespimycin (25 mg/kg twice/week) or the combination of the 2 agents (Ime. 30 mg/kg twice/week + Alv25 mg/kg twice/week). 10–14 mice were treated in each group. (A) Telomerase activity. After one month of treatment, 2 mice in each group were sacrificed and tissue was harvested for telomerase activity measured by TRAP assays. A range of protein concentrations from 250 ng, 25 ng and 2.5ng were used. The lower panel shows the fluorescence intensity corresponding to the lanes of 25ng protein in the gel electrophoresis images above. Buf: cell lysate buffer as a negative control. TSR8: telomerase quantitation template control. Intern: 36 bp internal PCR standard control. Marker, 10 bp DNA ladder. (B) The curves show the mean volumes of tumor size at different time points. Compared to PBS group, 3 treatment groups significantly inhibited tumor growth (imetelstat, p = 0.045, alvespimycin, p = 0.034; imetelstat plus alvespimycin, Imet+Alv, p = 0004). (C) Tumor growth curves in individual mice in control and combination-treated groups. The dashed lines represent mice in PBS group, the solid lines represent the combination treatment group.
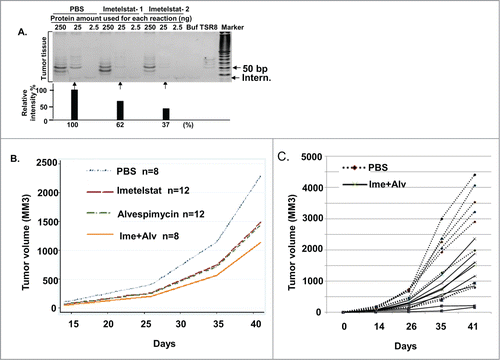
Tumor volumes were measured at days 0, 14, 26, 35, 41 after treatment. As shown in , all 3 treatment groups (8–12 mice for each group) significantly inhibited tumor growth compared with the PBS group (imetelstat, p = 0.045, alvespimycin, p = 0.034; imetelstat and alvespimycin, p = 0004). The combined treatment group showed the greatest degree of growth inhibition, though the differences between the 3 treatment groups were not statistically significant. illustrates the tumor growth curve for each mouse in the PBS group (dashed line) and the combined alvespimycin/imetelstat treated group (solid line). The rate of lung metastasis was reduced in the combination treatment group (14%) compared with the PBS group (25%), imetelstat group (25%), and alvespimycin group (36%), though the numbers were too small to draw firm conclusions about the preventative effect of combination treatment on metastatic potential.
Discussion
Our data demonstrate that long-term inhibition of telomerase activity by imetelstat led to progressive telomere shortening, growth inhibition, and decreased colony formation in osteosarcoma cells. However, the cell cultures did not manifest complete growth arrest or apoptosis after exposure to imetelstat as a single agent. The combination of imetelstat and alvespimycin reduced telomerase activity and telomere length compared to imetelstat alone and effectively induced cell growth arrest and apoptosis of cell cultures. Pre-treatment with imetelstat sensitized cells to alvespimycin, indicated by reduced clonogenic survival and enhanced activation of the ATM-p53 apoptosis pathway. In xenograft models, all 3 treatment groups significantly inhibited tumor growth compared with the placebo-treated control group, with the greatest effect seen in the combined treatment group.
Several factors may explain the resistance of cell lines to single-agent telomerase inhibition. First, cells may develop mechanisms to overcome the inhibitory effect of imetelstat, such as increased expression of telomerase components or efflux of the telomerase inhibitor from cells. However, there was no evidence of such resistance mechanisms because telomerase activity remained suppressed even after prolonged exposure to imetelstat (data not shown). It is also possible that osteosarcoma cells could activate the alternative lengthening of telomeres (ALT) pathway as a resistance mechanism.Citation31,32 ALT is of particular interest in osteosarcoma because up to 50% of human osteosarcoma tumors use ALT, rather than telomerase, to maintain telomeres.Citation8,9 Given the propensity to ALT in sarcomas without defining chromosomal translocations,Citation33,34 activation of the ALT pathway was a theoretical concern for targeting telomerase in osteosarcoma, but telomere length analysis conducted after prolonged exposure to imetelstat did not show the long and heterogenous telomere lengths that are characteristic of ALT. A third possibility is that the residual telomerase activity with imetelstat treatment, estimated to be about 40% of the baseline level, was sufficient to maintain the integrity of the shortest telomeres, which are thought to be the determinants of cellular viability and chromosome stability.Citation35 When we attempted to achieve a more complete telomerase blockade by increasing the imetelstat concentration (8 and 16 μM), cells showed immediate toxicity, likely the result of extra-telomeric effects of imetelstat such as decreased cell adhesion and disruption of the cytoskeleton.Citation36,37 Such effects may be desirable for treating cancer cells, but for the purpose of the current investigation, we chose to limit the concentration of imetelstat to focus on effects related to telomere shortening.
