
ABSTRACT
Significant improvement in targeted therapy for colorectal cancer (CRC) has occurred over the past few decades since the approval of the EGFR inhibitor cetuximab. However, cetuximab is used only for patients possessing the wild-type oncogene KRAS, NRAS, and BRAF, and even most of these eventually acquire therapeutic resistance, via activation of parallel oncogenic pathways such as RAS-MAPK or PI3K/Akt/mTOR. The two aforementioned pathways also contribute to the development of therapeutic resistance in CRC patients, due to compensatory and feedback mechanisms. Therefore, combination drug therapies (versus monotherapy) targeting these multiple pathways may be necessary for further efficacy against CRC. In this study, we identified PIK3CA mutant (PIK3CA MT) as a determinant of resistance to SMI-4a, a highly selective PIM1 kinase inhibitor, in CRC cell lines. In CRC cell lines, SMI-4a showed its effect only in PIK3CA wild type (PIK3CA WT) cell lines, while PIK3CA MT cells did not respond to SMI-4a in cell death assays. In vivo xenograft and PDX experiments confirmed that PIK3CA MT is responsible for the resistance to SMI-4a. Inhibition of PIK3CA MT by PI3K inhibitors restored SMI-4a sensitivity in PIK3CA MT CRC cell lines. Taken together, these results demonstrate that sensitivity to SMI-4a is determined by the PIK3CA genotype and that co-targeting of PI3K and PIM1 in PIK3CA MT CRC patients could be a promising and novel therapeutic approach for refractory CRC patients.
Introduction
Since the approval of the EGFR-targeting antibody cetuximab in 2004, much progress has occurred in targeted therapy for colorectal cancer (CRC).Citation1 Both cetuximab and another anti-EGFR antibody, panitumumab, are now recommended as first-line therapies for patients possessing wild-type (wt) oncogenes of RAS-MAPK pathway such as KRAS, NRAS, and/or BRAF.Citation2,Citation3 However, not all patients respond to cetuximab, due to mutation of many oncogenic drivers of the RAS-MAPK pathway, amplification of EGFR and other tyrosine kinase receptors (e.g., HER2 and c-MET), and/or activation of other oncogenic kinases such as PI3K or Src.Citation4–7 Also, for KRAS or NRAS mutated CRC patients, chemotherapy is the first-line treatment, since mutated RAS was considered as undruggable until the recent development of the mt KRAS-G12C inhibitor, AMG-510.Citation8 Before the development of AMG-510, numerous RAS-MAPK pathway-targeting agents have undergone clinical trials. However, due to compensatory and feedback mechanisms, monotherapy of these agents has proved disappointing,Citation9,Citation10 resulting in exploration of combinatorial therapies.Citation11–13
Phosphoinositide 3-kinases (PI3Ks), activated by growth factor receptors, phosphorylate lipid substrates to effect downstream signal transduction. Effectors of activated PI3K regulate numerous cellular signaling pathways such as metabolism, proliferation, survival, differentiation, motility, and immunity.Citation14,Citation15 It is well known that PI3K mutation leads to growth factor-independent PI3K signal activation which in turn increases proliferation and invasion and decreases apoptotic cell death.Citation16 Class 1 PI3K members have been most actively studied, as drug targets, due to their frequent alterations by mutation or amplification in human solid tumors, with the most frequent of these being PIK3CA which encodes the p110a subunit of PI3K.Citation14,Citation16–19
The PIM (proviral integration site for Moloney murine leukemia virus) family of serine/threonine kinases consists of three isoforms, PIM1, PIM2, and PIM3, which are highly conserved and highly homologous.Citation20 Rather than showing activity by conformational difference, PIM kinases themselves are expressed as short-lived, constitutively active forms. Therefore, their activity is represented by their expression levels, which are mainly regulated by the JAK/STAT pathway.Citation20,Citation21 PIM kinases participate in various cellular signaling pathways including metabolism, cell cycle, survival, proliferation, and metastasis,Citation22,Citation23 and these various effects of PIM kinases are known to be associated with the development and progression of cancer.
In addition to its own effects, PIM kinase signaling has been shown to overlap with PI3K/Akt/mTOR signaling, and elevated PIM kinase levels are considered as a mechanism of resistance to PI3K inhibitors in non-small cell lung cancer.Citation24 Also, triple-negative breast cancer (TNBC) cell lines showed significantly increased levels of PIM, with the PI3K/AKT/mTOR triple inhibitor IBL-302 demonstrating anti-TNBC activity.Citation25 Also, suppression of mTOR signaling in a preclinical study was shown by the PIM inhibitors SMI-4a25 and AZD-1208.Citation26 Another PIM inhibitor, INCB053914, showed additive or synergistic effects when combined with a selective PI3Kδ inhibitor in an in vivo tumor xenograft model.Citation27 Thus, while these results support combinational approaches of PIM- and PI3K-inhibitors,Citation26,Citation28 this possibility has not been evaluated in CRC.
In this study, we identified PIK3CA (encoding the p110α catalytic subunit of PI3K)Citation19 mutant (PIK3CA MT) as a determinant of CRC cell line sensitivity to SMI-4a, a highly selective PIM1 kinase inhibitor.Citation29 In our study, SMI-4a showed its efficacy against PIK3CA wild type (PIK3CA WT) expressing CRC cells. However, PIK3CA MT CRC cells did not show response to SMI-4a treatment as shown by cell death assays. Likewise, ectopic overexpression of PIK3CA MT in PIK3CA WT cells abrogated the response to SMI-4a in in vitro cell death assays, and in vivo xenograft and PDX (patient-derived xenograft) results confirmed that mutation in PIK3CA drives primary resistance to PIM1 inhibition in CRC. Analogously, inhibition of PIK3CA MT by PI3K inhibitors restored the response to SMI-4a in PIK3CA MT CRC cell lines.
Taken together, these results demonstrate that the sensitivity to SMI-4a, a highly selective PIM1 inhibitor, is determined by the presence of PIK3CA mutation and when PIK3CA mutation exists, CRC cells lose its responsiveness to SMI-4a. Therefore, PIK3CA mutation could be a negative predictive biomarker to inhibition of PIM1 in CRC, and co-targeting of PI3K and PIM1 in PIK3CA MT CRC patients could be a promising and novel therapeutic approach for refractory CRC patients.
