
ABSTRACT
Pancreatic ductal adenocarcinoma (PDAC) is a type of cancer with high morbidity and mortality rates worldwide. Owing to a lack of therapeutic options, the overall survival rate of patients with pancreatic cancer is low. Gemcitabine has been mainly used to treat patients with pancreatic cancer, but its efficacy is limited by chemoresistance. Therefore, a novel therapeutic agent for PDAC therapy is urgently needed. An anthelminthic drug, niclosamide, has already been researched in breast, lung, colon, and pancreatic cancer as an anti-cancer purpose by re-positioning its original purpose. However, combination therapy of gemcitabine and niclosamide was not informed yet. Here, we found that niclosamide co-administered with gemcitabine significantly inhibited tumorigenesis of pancreatic cancer compared to gemcitabine alone. Further, combining niclosamide and gemcitabine inhibited cell proliferation and induced apoptosis. Niclosamide induced cell cycle arrest at the G1 phase, and the levels of CDK4/6 and cyclin D1 were lowered after gemcitabine treatment. In addition, the combination of these chemical compounds more effectively increased the binding level of activated β-catenin destruction complex and β-catenin to enable phosphorylation, compared to gemcitabine alone. After phosphorylation, niclosamide – gemcitabine upregulated the ubiquitin level, which caused phosphorylated β-catenin to undergo proteasomal degradation; the combination was more potent than gemcitabine alone. Finally, the combination more effectively suppressed tumor growth in vivo, compared to gemcitabine alone. Altogether, our results indicate that niclosamide synergistically enhances the antitumor effect of gemcitabine in pancreatic cancer, by inducing the degradation of β-catenin with ubiquitination. Therefore, this drug combination can potentially be used in PDAC therapy.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is an insidious type of cancer, which is commonly diagnosed at stage IV.Citation1 Even if PDAC is diagnosed early, complete elimination cannot be achieved due to its high rate of recurrence and chemoresistance.Citation2 Currently, gemcitabine is a standard chemotherapeutic drug used to treat pancreatic cancer.Citation3 However, treatment with gemcitabine alone is less effective against PDAC.Citation4 Therefore, exploring and improving novel therapeutic agents for PDAC therapy are of critical importance.
The Wnt/β-catenin signaling pathway is a key signaling pathway related to tumorigenesis.Citation5 A previous study revealed that the aberrant activation of Wnt/β-catenin signaling is often observed in pancreatic cancer cells.Citation6 Inhibition of Wnt ligands inactivates Frizzled (FZL) receptors, Disheveled (DVL) receptors, and LDL receptor-related protein (LRP) 5/6 complexes at the cell surface,Citation7 which leads to the activation of the β-catenin destruction complex comprising Adenomatous polyposis coli (APC), Glycogen synthase kinase-3β (GSK-3β), Axin, and Casein kinase 1 (CK1).Citation8 The destruction complex phosphorylates cytoplasmic β-catenin at residues Ser33, Ser37, and Thr41 for recognition by E3 ubiquitin ligase beta-transducin repeat-containing proteins (β-TrCP) to be ubiquitinated for the proteasomal degradation pathway.Citation9 Therefore, the Wnt/β-catenin signaling pathway may be a therapeutic target for regulating PDAC pathogenesis.
Niclosamide is an FDA-approved anthelminthic drug that blocks oxidative phosphorylation and triggers adenosine triphosphatase activity in the mitochondria of tapeworm to kill its head and body.Citation10 In the last 5 years, niclosamide has been acknowledged as a potential anti-cancer candidate by various high-throughput screening (HTS).Citation11 Previous studies found that niclosamide targets Wnt/β-catenin for its inhibition.Citation12–18 Moreover, niclosamide has been proven to be anti-tumorigenic in various types of cancers, such as breast, lung, and colon.Citation19 Previously, niclosamide was demonstrated to promote Wnt co-receptor LRP6 degradation with no effect on Dvl2 expression to inhibit the Wnt signaling pathway in prostate and breast cancer.Citation14 Another study also revealed that niclosamide promotes protein ubiquitination and correlates with poly ADP ribose polymerase (PARP) expression to prove its apoptotic effect in the glioblastoma U-87 MG cell line.Citation12 Although niclosamide is a known anti-cancer drug whether it plays a role in the ubiquitination level of Wnt/β-catenin signaling in pancreatic cancer remains unclear. Further, the effect of co-administering niclosamide and gemcitabine on the ubiquitination level of the Wnt/β-catenin pathway in pancreatic cancer has not been elucidated.
In this study, we sought to verify whether niclosamide can synergistically improve the anti-proliferative effect of gemcitabine in vitro and in vivo. Here, we found that co-treatment with niclosamide and gemcitabine mainly induced cell cycle arrest at the G1 phase compared to gemcitabine alone. Further, niclosamide and gemcitabine exhibited a synergistic effect on the level of apoptosis in pancreatic cancer cells. Based on further analysis, we determined that niclosamide – gemcitabine suppressed cell proliferation and manipulated the ubiquitin level to guide phosphorylated β-catenin to a proteasomal degradation pathway in pancreatic cancer. Further, co-treatment with the drugs more effectively induced binding between β-catenin and β-catenin destruction complex (Axin1, GSK-3β, and β-TrCP), compared to gemcitabine alone, leading to β-catenin phosphorylation. Herein, a xenograft model was established to determine whether niclosamide can enhance the anti-proliferative effect of gemcitabine on tumor growth in vivo. Niclosamide significantly improved the inhibitory effect of gemcitabine as a novel therapeutic agent for PDAC therapy.