To circumvent the limitations of imetelstat as a single agent, we tested the effect of combined treatment with imetelstat and the HSP90 inhibitor alvespimycin. HSP90 is a chaperone protein that stabilizes several proteins that play crucial roles in oncogenic pathways, including SRC, ERBB2, EGFR, FLT3, Raf-1, AKT, HIF1α, and MET.Citation26 One of the HSP90 “client proteins” is the telomerase catalytic subunit, TERT. HSP90 associates with TERT and promotes its assembly into the active telomerase enzyme complex.Citation38 Several studies demonstrated that telomerase activity was inhibited by HSP90 inhibitor 17AAG or radicicol in melanoma,Citation28 prostate cancerCitation29 and myeloma cells.Citation17 As predicted, dual treatment with imetelstat and alvespimycin resulted in more complete telomerase inhibition () and telomere shortening () compared to either agent alone. Traditional drug combination assays to assess for synergy were not conducted because telomerase inhibitors require a lag time to allow telomere shortening to occur before their maximal effect is observed. However, long-term treatment with both agents resulted in eradication of the cell cultures in 2 of the 3 cell lines tested ( and data not shown). Similar findings were reported with combined imetelstat and 17AAG treatment in myeloma cells.Citation17 Moreover, clonogenic survival assays assessing the effect of varying concentrations of alvespimycin in imetelstat pretreated cells indicated that the longer the cells were treated with imetelstat (and the shorter the telomeres became), the more sensitive they became to alvespimycin ().
Two lines of evidence indicated that apoptosis was the mechanism of decreased cell growth, viability and colony formation in cells treated with the combination of alvespimycin and imetelstat. First, flow cytometry of cells stained with annexin V and 7-AAD demonstrated that the percentage of apoptotic cells increased with combination therapy (). Second, cells treated with combination therapy, but not with either agent alone, showed increased levels of cleaved caspase 3, indicative of activation of the apoptotic pathway ().
The cellular response to telomere dysfunction parallels the response to ionizing radiation. Both are associated with formation of γH2AX foci, phosphorylation of ATM, phosphorylation of p53 and upregulation of its downstream targets, leading to cell growth arrest or apoptosis.Citation5,6,39 HSP90 inhibition has been shown to act as a radiosensitizer by delaying DNA repair.Citation40 Cells treated with an HSP90 inhibitor followed by ionizing radiation (IR) had delayed resolution of γH2AX foci compared to cells treated with ionizing radiation alone.Citation40 Likewise, cells treated with the combination of imetelstat and alvespimycin had increased levels of γH2AX compared to treatment with either agent alone (, lane 4 vs. lane 2 or 3). The increased γH2AX levels may represent increased DNA damage or delay in DNA damage repair. Given the pleiotropic effects of HSP90 inhibition, we cannot exclude the possibility that the HSP90 inhibitor enhanced the effect of the telomerase inhibitor in other ways.
Both imetelstat and alvespimycin inhibited tumor growth in osteosarcoma xenograft models compared to mock-treated controls. Combined telomerase and HSP90 blockade resulted in more growth inhibition than with either agent alone, but the difference was not statistically significant. Pharmacodynamic studies demonstrated telomerase and HSP90 inhibition in the xenografts, but the degree of telomerase inhibition in the xenografts was not as great as in the cell lines. Further studies are needed to optimize delivery of both agents in the xenograft models.