Results
Identification of PIK3CA mutation as the underlying factor for resistance to SMI-4a in various CRC cell lines
Since PIM kinases are overexpressed in many cancers, including hematological malignancies and solid tumors, and their oncogenic function has been shown in numerous cancer cell lines,Citation23,Citation30,Citation31 a number of PIM inhibitors have recently been developed and entered into clinical trials. However, most such inhibitors failed to prove efficacy as monotherapies, with more recent developmental approaches focusing on combinational strategies.Citation24,Citation32
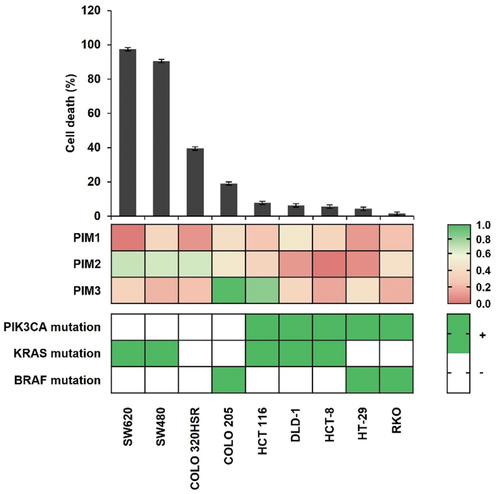
To identify a possible PIM1-cotherapeutic target in CRC, we chose SMI-4a, a highly selective PIM1 kinase inhibitor,Citation29 and evaluated its effects on cancer cell death. First, we assessed the expression levels of the PIM kinase family members PIM1, PIM2, and PIM3. As shown in , most CRC cell lines expressed PIM1, as well as PIM2 and PIM3, but with considerable variation. Analogously, the various CRC cells treated with 30 μM SMI-4a for 72 h, followed by trypan blue exclusion, also showed highly disparate response rates. As a result, CRC cell lines were divided into two groups according to cell death rates of over or under 10%. Therefore, we analyzed the genotype of representative oncogenic mutations in these CRC cell lines, noting that only PIK3CA mutation positively correlated with response to SMI-4a treatment (). Therefore, we hypothesized that PIK3CA mutation could be the underlying cause for primary resistance to SMI-4a.
Figure 1. Cell death efficacy of SMI-4a against various CRC cell lines. Cell death rates evaluated by trypan blue exclusion after 72 h treatment with 30 μM SMI-4a in nine CRC cell lines (top). Expression of PIM kinases by Western blot and RT-PCR (middle) was conducted to analyze the genotype of major oncogenic mutation in CRC cell lines (bottom). Experiments were conducted three times in triplicate.
PIK3CA mutation induces primary resistance to SMI-4a treatment in CRC cell lines
To further analyze the possibility of PIK3CA mutation as a negative predictive biomarker of SMI-4a response, we evaluated the effect of PIM inhibitors by colony-forming assays, and annexin V staining, in SW620 (PIK3CA WT) and HCT116 (PIK3CA MT) cell lines. As shown in (), SMI-4a treatment decreased colony numbers by 90% in SW620 cells, while HCT116 cells showed no response. Similar results were found with a pan-PIM inhibitor, AZD-1208, in PIK3CA WT and PIK3CA MT cell lines (Figure S1).
Figure 2. PIK3CA mutation as a negative predictive biomarker of SMI-4a treatment in CRC cell lines. (a) Colony-forming assays of SW620 (PIK3CA WT) and HCT116 (PIK3CA MT) after 30 μM of SMI-4a treatment. Cells were continuously exposed to SMI-4a for 16 (SW620) to 18 (HCT116) days. Colonies were stained with crystal violet and the number of colonies counted. (left) Representative image of colonies. (right) Quantification of colonies represented as relative colony formation ratios (%). ***p < .001 indicates significantly different from control group. (b) Annexin V-FITC/PI staining in SW620 (PIK3CA WT) and HCT116 (PIK3CA MT) after 30 μM SMI-4a treatment for 72 h. (left) Representative flow cytometry plots of annexin V-FITC/PI-stained apoptotic cells. (right) the percentages of apoptotic cells were statistically compared, with significant differences denoted by ***p < .001. (c) Expression of PI3K downstream effectors (left) and downstream signaling molecules related to cell proliferation and apoptosis (right) in SW480 (PIK3CA WT) and DLD-1 (PIK3CA MT) cell lines treated with 30 μM SMI-4a. (d) Cell death rates evaluated by trypan blue exclusion after 72 h SMI-4a (30 μM) treatment in DLD-1 PIK3CA WT (351) vs. MT (353) isogenic cell lines. ***p < .001 indicates significantly different from WT group. Experiments were repeated three times in triplicate.
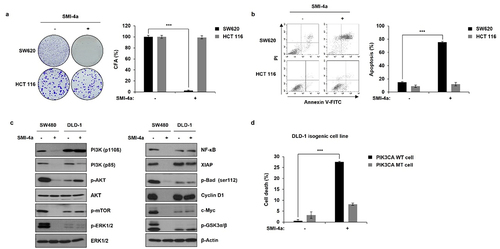
Also, as shown in , SMI-4a induced ~80% apoptotic cell death (i.e., annexin V staining) in SW620 cells, while HCT116 cells showed no notable response. In accord with these results, expression of the anti-apoptotic proteins XIAP and p-Bad was maintained in PIK3CA MT cell line, while PIK3CA WT cell line showed significant decrease in anti-apoptotic proteins when treated with SMI-4a ().
Since SMI-4a is known to exert its effect through PI3K/Akt/mTOR signaling in non-small cell lung cancer (NSCLC) cells,Citation33 we evaluated downstream effectors in PIK3CA WT and MT expressing cell lines, SW480 and DLD-1, respectively. Unlike PIK3CA WT cells, expression, and activation of the major downstream effectors of PIK3CA contributing to cell proliferation and survival was maintained upon SMI-4a treatment in PIK3CA MT cells (). Next, we evaluated the effects of SMI-4a in DLD-1 cell line, which is PIK3CA MT expressing CRC cell line and its isogenic cell line expressing PIK3CA WT. As shown in (), DLD-1 did not respond to SMI-4a; however, the isogenic cell line expressing PIK3CA WT responded to SMI-4a, demonstrating 27% efficacy, compared to 9% for PIK3CA MT DLD-1 cells.
Overexpression of PIK3CA mutants shows resistance to SMI-4a treatment
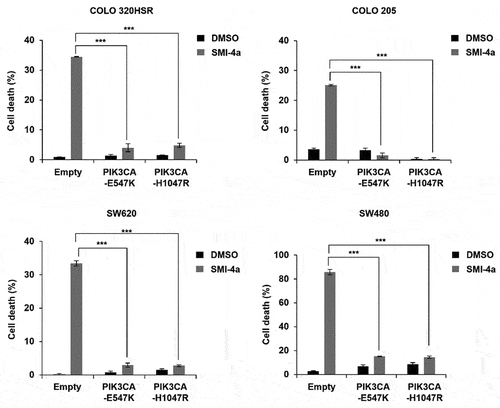
To further confirm the function of PIK3CA MT as a negative predictive biomarker for SMI-4a efficacy, SMI-4a-induced death was compared in cells overexpressing PIK3CA MT vs WT. As mutations in PIK3CA-E542K, E547K, and H1047R are the three major mutational hotspots in the PIK3CA protein, we overexpressed PIK3CA-E547K and PIK3CA-H1047R in PIK3CA WT CRC cell lines. As shown in , all cell lines with PIK3CA WT lost their response to SMI-4a by the introduction of PIK3CA-E547K and PIK3CA-H1047R, verifying that PIK3CA mutation is the underlying determinant of primary resistance to PIM1 inhibition.