Materials and methods
Cell lines and cell culture
Human pancreatic cancer cell lines (MIA PaCa-2, PANC-1, AsPC-1, and BxPC-3) and the kidney of human embryo (HEK-293A) were purchased from the American Type Culture Collection (ATCC, Manassas, VA). MIA PaCa-2, PANC-1, and HEK-293A cells exhibiting epithelial morphology were maintained as cultured in DMEM supplemented with 10% FBS (Biowest, MO, USA) and 1% antibiotic – antimycotic reagent (Gibco, MA, USA) at 37°C with 5% CO2. AsPC-1 and BxPC-3 cells exhibiting epithelial morphology were maintained as cultured in RPMI supplemented with 10% FBS (Biowest, MO, USA) and 1% antibiotic – antimycotic reagent (Gibco, MA, USA) at 37°C with 5% CO2.
Cell viability and proliferation assay
A water-soluble tetrazolium salt (WST) assay was used to assess cell viability. Cells were seeded into a 96-well plate at a density of 3 × 10Citation3 cells per well and incubated overnight. Thereafter, the cells were treated with various concentrations of gemcitabine (Yuhan, Seoul, Korea), niclosamide (Cayman Chemical Company, MI, USA), and Control (DMSO) for 72 h. A stock solution for niclosamide was prepared by dissolving the ethanolamine salt in DMSO as an organic solvent for indicated concentration. Growth medium was replaced with 10% WST-1 reagent (DoGenBio, Seoul, Korea) and incubated at 37°C with 5% CO2 for 1 h. The data are expressed as optical density (OD450) measurements and were obtained using a VersaMax microplate reader (Molecular Devices, CA, USA) with a 450-nm filter.
Patient-derived organoids and drug testing
Human pancreatic tissues were obtained from patients diagnosed with pancreatic cancer at the Gangnam Severance Hospital from 2018 to 2019. Written informed consent was obtained from each patient and the study was approved by the institutional review board (3–2018–0241). Briefly, tissues were sliced into 5–10 mm pieces, washed with advanced DMEM/F12 (Gibco) supplemented with 1% penicillin – streptomycin (Welgene, Gyeongsan, Korea), and then enzymatically digested with advanced DMEM/F12 supplemented with 0.125 mg/mL dispase II (Wako, VA, USA), 0.1 mg/mL DNase I (Millipore, MA, USA), 0.125 mg/ml collagenase II (Gibco), and 1% penicillin – streptomycin for 1 h at 37°C with shaking (150 rpm). After digestion, the supernatant was filtered through a 70-μm cell strainer (SPL, Gyeonggi-do, Korea) and pelleted by centrifugation at 200 × g for 5 min at RT. The pellet was resuspended in culture media and mixed with Matrigel (Corning, NY, USA) at a ratio of 1:1 (v:v), seeded in a 48-well plate at a density of 1 × 10Citation4 per well, and incubated with 5% CO2 at 37°C for 10 min to polymerize the matrices. After polymerization, the matrices were treated with 0.5 and 1 μM niclosamide, and control (DMSO) for 72 h.
Cell cycle
MIA PaCa-2 cells were incubated with gemcitabine, niclosamide, and control (DMSO) for 24 h, harvested using trypsin-EDTA (0.25%) with phenol red (Gibco) and washed twice in Dulbecco’s phosphate-buffered saline (Biowest). Cells were fixed at −20°C for 15 min, washed twice with cold DPBS, and then incubated with propidium iodide (PI) dye with RNase A (Sigma-Aldrich) for 30 min. Samples were analyzed on a FACScanto II flow cytometer (BD Biosciences) with a 488-nm ion laser. A minimum of 10,000 events were recorded for each sample. Cell cycle analysis of DNA histograms was performed using the FlowJo software (BD Biosciences).
Apoptosis
MIA PaCa-2 cells were incubated with the assigned drugs for 48 h, harvested by using Trypsin-EDTA (0.25%) with phenol red (Gibco) and washed twice in Dulbecco’s phosphate-buffered saline (Biowest). The cells were incubated with 10% binding buffer, propidium iodide (PI), and FITC (BD Biosciences) for 15 min. The samples were then analyzed for phosphatidylserine exposure by annexin-V FITC/propidium iodide double-staining on a FACScanto II flow cytometer (BD Biosciences) with a 488-nm ion laser. A minimum of 10,000 events were recorded for each sample.
RT-PCR
The cells were treated with the assigned drug compounds for 24 h. Thereafter, the cells were harvested using TRIZOL Reagent (Sigma-Aldrich). A total of 0.4 μg of total isolated RNA was analyzed via reverse transcription PCR using the Maxime™ RT-PCR premix kit (Intron, Gyeonggi-do, Korea).