Several phase 1 and 2 clinical trials of imetelstat have been conducted (www.clinicaltrials.gov). The most promising clinical effects were observed in hematological malignancies such as multiple myeloma and essential thrombocythenia.Citation19,20 Less promising results were observed with solid tumors, including breast cancer and non-small cell lung cancer.Citation21,22 However, a recent Children's Oncology Group phase 1 study of imetelstat for children with recurrent solid tumors observed partial responses in one patient with Ewing sarcoma and one patient with osteosarcoma.Citation23 Our results indicate that combination therapy may enhance the effect of imetelstat as a single agent.
In summary, our data indicate that the HSP90 inhibitor alvespimycin potentiates the effect of the telomerase inhibitor imetelstat in pre-clinical models of human osteosarcoma. Compared to either agent alone, the use of both agents resulted in more complete inhibition of telomerase activity, greater telomere shortening, greater DNA damage, and increased apoptosis. Dual targeting of telomerase and HSP90 warrants further investigation as a strategy to augment the effect of telomerase inhibition.
Materials and Methods
Reagents
The telomerase inhibitor, imetelstat (GRN163L), 5′-Palm-TAGGGTTAGACAA-NH2–3′, has sequence complementary to the TERC RNA template component of the telomerase ribonucleoprotein enzyme complex. The mismatched oligonucleotide GRN140S with the mismatched bases as underlined, 5′-Palm-TAGGTGTAAGCAA-NH2–3′, served as a negative control. Both oligonucleotides were provided by Geron Corporation (Menlo Park, CA). The HSP90 inhibitor, alvespimycin (or 17-NN-Dimethyl Ethylene Diamine-Geldanamycin, 17-DMAG), was purchased from LC Laboratories (Woburn, MA).
Cell lines
The three telomerase-positive cell lines, 143B, HOS and MG-63, were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and were authenticated by Research Animal Diagnostic Laboratory (Columbia, MO). 143B and HOS cells were maintained in Eagle's Minimum Essential (EME) medium containing 10% fetal bovine serum (FBS). MG-63 cells were cultured in EME medium containing 10% heat-inactivated FBS. All cells were maintained at 37°C in humidified air with 5% CO2. Culture mediums were purchased from ATCC. FBS was purchased from HyClone (South Logan, UT).
Telomerase activity assay
Telomerase activity was measured with the TRAP-eze kit by following the protocol provided by the manufacturer (Millipore, Billerica, MA). Briefly, harvested cell pellets were washed once in cold phosphate-buffered saline (PBS), and then lysed in 1x cold CHAPS buffer for 30 min on ice. 100 cells for cell lines or 2.5 to 250 ng of total protein from tumor tissues were utilized for each reaction. Reactions consisted of a 30-minute telomere extension step at 30°C followed by heating at 95°C for 10 minutes to activate ampliTaq Gold enzyme (Roche, Basel, Switzerland), followed 33 cycles of PCR reaction with denaturing at 94°C/30 seconds, annealing at 59°C/30 seconds and elongation at 72°C/1 minute. PCR products were separated in 12.5% PAGE gels and stained with SYBR green dye (Life Technologies, Washington, DC, USA). The BioRad Gel Doc XR documentation system (Hercules, CA, USA) was employed to visualize and quantitate DNA bands.
Long-term cell culture assays
For each passage, a fixed number of cells were seeded in triplicate in 6-well cell culture plates. The number of seeded cells was based on the cell doubling times of untreated cells, with the goal of reaching 90% confluence after one week in culture. The number of seeded cells was 1 × 104 cells for HOS and 143B, and 2.5 × 104 cells for the MG-63 line. Cells were split weekly and reseeded at a fixed number. Medium containing different drugs was added immediately after cells were seeded and replaced every 3 to 4 days. At selected passages, cells were collected for analysis of telomerase activity, telomere length and various other assays.