Figure 3. Overexpression of mt PIK3CA induces resistance to SMI-4a treatment. PIK3CA-E547K and PIK3CA-H1047R were overexpressed in four (COLO 320HSR, COLO 205, SW620, and SW480) PIK3CA WT CRC cell lines, and cell death rates evaluated after 72 h of 30 μM SMI-4a treatment, by trypan blue exclusion assay. ***p < .001 indicates significantly different from control group. Experiments were repeated three times in triplicate.
Differences in response to SMI-4a in tumor xenografts, depending on PIK3CA genotype
We next sought to test our hypothesis by in vivo experiments, including mouse xenograft and PDX models. For in vivo mouse xenografts, we chose COLO 205 (PIK3CA WT) and HT-29 (PIK3CA MT) human CRC cell lines. Both cell lines have mutated BRAF. As shown in (), 100 mg/kg of the PIM1 inhibitor SMI-4a showed 43.2% efficacy against COLO 205 xenografts, while HT-29 xenograft showed no growth inhibition. As a general cytotoxicity control, no noticeable differences in weight change were observed in these xenograft models (Figure S2).
Figure 4. Sensitivity to SMI-4a is determined by PIK3CA genotype in in vivo tumor xenograft and PDX models. (a) COLO 205 and HT-29 ×enografts were subcutaneously infected into nude mice and 50- and 100-mg/kg SMI-4a administered daily by P.O. injection. Representative images of tumors and relative tumor sizes in COLO 205 (top) and HT-29 (bottom) xenograft models. (b) Colon tumors from patients grown in nude mice followed by 50- or 100-mg/kg SMI-4a injected daily by oral gavage. Representative images of tumors and relative tumor sizes in PIK3CA WT (left) and PIK3CA MT (right) mouse models. *p < 0.05 and ***p < 0.001 indicate significantly different from control group, respectively.
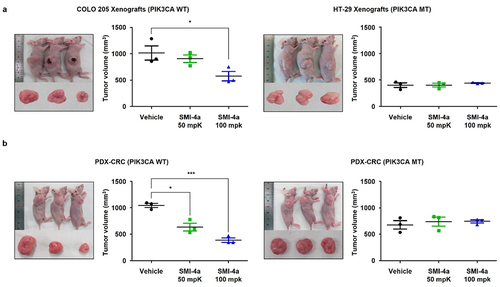
Similar results were seen in colon cancer PDX models with PIK3CA WT versus MT expression (), with SMI-4a being effective against PIK3CA WT, but not PIK3CA MT expressing PDX models, without noticeable differences in body weight change (Figure S2).
Taken together, these results indicate that PIK3CA mutations are responsible for primary resistance to PIM1 inhibition, thus representing a potentially novel negative predictive biomarker for PIM1-positive CRC targeting drugs.
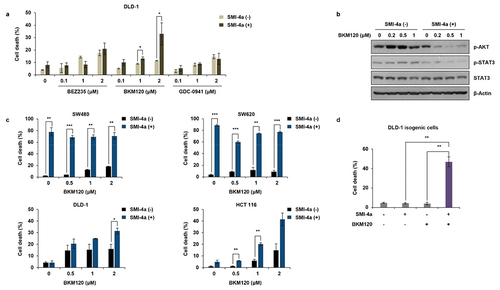
Co-treatment of NVP-BKM120 in PIK3CA mutated CRC cell lines restores SMI-4a sensitivity
Since PIK3CA mutation was responsible for primary resistance to PIM1 inhibitors in CRC, we assessed whether PIK3CA inhibitors could overcome SMI-4a resistance in PIK3CA MT CRC cell lines. First, we evaluated the mono therapeutic effects of three PI3K inhibitors, BEZ235, NVP-BKM120, and GDC-0941, in PIK3CA MT DLD-1 human CRC cells. BEZ235 is a dual ATP-competitive PI3K and mTOR inhibitor for p110α/γ/δ/β and mTOR,Citation34 NVP-BKM120 is a selective PI3K inhibitor of p110α/β/δ/γ,Citation35 and GDC-0941 is a potent inhibitor of PI3Kα/δ.Citation36 As shown in , co-treatment of DLD-1 cells with NVP-BKM120 and SMI-4a synergistically increased cell death rates (), compared to the negligible effect of NVP-BKM120 alone (Figure S3a). Similar results were shown with HCT-116, another PIK3CA MT CRC cell line (). However, the synergistic effect of BKM120 and SMI-4a was not shown in PIK3CA WT cell lines, SW480 and SW620 (), showing that co-targeting PIK3CA MT and PIM1 could be a possible therapeutic approach in PIM1-positive, PIK3CA-mutated CRC patient tumors. In addition, the expression levels of p-STAT3, a PIM1 effector molecule, and p-Akt, a PI3K effector molecule, were concomitantly decreased by BKM120 and SMI-4a co-treatment of DLD cells, in a dose-dependent manner, showing that “crosstalk” between the two pathways induced synergistic cell death (). Similar results were shown in other PIK3CA MT cell lines (Figure S3b).
Figure 5. Combinational effect of SMI-4a and PIK3CA inhibitors on CRC cell lines. (a) DLD-1 (PIK3CA MT) cells were treated with the PIK3CA inhibitors BEZ235, BKM120, and GDC-0941 for 72 h, and cell death rates evaluated by trypan blue exclusion. (b) Western blot analysis after BKM120 and/or SMI-4a in DLD-1 cells treated with increasing doses of BKM120 and/or SMI-4a for 48 h and lysed with RIPA buffer. Lysates were subjected to western blot analysis and expression levels of p-Akt, p-STAT3, and STAT3 was anlalyzed. Beta-actin was used as a loading control. (c) HCT116 (PIK3CA MT), DLD-1 (PIK3CA MT), SW480 (PIK3CA WT), and SW620 (PIK3CA WT) CRC cell lines were treated with increasing doses of BKM120 and/or 30 μM SMI-4a for 72 h. Cell death rates were evaluated by trypan blue exclusion assay. (d) DLD-1 (PIK3CA MT) isogenic cell line (353) was treated with 2 μM of BKM120 and/or 30 μM SMI-4a for 72 h. Cell death rates were evaluated by trypan blue exclusion assay. *p < .05, **p < .01, and ***p < .001 indicate significantly different from control group, respectively. Experiments were conducted three times in triplicate.