Western blot analysis
The cells treated with the assigned compounds were washed with DPBS, collected, and lysed in RIPA buffer. The cell lysates were then separated on SDS polyacrylamide gels and transferred to polyvinylidene fluoride membranes (Merck Millipore, MA, USA). After blocking nonspecific binding with TBS-T (0.1% Tween) containing 5% skim milk for 1 h at room temperature, the membranes were immunoblotted with primary antibodies at 4°C overnight. Subsequently, the membranes were incubated with HRP-conjugated goat anti-rabbit secondary antibody or goat anti-mouse secondary antibody for 1 h at room temperature. The protein bands were exposed to an enhanced chemiluminescent (ECL) horseradish peroxidase (HRP) substrate (Thermo Fisher Scientific, USA) and detected on X-ray films. Primary antibodies against anti-Wnt5a (# 174963) were purchased from Abcam (Cambridge, UK). Anti-cleaved/pro-caspase 9 (# 56076), anti-cleaved/pro-caspase 3 (# 9662/9661), anti-ubiquitin (# 8017), anti-c-Myc (# 40), anti-Bcl2 (# 509), anti-β-actin (# 47778), anti-p21 (# 6246), anti-CDK4 (# 601), anti-CDK6 (# 7961), and anti-CDK2 (# 163) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Anti-Rb (# 9309), anti-Phospho-Rb (# 9307), anti-Axin1 (# 2087T), anti-Phospho-β-catenin (S33/37/T41) (# 9561S), anti-β-catenin (# 9582S), anti-β-TrCP (#4394S), anti-Cyclin D1 (# 2922S), anti-Phospho-Akt (T308) (# 9275S), anti-Akt (# 9272S), anti-Bax (# 2772T), anti-Phospho-JAK2 (# 3771), anti-JAK2 (# 3230), anti-Phospho-STAT3 (# 9145), anti-STAT3 (# 9139), anti-Phospho-GSK3β (# 9323), and anti-GSK3β (# 9315) were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-γ-tubulin (# T6557) was purchased from Sigma-Aldrich (St. Louis, MO, USA). HRP-conjugated goat anti-mouse secondary (# 7076S) and HRP-conjugated goat anti-rabbit secondary (# 7074S) antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA).
Immunoprecipitation assay
After treatment, the cells were washed once with Dulbecco’s phosphate-buffered saline and lysed for 30 min on ice. The cell lysates were incubated with the indicated antibodies overnight at 4°C. After incubation, magnetic A/G beads (Burlington, MA, USA) were precleared with Dulbecco’s phosphate-buffered saline (Biowest). The A/G beads were incubated with antigen-antibody complexes for 30 min. The protein A/G beads were washed three times with DPBS and the complexes were boiled with denaturing elution buffer (2X Laemmli sample buffer, β-mercaptoethanol, and Dulbecco’s phosphate-buffered saline) at 95°C for 5 min. The IP samples were subjected to SDS-PAGE followed by western blotting using the indicated antibodies.
Immunofluorescence assay
Tissue sections were deparaffinized in xylene for 5 min and rehydrated using ethanol. Antigen retrieval was performed by boiling in preheated buffer 10 mM citrate buffer pH 7.6 for 10 min at 200 W in a microwave. Next, slides were blocked by 10% goat serum in phosphate-buffered saline tween pH 7.4 for 1 h at room temperature. Primary antibodies rabbit monoclonal anti-phospho-β-catenin (S33/37/T41), anti-β-catenin, anti-GSK3β, anti-β-TrCP (1:100, Cell Signaling Technology, Danvers, MA, USA) were added and incubated at 4°C overnight. Texas Red goat anti-rabbit IgG (H+L) (Molecular Probes, Eugene, Oregon, USA) was used as secondary antibody. Pictures were taken using the ZEISS LSM 980 with Airyscan 2.
Xenograft tumor model
Five-week-old male BALB/c mice were purchased from the Model Animal Research Center of Yonsei University (Seoul, Korea). For the tumor xenograft assay, 4 × 10Citation6 PANC-1 cells were suspended in 100 μL of DPBS and subcutaneously injected into the left flank of mice. When the tumor volume reached approximately 100 mm3, mice were randomly divided into six groups. Gemcitabine (10 mg/kg), niclosamide (10 mg/kg), and control (DMSO) were intraperitoneally administered twice per week. After four weeks, all mice were euthanized. Tumors in each group were collected and fixed in 4% paraformaldehyde. All animal experimental procedures followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) of Seoul Yonsei Pharmaceutical University Experimental Animal Center.
Statistical analysis
Statistical analysis was performed with an unpaired t-test using GraphPad Prism version 8.0 (GraphPad Software, CA, USA). Data are presented as mean ± SD. Statistical significance was set at p < .05; a p value ≤ 0.01 was considered significant and indicated as **.
Results
Niclosamide enhances the inhibitory effect of gemcitabine on cell proliferation
To determine whether gemcitabine and niclosamide could effectively inhibit cancer cell proliferation, we administered both drugs to the PDAC cells, PANC-1 and MIA PaCa-2, AsPC BxPC3 HEK 293aat increasing dosages in the WST-1 assay. To determine the drug treatment concentrations of both chemical compounds, the IC50 value was first calculated by verifying both niclosamide and gemcitabine treatment in PANC-1 and MIA PaCa-2 cells (). After determining the drug concentration via IC50 analysis, we proceeded to determine whether niclosamide alone could hinder the cell proliferative effect at the organoid scale. The total effectiveness of niclosamide was calculated by measuring the spreading area of each organoid. Niclosamide significantly suppressed organoid proliferation compared to levels of the control ()
Figure 1. Niclosamide enhances the inhibitory effect of gemcitabine on cell proliferation.