Telomere length measurement
Telomere restriction fragment Southern blots were utilized to assess telomere length as previously described.Citation5 Briefly, genomic DNA was prepared with the Qiagen DNeasy Blood & Tissue kit (Valencia, CA). 3–20 μg DNA from each sample was digested with a mixture of 6 enzymes (AluI, CfoI, HaeIII, Hinf I, MspI, and RsaI, New England Biolabs, Ipswich, MA, USA), and then the digested DNA fragments were separated on a 1.0% agarose gel, blotted on nylon membranes, and hybridized by a digoxigenin-labeled (TTAGGG)6 probe. The signal was detected by enzyme immunoassay and visualized by exposure to film.
Colony formation assay
Assays for colony formation and clonogenic survival were performed as previously described.Citation41 In brief, 500 cells were plated on 100 mm2 dishes and grown for 7–11 days. Treatment medium was refreshed every 3 to 4 days. Cells were washed once in PBS and stained with 0.1% Giemsa solution in 70% ethanol. Colonies with more than 50 cells were counted. Colony forming efficiency (%) was indicated by the ratio of number of colonies in the treated cells divided by the number of colonies in the untreated cells multiplied by 100.
Apoptosis assay
For each assay, 20,000 cells were stained with APC-Annexin V or 7-Amino-Actinomycin D (7-AAD) and analyzed with a Becton-Dickinson FACSClibur flow cytometer within 1 hour, according to the manufacturer's instructions (BD, San Jose, CA, USA).
Western blotting
Cells or minced tumor tissues were lysed in lysis buffer (PBS, pH 7.4, containing 1% Triton X-100, 0.1 mmol/l EDTA and complete proteinase inhibitor cocktail) (Roche, Basel, Switzerland). Proteins were separated in polyacrylamide gel by electrophoresis, and then protein bands were transferred to nitrocellulose membranes. The membranes were incubated in the cold overnight with primary antibodies in 1:1000 dilution, anti-α tubulin mouse monoclonal antibody (mAb) (Sigma, St. Louis, MO, USA), Anti- HSP72 mAb (Millipore, Billerica, MA, USA), anti-γH2AX pS139 (Millipore, Billerica, MA, USA), anti-p53 and anti-p53 pS15 (Cell Signaling, Danvers, MA, USA), anti-cleaved caspase 3 (Cell Signaling, Danvers, MA, USA), anti-ATM pS1981 (Rockland, Gilbertsville, PA, USA), and anti-p21 mAb (Thermo Scientific, Fremont, CA, USA). Membranes were incubated with secondary antibodies, goat anti-mouse or anti-rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA), at room temperature for one hour. SuperSignal West Dura extended duration chemi-luminescent substrate was used to visualize the protein bands (Thermo Scientific, Waltham, MA).
Xenograft experiments
Six-week old CB17SC-SCID male mice were purchased from Taconic (Germantown, NY, USA). Mice were maintained in a pathogen-free area according the guidelines of the Institutional Animal Care and Use Committee (IACUC). All experimental procedures were approved by the IACUC and the Occupational Health and Safety Department of Children's National Medical Center. One million 143B cells suspended in 100 μL PBS were injected subcutaneously into the right flank of each mouse. One week after injection, all mice had palpable tumors and the baseline tumor sizes were measured (day 0). The mice were randomly divided into 4 treatment groups: PBS control, imetelstat, alvespimycin, and combined therapy with imetelstat and alvespimycin. The administration of drugs was given by intraperitoneal injection twice a week. The dosages were 30 mg/kg mouse for imetelstat and 25 mg/kg mouse for alvespimycin. Tumor sizes were measured at days 14, 26, 35, and 41 after treatment. Tumor volume was calculated according to the formula 3.14/6*(length*width*height). The primary tumors, livers and lungs were harvested at necropsy. A portion of the tumor samples were used for assays of telomerase activity and HSP71/72 expression, as a read-out of alvespimycin activity. Samples from tumor and lungs were fixed in 4% 4-para-formaldehydrate for histological analysis.
Statistical Methods
Longitudinal analysis was used to compare the tumor volumes at different timepoints among the different groups with adjustment for the baseline tumor volume. The analysis was done by Stata 11 SE (StataCorp, College Station, TX, USA). A T-test was used to compare groups, analyzed with SPSS Ver. 13.0 for Windows (SPSS Institute, Inc., Chicago, IL, USA). A significance level was established at P < 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
Grant sponsors: The Children's Cancer Foundation and the Thomas Willson and Lenore Williams McKnew Endowment fund at Children's National Medical Center.