Taken together, these results indicate that resistance to PIM inhibitors, due to mutations in PIK3CA, can be overcome by combinational treatment with PI3K inhibitors.
Discussion
PIM proteins make up a small serine/threonine kinase family initially identified after Moloney-murine leukemia virus (MMLV) infection,Citation20,Citation30 where the Proviral Integration site for MMLV (PIM) gene locus showed altered expression in early T cell lymphoma.Citation37 Oncogenic functions of PIM kinases have been further shown in many cancer cell types. For example, PIM1 has been associated with IL-6 induction of cancer stemness and the epithelial-to-mesenchymal transition (EMT), in breast cancer,Citation38 and the PIM1 inhibitor, SMI-4a reversed EMT in alveolar epithelial cells to ameliorate bleomycin-induced pulmonary fibrosis.Citation39 In CRC, PIM1 also facilitates the Warburg effect, which shifts energy production to anaerobic glycolysis.Citation40 Analogously, attenuation of PIM1 leads to decreased oncogenicity (i.e., decreased cell proliferation and enhanced cell death) in CRC, triple-negative breast cancer, liver cancer, and ovarian cancer cell lines.Citation41–44 PIM2 is well associated with hematological malignancies and lung cancer,Citation45 and loss of PIM3 by PIM3 shRNA induced cell death, in pancreatic cancer and colon cancer cell lines.Citation46,Citation47 Due to these oncogenic functions, PIM kinases represent potential anticancer targets for drug development.Citation40,Citation48,Citation49
Despite this potential, most PIM inhibitors have failed as human antineoplastic monotherapies.Citation42 For example, phase I clinical trial results of the most advanced pan-PIM inhibitor, AZD1208, showed no clinical response, even at a 900-mg dose, in patients with AML or solid tumors.Citation26 Moreover, a clinical trial of another pan PIM inhibitor, PIM447, was discontinued due to disease progression in most patients.Citation28
Herein, we showed that the PIK3CA genotype can underlie resistance to PIM inhibitors, at least in CRC cells, which showed resistance to SMI-4a (i.e., significantly decreased cell death rates) in PIK3CA MT cell lines, independent of PIM1 expression levels. SMI-4a is modestly potent against PIM2 (IC50 = 100 nM); therefore, these results indicate that the activity of PIM1, but not PIM2, is related to the synergistic effects of SMI-4a and the PIK3CA MT targeting inhibitor BKM120.
The crosstalk between PIM and PI3K signaling pathways and investigation of combination strategies have been reported by many other studies.Citation24–28,Citation33,Citation50 PIM and PI3K signaling pathway both regulate mTOR signaling; therefore, inhibition of either pathway could be compensated by each other. Also, PIM1 is known to increase the expression of ROS scavengers, which prevents the cellular response to PI3K inhibitorsCitation50 in prostate cancer cells. There are also reports that PIM1 could phosphorylate IRS1/2 which results in direct activation of PI3K.Citation51,Citation52 In our study, in concordance with other reports, possible regulation of PI3K activation by PIM was shown in PIK3CA WT CRC cell lines. In , the expression of PI3K was decreased by SMI-4a in SW480 cell line. These results mentioned above support combinational approaches of PIM and PI3K-inhibitors; however, this possibility has not been evaluated in CRC, especially when PIK3CA is mutated.
In PIK3CA MT CRC cells, activity of PI3K, as well as its downstream effectors Akt and p-mTOR, was unaffected by SMI-4a treatment (). This finding is consistent with another report in non-small cell lung cancer showing that SMI-4a antitumor activity depends on PI3K/mTOR/Akt signaling (which would thus be abrogated by activating mutations of PI3K).Citation33 These results indicate that PIM1 inhibition alone cannot exert sufficient anticancer effects to oppose the strong oncogenic signaling of PIK3CA mutations. In accordance with those results, downstream signaling pathways of PIM and PI3K, both leading to cell survival and proliferation, were sustained even after SMI-4a treatment, indicating the drug’s ineffectiveness against such redundant signal transduction. Cell death effects were shown only upon cotreatment with combined treatment of PI3K inhibitor and PIM inhibitor, leading to suppression of their downstream effectors (), showing promising results for combinational therapy in PIK3CA-mutated, PIM-positive CRC patients. We would note, however, that inactivation of PTEN also causes resistance to PI3K inhibition,Citation53 and SIM-4a also inactivates JAK3/STAT2 signaling;Citation54 these relationships also merit consideration. Thus, further studies with CRC and other cancer cells are necessary to further generalize our results.
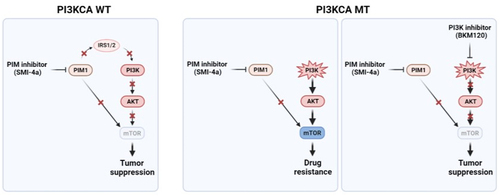
Although many PI3K inhibitors have been developed and entered CRC clinical trials, PI3K inhibitors alone did not show promise due to the development of resistance by activation of MAPK and β-catenin,Citation55,Citation56 consistent with SIM-4a inhibition of upstream effectors of β-catenin nuclear translocation (and its ensuing downstream gene activation).Citation57 Therefore, combinational approaches appear to be the best strategy for PI3K inhibitors, and such studies are ongoing with EGFR and VEGFR inhibitors.Citation58,Citation59 Our results indicate that another combinational strategy, i.e., co-targeting PIM1 and PIK3CA mutation, could be an attractive therapeutic option for recalcitrant CRC patients ().
Figure 6. Schematic illustration of mutant PIK3CA genotype as a negative predictive biomarker of SMI-4a efficacy against PIK3CA mutated CRC.
Metastatic CRC has a dismal prognosis with few therapeutic options. While EGFR inhibitors have shown some efficacy, these are largely limited to patients with wild type KRAS; however, this oncogene is mutated in ~40% of all CRC cases. Consequently, other CRC-oncogenic signal pathways require in-depth study. PIM kinases are well-established oncoproteins in numerous cancer types, including CRC. The efficacy of PIM inhibitors, however, has been limited as monotherapies, being largely limited to CRC tumor expression of wild type but not mutant type (and activated), PIK3CA.