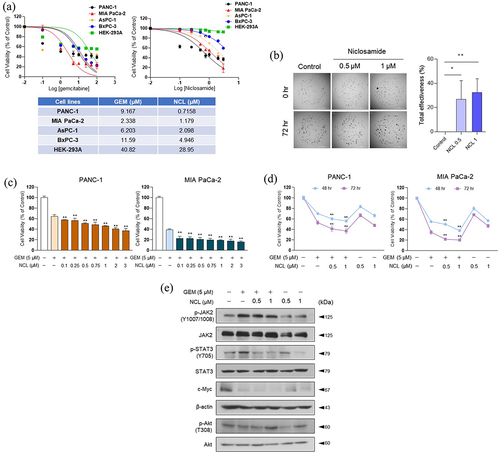
After examining the anti-proliferative effect of niclosamide in the organoid scale, we treated niclosamide on PANC-1 and MIA PaCa-2 cells to determine its regulatory effect on cell proliferation via an increase in its concentration with a fixed dose of gemcitabine (5 μM). Co-treatment with the two drugs led to a significantly enhanced inhibitory effect on cell proliferation compared to treatment with gemcitabine alone (). To determine the anti-proliferative effect of niclosamide on cell proliferation, PANC-1 and MIA PaCa-2 cells were treated with niclosamide with or without 5 μM gemcitabine and the drug-exposure time was varied. As the exposure time increased, niclosamide – gemcitabine significantly lowered the viability of PANC-1 and MIA PaCa-2 cells ().
As the co-administration of niclosamide – gemcitabine markedly induced cell cytotoxicity of pancreatic cancer cells, we investigated genes affect cell proliferation via the JAK-STAT signaling pathway in MIA PaCa-2 cells (). In a prior study, niclosamide was not found to affect JAK2, but could inhibit STAT3.Citation20 By confirming the phosphorylation level of STAT3, gemcitabine alone was not found to effectively lower the protein expression. However, when gemcitabine was combined with niclosamide, its anti-proliferative effect was enhanced to inhibit the phosphorylation of STAT3 at the Tyr705 residue (). Moreover, c-Myc, a downstream target of STAT3, was markedly weakened by niclosamide – gemcitabine. Therefore, niclosamide enhanced the inhibitory effect of gemcitabine via the JAK-STAT proliferation pathway.
Such findings suggest that niclosamide may exhibit significantly enhanced anti-proliferative effect on cell growth when co-treated with gemcitabine at both the cell and organoid levels via examining JAK-STAT proliferation signaling pathway.
Niclosamide induces cell cycle arrest at the G1 phase
As niclosamide enhanced the inhibitory effect of gemcitabine on cell proliferation, we assessed their ability of cell cycle modulation using various concentrations of gemcitabine and niclosamide. Based on FACS analysis, both niclosamide and gemcitabine only induced G1 and S-phase arrest, respectively. However, co-administering the two drugs caused a significantly higher percentage of arrest at the G1 -phase than gemcitabine and niclosamide alone (). Therefore, both drugs synergistically elevated early phase arrest in the cell cycle to prevent cell proliferation.
Figure 2. Niclosamide induces cell cycle arrest at the G1 -phase.
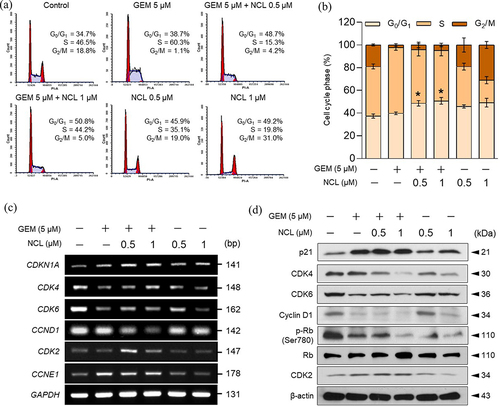
We assessed the genes involved in the G1 to S-phase transition at the mRNA and protein levels to verify the role of co-treatment with niclosamide – gemcitabine (). A previous study proved that the activation of CDK4/6 complexes with cyclin D1 binding initiates entry into the G1 -phase of the cell cycle.Citation21 After passing through the G1 phase, cyclin D1 phosphorylates Rb (retinoblastoma protein) to activate CDK2 with cyclin E1 binding to activate the S phase.Citation22 Interestingly, the protein level of CDK4 was markedly reduced by the combination of the two drugs (). In addition, both the mRNA and protein expression levels of CDK6 and cyclin D1 were strongly reduced by treatment with niclosamide and gemcitabine (). As niclosamide – gemcitabine reduced the level of CDK4/6-cyclin D1 complexes, the phosphorylation level of Rb was also reduced. Further, the level of p21 was markedly increased to enhance the formation of the inactive trimeric complexes of CDK4/6-cyclin D1.Citation21 However, the level of the S-phase entry-related genes, CDK2 and CCNE1, revealed that niclosamide had no significant effect on their mRNA and protein expression.
Altogether, niclosamide – gemcitabine induced cell cycle arrest at the G1 -phase by inhibiting the formation of CDK-cyclin complexes and the phosphorylation of Rb in the G1 -phase by inducing the escalation of p21 levels in pancreatic cancer cells.
Niclosamide increases the level of apoptosis
As both niclosamide and gemcitabine induced cell cycle arrest at the G1 -phase (), the combination of the two compounds was expected to induce apoptosis of PDAC cells. To determine whether the compounds synergistically induced apoptosis, MIA PaCa-2 cells were treated with gemcitabine and niclosamide. Based on the results, the percentage of both early apoptosis and late apoptosis was significantly increased by utilizing a dual staining system of FITC and PI via FACS analysis ().
Figure 3. Niclosamide increases the level of apoptosis.