References
- Howlader N, Noone A, Krapcho M, Neyman N, Aminou R, Waldron W, Altekruse S, Kosary C, Ruhl J, Tatalovich Z, et al. SEER Cancer Statistics Review, 1975–2008, 2011; http://seer.cancer.gov/csr/1975_2011/
- Gorlick R, Anderson P, Andrulis I, Arndt C, Beardsley GP, Bernstein M, Bridge J, Cheung NK, Dome JS, Ebb D, et al. Biology of childhood osteogenic sarcoma and potential targets for therapeutic development: meeting summary. Clin Cancer Res 2003; 9:5442-53; PMID:14654523
- de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 2005; 19:2100-10; PMID:16166375; http://dx.doi.org/10.1101/gad.1346005
- Harley CB. Telomere loss: mitotic clock or genetic time bomb. Mutat Res 1991; 256:271-82; PMID:1722017; http://dx.doi.org/10.1016/0921-8734(91)90018-7
- Bakkenist CJ, Drissi R, Wu J, Kastan MB, Dome JS. Disappearance of the telomere dysfunction-induced stress response in fully senescent cells. Cancer Res 2004; 64:3748-52; PMID:15172978; http://dx.doi.org/10.1158/0008-5472.CAN-04-0453
- d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003; 426:194-8; PMID:14608368; http://dx.doi.org/10.1038/nature02118
- Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer [see comments]. Science 1994; 266:2011-5; PMID:7605428; http://dx.doi.org/10.1126/science.7605428
- Ulaner GA, Huang HY, Otero J, Zhao Z, Ben Porat L, Satagopan JM, Gorlick R, Meyers P, Healey JH, Huvos AG, et al. Absence of a telomere maintenance mechanism as a favorable prognostic factor in patients with osteosarcoma. Cancer Res 2003; 63:1759-63; PMID:12702558
- Sanders RP, Drissi R, Billups CA, Daw NC, Valentine MB, Dome JS. Telomerase expression predicts unfavorable outcome in osteosarcoma. J Clin Oncol 2004; 22:3790-7; PMID:15365076; http://dx.doi.org/10.1200/JCO.2004.03.043
- Harley CB. Telomerase and cancer therapeutics. Nat Rev Cancer 2008; 8:167-79; PMID:18256617; http://dx.doi.org/10.1038/nrc2275
- Herbert BS, Gellert GC, Hochreiter A, Pongracz K, Wright WE, Zielinska D, Chin AC, Harley CB, Shay JW, Gryaznov SM. Lipid modification of GRN163, an N3′→P5′ thio-phosphoramidate oligonucleotide, enhances the potency of telomerase inhibition. Oncogene 2005; 24:5262-8; PMID:15940257; http://dx.doi.org/10.1038/sj.onc.1208760
- Gellert GC, Dikmen ZG, Wright WE, Gryaznov S, Shay JW. Effects of a novel telomerase inhibitor, GRN163L, in human breast cancer. Breast Cancer Res Treat 2005:1-9; PMID:16319992
- Dikmen ZG, Wright WE, Shay JW, Gryaznov SM. Telomerase targeted oligonucleotide thio-phosphoramidates in T24-luc bladder cancer cells. J Cell Biochem 2008; 104:444-52; PMID:18044713; http://dx.doi.org/10.1002/jcb.21635
- Marian CO, Cho SK, McEllin BM, Maher EA, Hatanpaa KJ, Madden CJ, Mickey BE, Wright WE, Shay JW, Bachoo RM. The telomerase antagonist, imetelstat, efficiently targets glioblastoma tumor-initiating cells leading to decreased proliferation and tumor growth. Clin Cancer Res 2010; 16:154-63; PMID:20048334; http://dx.doi.org/10.1158/1078-0432.CCR-09-2850
- Asai A, Oshima Y, Yamamoto Y, Uochi TA, Kusaka H, Akinaga S, Yamashita Y, Pongracz K, Pruzan R, Wunder E, Piatyszek M, Li S, et al. A novel telomerase template antagonist (GRN163) as a potential anticancer agent. Cancer Res 2003; 63:3931-9; PMID:12873987