Consequently, we comprehensively showed, both in vitro and in vivo, that response to PIM inhibitors is determined by PIK3CA genotype. PI3KCA mutated CRC cells did not respond well to PIM1 selective inhibitor, SMI-4a, while PIK3CA wild-type CRC cells responded to SMI-4a. The introduction of PIK3CA mutants into PIK3CA WT CRC cells induced resistance to SMI-4a confirming that PIK3CA genotype is important for the response to SMI-4a. This was further confirmed in xenograft and PDX models, demonstrating that PIK3CA mutation could act as a negative predictive biomarker for PIM1 inhibitors. Further studies to define the detailed mechanism of crosstalk between PIM kinase and PI3K signaling pathway in CRCs and with other cancer cells are necessary to further generalize our results. Combinations of PIM and PIK3CA inhibitors might represent a potentially effective approach against metastatic CRC with PIK3CA mutations.
Material and methods
Cell culture and reagents
The human CRC cell lines, COLO 205, COLO 320HSR, DLD-1, HCT 116, HCT-8, HT-29, SW480, SW620, and LoVo were purchased from the Korean Cell Line Bank (KCLB, Seoul, Republic of Korea). The RKO CRC cell line was from American Type Culture Collection (ATCC, Manassas, VA, USA). Cell lines were cultured in RPMI 1640 (Welgene, Daegu, Korea) or DMEM (Welgene) medium containing 10% heat-inactivated fetal bovine serum (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) in a 37°C incubator with a humidified atmosphere of 5% CO2. DLD-1 isogenic cell lines wt PIK3CA (351) and mt PIK3CA (353) were provided by Dr Bert Vogelstein, and cultured in McCoy’s medium (Welgene) supplemented with 10% FBS and penicillin/streptomycin (Welgene).
DNA constructs and transfection
PIKCA plasmids were purchased from OriGene (Rockville, MD, USA). PIK3CA-H1047R- and PIK3CA-E545K-expressing plasmids were generated by Muta-Direct site-directed mutagenesis kit (iNtRON Biotechnology, Seongnam-si, Republic of Korea) and transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) reagent, according to the manufacturer’s instructions.
Trypan blue cell exclusion assay (cell death assay)
Twenty-four hours before drug treatment, 1 × 105 cells/60-mm dishes were plated. After drug treatments for the indicated times, cells were harvested by trypsinization and centrifuged in a swinging bucket rotor at 300 × g for 5 min at 4°C. The pelleted cells were then resuspended in cold PBS and mixed with trypan blue solution (Gibco) at 1:1 ratio. Live and dead cells were counted using a hemocytometer, allowing the calculation of % dead cells out of total cells.
In vitro colony-forming assays
Seeds at a density of 100–200 cells per well in 6-well plates and drugs were added the following day. Cells were cultivated for an additional 16–18 days (until colonies formed), with culture media and drugs replaced every 2–3 days. Cell colonies were then fixed with 4% paraformaldehyde, stained with 0.01% crystal violet (Sigma Aldrich, St Louis, MO, USA) staining solution, and photographed. Numbers of colonies were counted by eye.
Annexin V-FITC/PI staining (apoptosis assay)
Cells were seeded in 60-mm dishes and cultured for 24 h. Test articles were treated at the described drug doses and incubated for 72 h. After 72 h, cells were harvested by trypsinization and centrifuged in a swinging bucket rotor at 300 × g for 5 min at 4°C and stained with propidium iodide (PI) and annexin-V solution (Becton Dickinson (BD), Franklin Lakes, NJ, USA) and analyzed by flow cytometry using a FACSCanto (BD) instrument.
Sample preparation and Western blot analysish
Cells were lysed with RIPA lysis buffer (T&I, Chuncheon, Republic of Korea), with phosphatase and protease inhibitors, followed by centrifugation for 15 min at 4°C. Protein concentrations were determined using a protein assay dye reagent (Bio-Rad, Hercules, CA, USA). About 20–40 µg of total lysate was boiled with a loading buffer (Elpis Biotech, Daejeon, Republic of Korea), separated by SDS-PAGE, and transferred to PVDF membranes for Western blot analysis. Membranes were blocked using 5% skim milk (BD) for 1 h, followed by incubation with the appropriate primary antibodies at 4°C, overnight. Membranes were washed three times with TBST and incubated with the appropriate secondary antibody for 2 h. The membranes were washed 3X with TBST, incubated with ECL solution (GE HealthCare, Chicago, IL, USA), and chemiluminescence detected by exposing the membranes to X-ray film (AGFA, Mortsel, Belgium). The X-ray films were then developed, fixed, and scanned for analysis.
Antibodies for PIM1, PIM2, PIM3, Akt, p-Akt, p-Bad, Erk, p-Erk, p-GSK-3α, PI3K, p-PI3K, p-mTOR, STAT, p-STAT, and secondary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies for XIAP, Nf-κB, cyclin D1, c-Myc, and beta-actin were from Santa Cruz Biotechnology (Dallas, TX, USA).
In vivo xenograft and PDX models
Xenograft tumor growth in BALB/c nude mice was used as a model for in vivo experiments. All animal studies were conducted in compliance with animal protocols approved by the Asan Medical Centre Institutional Animal Care and Use Committee. Xenografted tumors were established by subcutaneous injection of 1 × 107 COLO 205 cells resuspended in 100 µl PBS, or 1 × 107 HT-29 resuspended in 200 µl PBS and Matrigel at a 1:1 ratio. Mice were randomized when tumors reached a volume of approximately 100 mm3 and treated with vehicle control or SMI-4a, via oral gavage, daily. Tumor volumes were measured twice a week by a digital caliper and quantified by the formula: tumor volume = 1/2 (length × width2). For pre-transfer PDX tissue, fresh tumor fragments were transplanted subcutaneously (s.c.) into the right flanks of anaesthetized BALB/c-nude mice. When pre-transferred tumor sizes reached 800–1200 mm3, mice were sacrificed and tumor tissues harvested. For efficacy studies of PDX models, 1–2 mm3 tumor fragments were implanted s.c. into the lateral flanks of female BALB/c nude mice, and tumor volume measured twice a week by a digital caliper and quantified.
statistical analysis
Significance of statistical analysis was analyzed using the Student’s t-test. p < .05 was considered statistically significant.
Author contributions
Y.P., J.K., and D.J. conceived and designed the study. Y.P., J.K., Y.R., and J.M. contributed to setting the experimental lists. Y.P., J.K., Y.S., J.K., S.H., S.J, S.L., S.K., and D.L. contributed to the conduction of in vitro assay. Y.P., J.K., D.K., H.Y., J.Y., D.Y., and C.K. contributed to the conduction of in vivo assay. Y.R., and J.M. contributed to the conduction of related statistical analysis. Y.R., J.M., and D.K. contributed to preparing the figures and tables. Y.P wrote the original draft manuscript. All authors reviewed the manuscript.