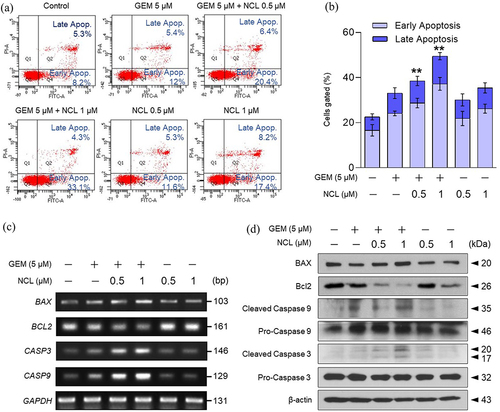
Treatment with gemcitabine and niclosamide had a significantly higher apoptotic effect than treatment with each drug alone (). To verify the effects of the chemical compounds at both the mRNA and protein levels, apoptotic genes were confirmed via RT-PCR and western blotting. Apoptosis-inducing genes were increased at both the mRNA and protein levels. In particular, co-treatment with niclosamide and gemcitabine triggered the expression of apoptosis-inducing genes (Bax, CASP9, and CASP3). In addition, co-administration of the two drugs mainly blunted the expression of Bcl-2, an apoptosis-inhibiting gene, at the mRNA and protein levels (, d). Consequently, niclosamide promoted the induction of apoptosis by gemcitabine in pancreatic cancer cells.
Niclosamide decreases cell proliferation through the Wnt/β-catenin signaling pathway
Wnt/β-catenin signaling pathway is a signaling pathway that contributes to tumorigenesis.Citation5 As niclosamide enhanced the anti-proliferative effect of gemcitabine, downstream targets of β-catenin were assessed at the mRNA expression level (). Niclosamide – gemcitabine showed the highest inhibitory effect on most of the Wnt/β-catenin signaling genes in MIA PaCa-2 cells.
Figure 4. Niclosamide decreases cell proliferation and inhibits linear ubiquitination of β-catenin.
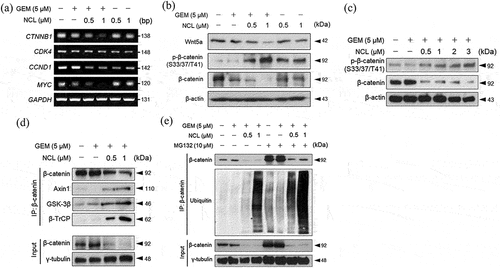
We proceeded to verify the protein level of Wnt5a to determine whether the drugs synergistically inhibit Wnt signaling when administered to MIA PaCa-2 cells. The co-administration of the drugs downregulated the protein levels of Wnt5a. In addition, as niclosamide promoted the intensity of induced phosphorylation of β-catenin at Ser33/37/Thr41, the level of β-catenin was simultaneously lowered ().
To determine whether increasing the dosage of niclosamide would have a negative impact on the cytoplasmic level of β-catenin, niclosamide was administered to MIA PaCa-2 cells at increasing concentrations with 5 μM gemcitabine (). As expected, increasing the dosage of niclosamide significantly induced the phosphorylation of β-catenin and lowered the level of β-catenin based on verification of the expression of each protein. Therefore, niclosamide was shown to affect the level of phosphorylated β-catenin.
As niclosamide increased the level of phosphorylated β-catenin, it also triggered the binding of endogenous Axin1, GSK-3β, and β-TrCP on phosphorylated β-catenin by co-immunoprecipitation (). β-TrCP (E3-ubiquitin ligase) is known to bind to phosphorylated β-catenin to induce ubiquitination.Citation7 Therefore, verifying the ability of niclosamide to induce the level of β-catenin-ubiquitin binding for the proteasomal degradation pathway proved important. To determine whether niclosamide enhances the level of ubiquitination, β-catenin was immunoprecipitated to confirm its combination with ubiquitin by increasing the dosage of niclosamide (). When niclosamide was administered to MIA PaCa-2 cells in the presence of MG132, niclosamide was identified to enhance the anti-proliferative effect on PDAC cells by inducing ubiquitination.
Overall, treatment with niclosamide and gemcitabine markedly promoted the binding of the Wnt/β-catenin destruction complex with β-catenin to be led by ubiquitin to the proteasomal degradation pathway.
Niclosamide significantly inhibits tumorigenesis in vivo
To determine whether co-treatment with niclosamide and gemcitabine weakens the proliferation of PDAC tumors, PANC-1 cells were xenografted in BALB/c nude mouse models to generate PDAC tumors for four weeks in vivo (). After the tumor volume reached 100 mm3, the drugs were intraperitoneally administered twice per week (); body weight and tumor size were recorded after intraperitoneal injection.
Figure 5. Niclosamide inhibits pancreatic cancer growth in vivo.
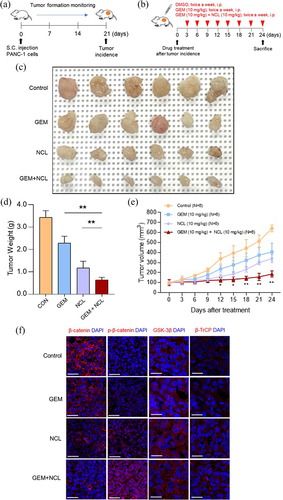
Tumor formation was more observably receded when both gemcitabine and niclosamide were administered (). Moreover, as tumor growth was suppressed in the xenograft model bearing PANC-1 cells, tumor weight was significantly lower in the co-treatment group than in the other two groups (). Although tumor growth was expanded in vivo, all three groups had no significant difference in body weight (data not shown). In addition, to compare the expression of phospho-β-catenin (S33/37/T41), β-catenin, GSK3β, and β-TrCP for each group in tumors, all the four different groups of tumors were collected to analyze via immunofluorescence assay (). The co-injected group showed the most upregulated level for both phosphorylated β-catenin and β-TrCP, but downregulated in β-catenin than any other groups. As expected, no significant difference was observed in GSK-3β as one of β-catenin destruction complex. Therefore, by comparing the different treatments, niclosamide was found to enhance the anti-tumor capacity of gemcitabine in vivo.