- Wang ES, Wu K, Chin AC, Chen-Kiang S, Pongracz K, Gryaznov S, Moore MA. Telomerase inhibition with an oligonucleotide telomerase template antagonist: in vitro and in vivo studies in multiple myeloma and lymphoma. Blood 2004; 103:258-66; PMID:12969977; http://dx.doi.org/10.1182/blood-2003-02-0546
- Shammas MA, Koley H, Bertheau RC, Neri P, Fulciniti M, Tassone P, Blotta S, Protopopov A, Mitsiades C, Batchu RB, et al. Telomerase inhibitor GRN163L inhibits myeloma cell growth in vitro and in vivo. Leukemia 2008; 22:1410-8; PMID:18449204; http://dx.doi.org/10.1038/leu.2008.81
- Hu Y, Bobb D, Lu Y, He J, Dome JS. Effect of telomerase inhibition on preclinical models of malignant rhabdoid tumor. Cancer Genet 2014; 207:403-11; PMID:25441685; http://dx.doi.org/10.1016/j.cancergen.2014.09.002
- Huff CA, Wang Q, Badros AZ, Jones RJ, Reddy A, Nishimoto K, Stuart MJ, Kelsey SM, Matsui W. The telomerase inhibitor, imetelstat, rapidly reduces myeloma cancer stem cells (CSCs) in a phase II trial. ASH Ann Meet Abstr 2012; 120:4898
- Baerlocher GM, E.O. L, Ayran C, Blaney M, Burington B, Morfeld D, Olatoyosi O, Ottman O, Reddy A, Roeth A, et al. Imetelstat rapidly induces and maintains substantial hematologic and molecular responses in patients with essential thrombocythemia (ET) who are refractory or intolerant to prior therapy: preliminary phase II results. Congr Eur Hematol Assoc 2013; abstract # 4398
- Williams SC. No end in sight for telomerase-targeted cancer drugs. Nat Med 2013; 19:6; PMID:23295993; http://dx.doi.org/10.1038/nm0113-6
- Chiappori AA, Kolevska T, Spigel DR, Hager S, Rarick M, Gadgeel S, Blais N, Von Pawel J, Hart L, Reck M, et al. A randomized phase II study of the telomerase inhibitor imetelstat as maintenance therapy for advanced non-small-cell lung cancer. Annals Oncol 2015; 26:354-62; PMID:25467017; http://dx.doi.org/10.1093/annonc/mdu550
- Thompson PA, Drissi R, Muscal JA, Panditharatna E, Fouladi M, Ingle AM, Ahern CH, Reid JM, Lin T, Weigel BJ, et al. A phase I trial of imetelstat in children with refractory or recurrent solid tumors: a Children's Oncology Group Phase I Consortium Study (ADVL1112). Clin Cancer Res 2013; 19:6578-84; PMID:24097866; http://dx.doi.org/10.1158/1078-0432.CCR-13-1117
- Aisner DL, Wright WE, Shay JW. Telomerase regulation: not just flipping the switch. Curr Opin Genet Dev 2002; 12:80-5; PMID:11790559; http://dx.doi.org/10.1016/S0959-437X(01)00268-4
- Keppler BR, Grady AT, Jarstfer MB. The biochemical role of the heat shock protein 90 chaperone complex in establishing human telomerase activity. J Biol Chem 2006; 281:19840-8; PMID:16714764; http://dx.doi.org/10.1074/jbc.M511067200
- Bagatell R, Whitesell L. Altered Hsp90 function in cancer: a unique therapeutic opportunity. Mol Cancer Ther 2004; 3:1021-30; PMID:15299085; http://dx.doi.org/10.4161/cbt.3.10.1142
- Jego G, Hazoume A, Seigneuric R, Garrido C. Targeting heat shock proteins in cancer. Cancer Lett 2013; 332:275-85; PMID:21078542; http://dx.doi.org/10.1016/j.canlet.2010.10.014
- Villa R, Folini M, Porta CD, Valentini A, Pennati M, Daidone MG, Zaffaroni N. Inhibition of telomerase activity by geldanamycin and 17-allylamino, 17-demethoxygeldanamycin in human melanoma cells. Carcinogenesis 2003; 24:851-9; PMID:12771028; http://dx.doi.org/10.1093/carcin/bgg028