Supplemental Material
Download MS Word (15.1 KB)Supplemental Material
Download MS Word (343.3 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data generated can be found within the published article.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15384047.2023.2246208
Additional information
Funding
Notes on contributors
Yoon Sun Park
Yoon Sun Park Graduated in 2010 with a master's degree from Ewha Woman’s University. Her research focuses on discovering and studying the function of novel targets related to cancer.
Joseph kim
Joseph Kim Graduated in 2013 with a master's degree from Handong Global University. His research focuses on discovering and studying the function of novel targets related to cancer.
Yea Seong Ryu
Yea Seong Ryu Graduated in 2015 with a Doctor's degree from Duksung Women’s University. Her research focuses on cell death induced by novel compound or target.
Jai-Hee moon
Jai-Hee Moon Graduated in 2011 with a Doctor's degree from Korea University. Her research focuses on overcoming drug resistance in cancer.
Yu Jin shin
Yu Jin Shin Graduated in 2013 with a Doctor's degree from Humboldt University of Berlin. Her research focuses on discovering and studying the function of novel targets related to cancer.
Jeong Hee kim
Jeong Hee Kim Graduated in 2014 with a Doctor's degree from Yonsei University. Her research focuses on discovering and studying the function of novel targets related to cancer.
Seung-Woo hong
Seung-Woo Hong Graduated in 2009 with a Doctor's degree from Myongji University. His research focuses on overcoming anticancer drug resistance and discovering and studying the function of novel targets related to cancer.
Soo-A jung
Soo-A Jung Graduated in 2017 with a Doctor's degree from University of Ulsan College of Medicine University. Her research focuses on discovering and studying the function of novel targets related to cancer.
Seul lee
Seul Lee Graduated in 2017 with a master's degree from University of Ulsan College of Medicine University. Her research focuses on discovering and studying the function of novel targets related to cancer.
Seung-Mi kim
Seung-Mi Kim Graduated in 2016 with a Doctor's degree from Myongji University. Her research focuses on overcoming anticancer drug resistance.
Dae Hee lee
Dae Hee Lee Graduated in 2017 with a Doctor's degree from Korea Advanced Institute of Science and Technology (KAIST). His research focuses on overcoming anticancer drug resistance and discovering and studying the function of novel targets related to cancer.
Do Yeon kim
Do Yeon Kim Graduated in 2020 with a master's degree from University of Ulsan College of Medicine. Her research focuses on overcoming drug resistance in cancer.
Hyeseon yun
Hyeseon Yun Graduated in 2019 with a master's degree from Chung-ang University. Her research focuses on the anti-tumor effects related to DNA damage targets in cancer.
Ji-Eun you
Ji-Eun You Graduated in 2021 with a master's degree from Konyang University. Her research focuses on functional studies in cancer through soluble form target.
Dong Il yoon
Dong Il Yoon Graduated in 2022 with a master's degree from University of Ulsan College of Medicine. His research focuses on overcoming drug resistance in cancer.
Chul Hee kim
Chul Hee Kim Graduated in 2022 with a master's degree from University of Ulsan College of Medicine. His research focuses on overcoming drug resistance in cancer and efficacy evaluation in animal models.
Dong-In koh
Dong-In Koh Graduated in 2014 with a Doctor's degree from Yonsei University. Her research focuses on inducing cell apoptosis through targets regulated by p53 in cancer.
Dong-Hoon jin
Dong-Hoon Jin Graduated in 2004 with a Doctor's degree from Sogang University. His research focuses on the molecular mechanism of p53 in cell apoptosis and aging, overcoming anticancer drug resistance, and discovering and studying the function of novel targets related to cancer.
References
- Xie Y, Chen Y, Fang J. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduction Targeted Ther. 2020;5(1):1–10. doi:10.1038/s41392-020-0116-z.
- Price TJ, Peeters M, Kim TW, Li J, Cascinu S, Ruff P, Suresh AS, Thomas A, Tjulandin S, Zhang K, et al. Panitumumab versus cetuximab in patients with chemotherapy-refractory wild-type KRAS exon 2 metastatic colorectal cancer (ASPECCT): a randomised, multicentre, open-label, non-inferiority phase 3 study. Lancet Oncol. 2014;15(6):569–579. doi:10.1016/S1470-2045(14)70118-4.
- Heinemann V, von Weikersthal LF, Decker T, Kiani A 4, Vehling-Kaiser U 5, Al-Batran S-E 6, Heintges T, Lerchenmüller C, Kahl C, Seipelt G, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014 Sep;15(10):1065–1075. doi:10.1016/S1470-2045(14)70330-4.
- Brand TM, Iida M, Wheeler D. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol Ther. 2011;11(9):777–792. doi:10.4161/cbt.11.9.15050.
- Cai W-Q, Zeng L-S, Wang L-F, Wang Y-Y, Cheng J-T, Zhang Y, Han Z-W, Zhou Y, Huang S-L, Wang X-W, et al. The latest battles between EGFR monoclonal antibodies and resistant tumor cells. Front Oncol. 2020;10:1249. doi:10.3389/fonc.2020.01249.
- Dienstmann R, Connor K, Byrne AT, Fridman W, Lambrechts D, Sadanandam A, Trusolino L, Prehn J, Tabernero J, Kolch W. Precision therapy in RAS mutant colorectal cancer. Gastroenterology. 2020;158(4):806–811. doi:10.1053/j.gastro.2019.12.051.
- Leto SM, Trusolino L. Primary and acquired resistance to EGFR-targeted therapies in colorectal cancer: impact on future treatment strategies. J Mol Med (Berl). 2014;92(7):709–722. doi:10.1007/s00109-014-1161-2.
- Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, Gaida K, Holt T, Knutson CG, Koppada N, et al. The clinical KRAS (G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575(7781):217–223. doi:10.1038/s41586-019-1694-1.
- El Bali M, Bakkach J, Bennani Mechita M. Colorectal cancer: From genetic landscape to targeted therapy. J Oncol. 2021;2021:1–17. doi:10.1155/2021/9918116.
- Yuan T, Cantley L. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27(41):5497–5510. doi:10.1038/onc.2008.245.
- Kim HR, Kang HN, Yun MR, Ju KY, Choi JW, Jung DM, Pyo KH, Hong MH, Ahn M-J, Sun J-M, et al. Mouse–human co-clinical trials demonstrate superior anti-tumour effects of buparlisib (BKM120) and cetuximab combination in squamous cell carcinoma of head and neck. Br J Cancer. 2020;123(12):1720–1729. doi:10.1038/s41416-020-01074-2.
- Seshacharyulu P, Ponnusamy MP, Haridas D, Jain M, Ganti AK, Batra S. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16(1):15–31. doi:10.1517/14728222.2011.648617.