Discussion
As the 5-year survival rate of pancreatic cancer patients is approximately 10%,Citation23 the discovery of therapeutic drugs to cure pancreatic cancer patients is urgently needed. Although a previous study revealed that gemcitabine cannot effectively kill cancer due to chemoresistance,Citation24 this drug had to be administered as the primary treatment for patients with unresectable locally advanced pancreatic adenocarcinoma.Citation25 To improve its anti-tumor effect, niclosamide, a drug approved by FDA and used for tapeworm infection, was repurposed as a cancer therapeutic drug candidate.Citation26
Of note, the effects of niclosamide on other types of cancer cells have already been studied via various signaling pathways. A previous study showed that niclosamide has an ability to inhibit β-catenin/TCF (T-cell factor) complex formation and interrupt downstream target gene transcription in colorectal cancer for regulating tumorigenesis and metastasis.Citation27 Also, another study found out that STAT3 is one of the main factors that mediate cancer cell survival by activating anti-apoptotic genes such as Bcl-2 and Bcl-XL in human head and neck cancer and non-small cell lung cancer.Citation28,Citation29 And, it turned out that niclosamide can block STAT3 phosphorylation and nuclear translocation in prostate cancer to halt cell proliferation and apoptosis.Citation20 Moreover, niclosamide triggers mitochondrial fission by causing mitochondrial fragmentation to induce apoptotic cell death.Citation30 There are some more studies that niclosamide suppresses the cell viability in osteosarcoma, head and neck squamous sarcoma, and thyroid cancer cells.Citation31–33
Therefore, we questioned that niclosamide may assist anticarcinogenic effect of gemcitabine in pancreatic cancer. Here, we found that the combination of niclosamide and gemcitabine had more significant antiproliferative effect than each of the single drug in pancreatic cancer.
To confirm the anti-proliferative effects of niclosamide on pancreatic cancer, we administered niclosamide with or without gemcitabine to PDAC cells. With increasing dosage of niclosamide and gemcitabine, cell viability was significantly lowered in both time- and concentration-dependent manners in PANC-1 and MIA PaCa-2 cells in vitro.
Niclosamide has been shown to effectively induce cell cycle arrest and apoptosis in other cancer cell types, such as hepatocellular carcinoma cells and thyroid cancer cells.Citation33,Citation34 Herein, by administering niclosamide to PDAC cells, we found that niclosamide significantly induced cell cycle arrest in the G1 phase and apoptosis. Moreover, its effect was found to be markedly improved when gemcitabine was co-administered to cancer cells. Therefore, we proved that niclosamide improves the effect of gemcitabine by blocking the cell cycle and inducing apoptosis in PDAC cells.
Abnormal activation of the Wnt/β-catenin signaling pathway contributes to tumorigenesis in pancreatic cancer.Citation5 We confirmed that both mRNA and protein levels of Wnt/β-catenin related genes were restrained by the combination of niclosamide and gemcitabine. Intriguingly, the phosphorylation of β-catenin was induced by niclosamide, while the protein level of β-catenin was concurrently lowered (). Therefore, we assumed that a change in the level of β-catenin phosphorylation could be a signal for ubiquitination. As the destruction complex of β-catenin induces the phosphorylation of β-catenin, β-TrCP may detect increased levels of phosphorylated β-catenin. Hence, niclosamide triggered an upregulation of the level of ubiquitin to be bound to the destabilized β-catenin, leading to proteasomal degradation. As niclosamide affected the proteasomal degradation of β-catenin in vitro, the anti-proliferation capacity of niclosamide was also verified in vivo by suppressing the proliferation of pancreatic tumor cells in the xenograft models. Our data support the hypothesis that niclosamide could improve the antiproliferative effect of gemcitabine, leading to the conversion from growth arrest to cell death. Further exploration of this combination therapy should be performed in a clinical trial.
In relation to ubiquitination, Wang et al. demonstrated that a genotoxic effect from chemotherapy could inhibit the phosphorylation of c-Abl2 and OTULIN (OTU Deubiquitinase with Linear Linkage Specificity) to encourage linear ubiquitination of β-catenin regardless of the FZD/LRP heterodimeric receptors and Wnt ligands in breast cancer.Citation35 Phosphorylated OTULIN is known to be bound to the ubiquitin ligase complex, LUBAC (Linear ubiquitin chain assembly complex: SHARPIN, HOIL-IL, and HOIP), which hinders the ubiquitination of β-catenin to stabilize the Wnt/β-catenin signaling pathway to promote tumorigenesis.Citation35 In our study, we proved that both gemcitabine and niclosamide enhance the activated levels of β-catenin destruction complex and ubiquitin to induce anti-tumorigenesis. Moreover, co-treatment with niclosamide – gemcitabine could be utilized as a chemotherapeutic therapy that triggers the genotoxic stress mentioned by Wang et al. Therefore, it would be intriguing to explore whether both niclosamide – gemcitabine and inhibition of OTULIN phosphorylation could synergistically strengthen the level of β-catenin ubiquitination with the induced genotoxic stress from niclosamide – gemcitabine.