- Compton SA, Elmore LW, Haydu K, Jackson-Cook CK, Holt SE. Induction of nitric oxide synthase-dependent telomere shortening after functional inhibition of hsp90 in human tumor cells. Mol Cell Biol 2006; 26:1452-62; PMID:16449656; http://dx.doi.org/10.1128/MCB.26.4.1452-1462.2006
- Maloney A, Clarke PA, Naaby-Hansen S, Stein R, Koopman JO, Akpan A, Yang A, Zvelebil M, Cramer R, Stimson L, et al. Gene and protein expression profiling of human ovarian cancer cells treated with the heat shock protein 90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Cancer Res 2007; 67:3239-53; PMID:17409432; http://dx.doi.org/10.1158/0008-5472.CAN-06-2968
- Bryan TM, Englezou A, Dallapozza L, Dunham MA, Reddel RR. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat Med 1997; 3:1271-4; PMID:9359704; http://dx.doi.org/10.1038/nm1197-1271
- Bryan TM, Englezou A, Gupta J, Bacchetti S, Reddel RR. Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J 1995; 14:4240-8; PMID:7556065
- Montgomery E, Argani P, Hicks JL, DeMarzo AM, Meeker AK. Telomere lengths of translocation-associated and nontranslocation-associated sarcomas differ dramatically. Am J Pathol 2004; 164:1523-9; PMID:15111298; http://dx.doi.org/10.1016/S0002-9440(10)63710-8
- Ulaner GA, Hoffman AR, Otero J, Huang HY, Zhao Z, Mazumdar M, Gorlick R, Meyers P, Healey JH, Ladanyi M. Divergent patterns of telomere maintenance mechanisms among human sarcomas: Sharply contrasting prevalence of the alternative lengthening of telomeres mechanism in Ewing's sarcomas and osteosarcomas. Genes Chromosomes Cancer 2004; 41:155-62; PMID:15287028; http://dx.doi.org/10.1002/gcc.20074
- Hemann MT, Strong MA, Hao LY, Greider CW. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell 2001; 107:67-77; PMID:11595186; http://dx.doi.org/10.1016/S0092-8674(01)00504-9
- Jackson SR, Zhu CH, Paulson V, Watkins L, Dikmen ZG, Gryaznov SM, Wright WE, Shay JW. Antiadhesive effects of GRN163L–an oligonucleotide N3'->P5' thio-phosphoramidate targeting telomerase. Cancer Res 2007; 67:1121-9; PMID:17283146; http://dx.doi.org/10.1158/0008-5472.CAN-06-2306
- Mender I, Senturk S, Ozgunes N, Akcali KC, Kletsas D, Gryaznov S, Can A, Shay JW, Dikmen ZG. Imetelstat (a telomerase antagonist) exerts offtarget effects on the cytoskeleton. Int J Oncol 2013; 42:1709-15; PMID:23545855
- Holt SE, Aisner DL, Baur J, Tesmer VM, Dy M, Ouellette M, Trager JB, Morin GB, Toft DO, Shay JW, et al. Functional requirement of p23 and Hsp90 in telomerase complexes. Genes Dev 1999; 13:817-26; PMID:10197982; http://dx.doi.org/10.1101/gad.13.7.817
- Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol 2003; 13:1549-56; PMID:12956959; http://dx.doi.org/10.1016/S0960-9822(03)00542-6
- Dote H, Burgan WE, Camphausen K, Tofilon PJ. Inhibition of hsp90 compromises the DNA damage response to radiation. Cancer Res 2006; 66:9211-20; PMID:16982765; http://dx.doi.org/10.1158/0008-5472.CAN-06-2181
- Gellert GC, Dikmen ZG, Wright WE, Gryaznov S, Shay JW. Effects of a novel telomerase inhibitor, GRN163L, in human breast cancer. Breast Cancer Res Treat 2006; 96:73-81; PMID:16319992; http://dx.doi.org/10.1007/s10549-005-9043-5