- Yang Z, Tam K. Combination strategies using EGFR-TKi in NSCLC therapy: learning from the gap between pre-clinical results and clinical outcomes. Int J Biol Sci. 2018;14(2):204. doi:10.7150/ijbs.22955.
- Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7(8):606–619. doi:10.1038/nrg1879.
- Martini M, De Santis MC, Braccini L, Gulluni F, Hirsch EJ. PI3K/AKT signaling pathway and cancer: an updated review. Ann Med. 2014;46(6):372–383. doi:10.3109/07853890.2014.912836.
- Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13(2):140–156. doi:10.1038/nrd4204.
- Janku F. Phosphoinositide 3-kinase (PI3K) pathway inhibitors in solid tumors: From laboratory to patients. Cancer Treat Rev. 2017;59:93–101. doi:10.1016/j.ctrv.2017.07.005.
- Liu S, Knapp S, Ahmed A. The structural basis of PI3K cancer mutations: from mechanism to therapy. Cancer Res. 2014;74(3):641–646. doi:10.1158/0008-5472.CAN-13-2319.
- Kandula M, Chennaboina KK, Ammi Raju Y, Raju S. Phosphatidylinositol 3-kinase (PIK3CA) oncogene mutation analysis and gene expression profiling in primary breast cancer patients. Asian Pac J Cancer Prev. 2013;14(9):5067–5072. doi:10.7314/APJCP.2013.14.9.5067.
- Narlik‐Grassow M, Blanco‐Aparicio C, Carnero A. The PIM family of serine/threonine kinases in cancer. Med Res Rev. 2014;34(1):136–159. doi:10.1002/med.21284.
- Blanco-Aparicio C, Carnero A. Pim kinases in cancer: diagnostic, prognostic and treatment opportunities. Biochem Pharmacol. 2013;85(5):629–643. doi:10.1016/j.bcp.2012.09.018.
- Santio NM, Koskinen P, Biology C. PIM kinases: From survival factors to regulators of cell motility. Int J Biochem Cell Biol. 2017;93:74–85. doi:10.1016/j.biocel.2017.10.016.
- Xu J, Zhang T, Wang T, You L, Zhao Y. PIM kinases: an overview in tumors and recent advances in pancreatic cancer. Future Oncol. 2014;10(5):865–876. doi:10.2217/fon.13.229.
- Moore G, Lightner C, Elbai S, Brady L, Nicholson S, Ryan R, O’Sullivan KE, O’Byrne KJ, Blanco-Aparicio C, Cuffe S, et al. Co-targeting PIM kinase and PI3K/mTOR in NSCLC. Cancers Basel. 2021;13(9):2139. doi:10.3390/cancers13092139.
- Kennedy SP, O’Neill M, Cunningham D, Morris PG, Toomey S, Blanco-Aparicio C, Martinez S, Pastor J, Eustace AJ, Hennessy BT. Preclinical evaluation of a novel triple-acting PIM/PI3K/mTOR inhibitor, IBL-302, in breast cancer. Oncogene. 2020;39(14):3028–3040. doi:10.1038/s41388-020-1202-y.
- Cortes J, Tamura K, DeAngelo DJ, De Bono J, Lorente D, Minden M, Uy GL, Kantarjian H, Chen LS, Gandhi V, et al. Phase I studies of AZD1208, a proviral integration Moloney virus kinase inhibitor in solid and haematological cancers. Br J Cancer. 2018;118(11):1425–1433. doi:10.1038/s41416-018-0082-1.
- Koblish H, Li Y-L, Shin N, Hall L, Wang Q, Wang K, Covington M, Marando C, Bowman K, Boer J, et al. Preclinical characterization of INCB053914, a novel pan-PIM kinase inhibitor, alone and in combination with anticancer agents, in models of hematologic malignancies. PLoS One. 2018;13(6):e0199108. doi:10.1371/journal.pone.0199108.
- Raab MS, Thomas SK, Ocio EM, Guenther A, Goh Y-T, Talpaz M, Hohmann N, Zhao S, Xiang F, Simon C, et al. The first-in-human study of the pan-PIM kinase inhibitor PIM447 in patients with relapsed and/or refractory multiple myeloma. Leukemia. 2019;33(12):2924–2933. doi:10.1038/s41375-019-0482-0.
- Liao Y, Feng Y, Shen J, Gao Y, Cote G, Choy E, Harmon D, Mankin H, Hornicek F, Duan Z. Clinical and biological significance of PIM1 kinase in osteosarcoma. J Orthop Res. 2016;34(7):1185–1194. doi:10.1002/jor.23134.
- Brault L, Gasser C, Bracher F, Huber K, Knapp S, Schwaller J. PIM serine/threonine kinases in the pathogenesis and therapy of hematologic malignancies and solid cancers. Haematologica. 2010;95(6):1004. doi:10.3324/haematol.2009.017079.
- Garcia PD, Langowski JL, Wang Y, Chen M, Castillo J, Fanton C, Ison M, Zavorotinskaya T, Dai Y, Lu J, et al. Pan-PIM kinase inhibition provides a novel therapy for treating hematologic cancers. Clin Cancer Res. 2014;20(7):1834–1845. doi:10.1158/1078-0432.CCR-13-2062.
- Luszczak S, Kumar C, Sathyadevan VK, Simpson BS, Gately KA, Whitaker HC, Heavey S. PIM kinase inhibition: Co-targeted therapeutic approaches in prostate cancer. Signal Transduction Targeted Ther. 2020;5(1):1–10. doi:10.1038/s41392-020-0109-y.
- Jiang W, Chen Y, Song X, Shao Y, Ning Z, Gu WJ. Pim-1 inhibitor SMI-4a suppresses tumor growth in non-small cell lung cancer via PI3K/AKT/mTOR pathway. Onco Targets Ther. 2019;12:3043. doi:10.2147/OTT.S203142.
- Maira S-M, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chene P, De Pover A, Schoemaker K, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7(7):1851–1863. doi:10.1158/1535-7163.MCT-08-0017.
- Burger MT, Pecchi S, Wagman A, Ni Z-J, Knapp M, Hendrickson T, Atallah G, Pfister K, Zhang Y, Bartulis S, et al. Identification of NVP-BKM120 as a potent, selective, orally bioavailable class I PI3 kinase inhibitor for treating cancer. ACS Med Chem Lett. 2011;2(10):774–779. doi:10.1021/ml200156t.
- Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles S, et al. The identification of 2-(1 H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno [3, 2-d] pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem. 2008;51(18):5522–5532. doi:10.1021/jm800295d.
- Cuypers HT, Selten G, Quint W, Zijlstra M, Maandag ER, Boelens W, van Wezenbeek P, Melief C, Berns A. Murine leukemia virus-induced T-cell lymphomagenesis: integration of proviruses in a distinct chromosomal region. Cell. 1984;37(1):141–150. doi:10.1016/0092-8674(84)90309-X.