In conclusion, niclosamide – gemcitabine significantly inhibited the viability of pancreatic cancer cells at physiologically relevant concentrations. Further, higher levels of cell cycle arrest at the G1 -phase and apoptosis were confirmed in the co-treatment group compared to the gemcitabine only group. Altogether, we verified the anti-proliferative effect of niclosamide – gemcitabine in both the JAK/STAT and Wnt/β-catenin signaling pathways. The degradation of β-catenin with ubiquitination facilitated the anti-proliferative effect of niclosamide – gemcitabine in vivo.
Author contributions
H.W.K., S.S.F., J.M.Y., H.J.K., and J.S.P. conceived and designed the experiments. J.H.K., D.E.L., Y.S.L., M.J.K, H.S.K., B.E.L., and K.J.L. performed the experiments. H.W.K. and H.J.K. analyzed data. H.W.K., H.J.K., and J.S.P. wrote the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Notes on contributors
Hyeon Woong Kang
Hyeon Woong Kang received his B.S. degree in Biochemistry from the Pennsylvania State University in 2015. Now, he studies an Integrated Master’s and Doctoral degree in Medical Science as a major at Yonsei University. He also works in Yonsei Biomedical Research Center at Gangnam Severance Hospital. His research focuses on the mechanisms and treatment of pancreatic cancer.
Ju Hyun Kim
Ju Hyun Kim received her B.S. degree in Integrative Bioscience and Biotechnology from Sejong University in 2018. At present, she studies a Master’s degree in medicine at Yonsei University. She also works in Yonsei Biomedical Research Center at Gangnam Severance Hospital. Her research focuses on the mechanisms and treatment of pancreatic cancer.
Da Eun Lee
Da Eun Lee received her B.S. degree in Molecular biology from Pusan National University in 2020. Currently, she obtained a Master’s degree in Medical Science from Yonsei University. She also works in Yonsei Biomedical Research Center at Gangnam Severance Hospital. Her research focuses on the mechanisms and treatment of pancreatic cancer.
Yun Sun Lee
Yun Sun Lee received a Ph.D. degree in Medical science from Yonsei University. She also works in Yonsei Biomedical Research Center at Gangnam Severance Hospital. Her research focuses on the mechanisms and treatment of pancreatic cancer.
Myeong Jin Kim
Myeong Jin Kim received his B.S. degree in Integrative Bioscience and Biotechnology from Sejong University in 2021. Now, he studies a Master’s degree in Medical science as a major at Yonsei University. He also works in Yonsei Biomedical Research Center at Gangnam Severance Hospital. His research focuses on the mechanisms and treatment of pancreatic cancer.
Hyung Sun Kim
Hyung Sun Kim received her B.S. degree in Biotechnology from Yonsei University in 2008. Moreover, she obtained MS degree in Kyungpook National University in 2012. Later, she received Ph.D. degree in Medical Science from Yonsei University in 2023. Now, Professor Kim is a clinical professor in the Hepatopancreatobiliary Surgery Department at Yonsei Medical Center in Gangnam Severance Hospital.
SungSoon Fang
SungSoon Fang received his B.S. degree in Molecular Biology from Seoul National University in 1999. Later, he obtained Ph.D. degree in Molecular Physiology at University of Illinois at Urbana-Champaign in 2008. Now, Professor Fang is an associate professor in the Department of Biomedical Sciences at Yonsei University College of Medicine.
Bo Eun Lee
Bo Eun Lee is currently in Ph.D. program at CHA university. Moreover, she is a part of the members in Organoid Laboratory. Her research focuses on the patient-derived pancreatic cancer organoid characterization.
Kyung Jin Lee
Kyung Jin Lee is graduated from the Korea Advanced Institute of Science and Technology. At present, he is a chief technical officer in Organoid Science Co., Ltd. His research focuses on recent progress in organoid based regenerative medicine.
Jongman Yoo
Jongman Yoo received B.S. degree in Korea University. Later, He obtained M.S. and Ph.D. degree in CHA University. Moreover, he was a medical director in Clinical Development Department, CHA Biotech, Ltd in 2019. Currently, he is an associate professor in the Department of Microbiology, School of Medicine at Cha University.
Hyo Jung Kim
Hyo Jung Kim was a professor of Basic Research in the Department of Biochemistry and Molecular Biology at Yonsei University College of Medicine in 2010-2020. Currently, Professor Kim is an associate professor in the Department of Surgery Research Division at Yonsei University College of Medicine. Her research focuses on metabolic disorders, cancer metabolism, development of new drugs for cancer and the discovery of early diagnostic markers for cancer, and the mechanisms of resistance to pancreatic cancer chemotherapy.
Joon Seong Park
Joon Seong Park received his B.S., M.S., and Ph.D. degree in Medical Science from Yonsei University in 1997, 2006, and 2012. Now, Professor Park is the chief of the Hepatopancreatobiliary Surgery Department at Yonsei University College of Medicine in Gangnam Severance Hospital. His research focuses on orchestrating novel cancer therapeutics development specially for therapy refractory pancreatic cancers.
References
- Kim Y, Kim SC, Song KB, Kim J, Kang DR, Lee JH, Park KM, Lee YJ. Improved survival after palliative resection of unsuspected stage IV pancreatic ductal adenocarcinoma. HPB: Offic J Int. 2016;18(4):325–11. doi:10.1016/j.hpb.2015.10.014.