- Gao X, Liu X, Lu Y, Wang Y, Cao W, Liu X, Hu H, Wang H. PIM1 is responsible for IL-6-induced breast cancer cell EMT and stemness via c-myc activation. Breast Cancer-Tokyo. 2019;26(5):663–671. doi:10.1007/s12282-019-00966-3.
- Zhang X, Zou Y, Liu Y, Cao Y, Zhu J, Zhang J, Chen X, Zhang R, Li J. Inhibition of PIM1 kinase attenuates bleomycin-induced pulmonary fibrosis in mice by modulating the ZEB1/E-cadherin pathway in alveolar epithelial cells. Mol Immunol. 2020;125:15–22. doi:10.1016/j.molimm.2020.06.013.
- Zhang M, Liu T, Sun H, Weng W, Zhang Q, Liu C, Han Y, Sheng W. Pim1 supports human colorectal cancer growth during glucose deprivation by enhancing the Warburg effect. J Cancer Prev. 2018;109(5):1468–1479. doi:10.1111/cas.13562.
- Brasó-Maristany F, Filosto S, Catchpole S, Marlow R, Quist J, Francesch-Domenech E, Plumb DA, Zakka L, Gazinska P, Liccardi G, et al. PIM1 kinase regulates cell death, tumor growth and chemotherapy response in triple-negative breast cancer. Nat Med. 2016;22(11):1303–1313. doi:10.1038/nm.4198.
- Leung CO, Wong CC, Fan DN, Kai AK, Tung EK, Xu IM, Ng IO, Lo RC. PIM1 regulates glycolysis and promotes tumor progression in hepatocellular carcinoma. Oncotarget. 2015;6(13):10880. doi:10.18632/oncotarget.3534.
- Weirauch U, Beckmann N, Thomas M, Grünweller A, Huber K, Bracher F, Hartmann RK, Aigner AJN. Functional role and therapeutic potential of the pim-1 kinase in colon carcinoma. Neoplasia. 2013;15(7):783–IN728. doi:10.1593/neo.13172.
- Wu Y, Deng Y, Zhu J, Duan Y, Weng W, Wu X. Pim1 promotes cell proliferation and regulates glycolysis via interaction with MYC in ovarian cancer. Onco Targets Ther. 2018;11:6647. doi:10.2147/OTT.S180520.
- Liu Z, Liu H, Yuan X, Wang Y, Li L, Wang G, Song J, Shao Z, Fu R. Downregulation of Pim-2 induces cell cycle arrest in the G0/G1 phase via the p53-non-dependent p21 signaling pathway. Oncol Lett. 2018;15(4):4079–4086. doi:10.3892/ol.2018.7865.
- Li Y-Y, Popivanova BK, Nagai Y, Ishikura H, Fujii C, Mukaida N. Pim-3, a proto-oncogene with serine/threonine kinase activity, is aberrantly expressed in human pancreatic cancer and phosphorylates bad to block bad-mediated apoptosis in human pancreatic cancer cell lines. Cancer Res. 2006;66(13):6741–6747. doi:10.1158/0008-5472.CAN-05-4272.
- Popivanova BK, Li YY, Zheng H, Omura K, Fujii C, Tsuneyama K, Mukaida N. Proto‐oncogene, Pim‐3 with serine/threonine kinase activity, is aberrantly expressed in human colon cancer cells and can prevent Bad‐mediated apoptosis. Cancer Science. 2007;98(3):321–328. doi:10.1111/j.1349-7006.2007.00390.x.
- Le BT, Kumarasiri M, Adams JR, Yu M, Milne R, Sykes MJ, Wang S. Targeting Pim kinases for cancer treatment: opportunities and challenges. Future Med Chem. 2015;7(1):35–53. doi:10.4155/fmc.14.145.
- Tursynbay Y, Zhang J, Li Z, Tokay T, Zhumadilov Z, Wu D, Xie Y. Pim-1 kinase as cancer drug target: An update. Biomed Rep. 2016;4(2):140–146. doi:10.3892/br.2015.561.
- Song JH, Singh N, Luevano LA, Padi SK, Okumura K, Olive V, Black SM, Warfel NA, Goodrich DW, Kraft AS. Mechanisms behind resistance to PI3K inhibitor treatment induced by the PIM kinase. Mol Cancer Ther. 2018;17(12):2710–2721. doi:10.1158/1535-7163.MCT-18-0374.
- Aziz AUR, Farid S, Qin K, Wang H, Liu B. PIM kinases and their Relevance to the PI3K/AKT/mTOR Pathway in the Regulation of ovarian cancer. Biomolecules. 2018 Feb 4;8(1):E7. doi:10.3390/biom8010007.
- Ciardiello F, Ciardiello D, Martini G, Napolitano S, Tabernero J, Cervantes A. Clinical management of metastatic colorectal cancer in the era of precision medicine. CA Cancer J Clin. 2022;72(4):372–401. doi:10.3322/caac.21728.
- Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011;11(4):289–301. doi:10.1038/nrc3037.
- Kuang X, Xiong J, Wang W, Li X, Lu T, Fang Q, Wang J. PIM inhibitor SMI-4a induces cell apoptosis in B-cell acute lymphocytic leukemia cells via the HO-1-mediated JAK2/STAT3 pathway. Life Sci. 2019;219:248–256. doi:10.1016/j.lfs.2019.01.022.
- Burris H. Overcoming acquired resistance to anticancer therapy: focus on the PI3K/AKT/mTOR pathway. Cancer Chemother Pharmacol. 2013;71(4):829–842. doi:10.1007/s00280-012-2043-3.
- Yang J, Nie J, Ma X, Wei Y, Peng Y, Wei X. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol Cancer Ther. 2019;18(1):1–28. doi:10.1186/s12943-019-0954-x.
- Fan RF, Lu Y, Fang ZG, Guo XY, Chen YX, Xu YC, Lei YM, Liu KF, Lin DJ, Liu LL. PIM-1 kinase inhibitor SMI-4a exerts antitumor effects in chronic myeloid leukemia cells by enhancing the activity of glycogen synthase kinase 3β. Mol Med Rep. 2017;16(4):4603–4612. doi:10.3892/mmr.2017.7215.
- He Y, Sun MM, Zhang GG, Yang J, Chen KS, Xu WW, Li B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduction Targeted Ther. 2021;6(1):1–17. doi:10.1038/s41392-021-00828-5.
- Vanhaesebroeck B, Perry MW, Brown JR, André F, Okkenhaug K. PI3K inhibitors are finally coming of age. Nat Rev Drug Discov. 2021;20(10):741–769. doi:10.1038/s41573-021-00209-1.