- Zeng S PM, Lan B, Grützmann R, Pilarsky C, Yang H. Chemoresistance in pancreatic cancer. Int J Mol Sci. 2019;20(18):4504. doi:10.3390/ijms20184504.
- SD NJ, Friess H, Bassi C, Dunn JA, Hickey H, Beger H, Fernandez-Cruz L, Dervenis C, Lacaine F, Falconi M, et al., A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med. 2004;350(12):1200–1210. 10.1056/NEJMoa032295
- Singh RR, O’Reilly EM. New treatment strategies for metastatic pancreatic ductal adenocarcinoma. Drugs. 2020;80(7):647–669. doi:10.1007/s40265-020-01304-0.
- Jung YS, Park JI. Wnt signaling in cancer: therapeutic targeting of Wnt signaling beyond β-catenin and the destruction complex. Experimental & Molecular Medicine. 2020;52(2):183–191. doi:10.1038/s12276-020-0380-6.
- Zeng G, Germinaro M, Micsenyi A, Monga NK, Bell A, Sood A, Malhotra V, Sood N, Midda V, Monga DK, et al. Aberrant Wnt/β-catenin signaling in pancreatic adenocarcinoma. Neoplasia. 2006;8(4):279–289. doi:10.1593/neo.05607.
- Gao C, Xiao G, Hu J. Regulation of Wnt/β-catenin signaling by posttranslational modifications. Cell & Bioscience. 2014;4(1):13. doi:10.1186/2045-3701-4-13.
- Stamos JL, Weis WI. The β-catenin destruction complex. Cold Spring Harb Perspect Biol. 2013;5:a007898. doi:10.1101/cshperspect.a007898.
- Chen C, Zhu D, Zhang H, Han C, Xue G, Zhu T, Luo J, Kong L. YAP-dependent ubiquitination and degradation of β-catenin mediates inhibition of Wnt signalling induced by physalin F in colorectal cancer. Cell Death Disease. 2018;9(6):591. doi:10.1038/s41419-018-0645-3.
- Al-Hadiya BM. Niclosamide: comprehensive profile. Profiles Drug Subst Excip Relat Methodol. 2005;32:67–96.
- Li Y, Li PK, Roberts MJ, Arend RC, Samant RS, Buchsbaum DJ. Multi-targeted therapy of cancer by niclosamide: a new application for an old drug. Cancer Lett. 2014;349(1):8–14. doi:10.1016/j.canlet.2014.04.003.
- Cheng B ML, Zhang Y, Mito S, Mito, Tsin S, Tsin A. A niclosamide induces protein ubiquitination and inhibits multiple pro-survival signaling pathways in the human glioblastoma U-87 MG cell line. PloS One. 2017;12(9):e0184324. doi:10.1371/journal.pone.0184324.
- Li Z, Li Q, Wang G, Huang Y, Mao X, Zhang Y, Wang X. Inhibition of Wnt/β-catenin by anthelmintic drug niclosamide effectively targets growth, survival, and angiogenesis of retinoblastoma. Am J Transl Res. 2017;9(8):3776–3786.
- Lu W, Lin C, Roberts MJ, Waud WR, Piazza GA, Li Y, Mei L. Niclosamide suppresses cancer cell growth by inducing Wnt co-receptor LRP6 degradation and inhibiting the Wnt/β-catenin pathway. PloS One. 2011;6(12):e29290. doi:10.1371/journal.pone.0029290.
- Chen M, Wang J, Lu J, Bond MC, Ren XR, Lyerly HK, Barak LS, Chen W. The anti-helminthic niclosamide inhibits Wnt/Frizzled1 signaling. Biochemistry. 2009;48(43):10267–10274. doi:10.1021/bi9009677.
- Mook RA Jr., Ren XR, Wang J, Piao H, Barak LS, Kim Lyerly H, Chen W. Benzimidazole inhibitors from the niclosamide chemotype inhibit Wnt/β-catenin signaling with selectivity over effects on ATP homeostasis. Bioorg Med Chem. 2017;25(6):1804–1816. doi:10.1016/j.bmc.2017.01.046.
- Mook RA Jr., Wang J, Ren XR, Piao H, Lyerly HK, Chen W. Identification of novel triazole inhibitors of Wnt/β-catenin signaling based on the niclosamide chemotype. Bioorg Med Chem Lett. 2019;29(2):317–321. doi:10.1016/j.bmcl.2018.11.022.
- Wang J, Ren XR, Piao H, Zhao S, Osada T, Premont RT, Mook RA, Morse MA, Lyerly HK, Chen W, et al. Niclosamide-induced Wnt signaling inhibition in colorectal cancer is mediated by autophagy. Biochem J. 2019;476(3):535–546. doi:10.1042/BCJ20180385.
- Kaushal JB, Bhatia R, Kanchan RK, Raut P, Mallapragada S, Ly QP, Batra S, Rachagani S. Repurposing niclosamide for targeting pancreatic cancer by inhibiting Hh/Gli Non-Canonical Axis of Gsk3β. Cancers Basel. 2021;13(13):3105. doi:10.3390/cancers13133105.
- Ren X, Duan L, He Q, Zhang Z, Zhou Y, Wu D, Pan J, Pei D, Ding K. Identification of niclosamide as a New small-molecule inhibitor of the STAT3 signaling pathway. ACS Med Chem Lett. 2010;1:454–459. doi:10.1021/ml100146z.
- Pack LR, Daigh LH, Chung M, Meyer T. Clinical CDK4/6 inhibitors induce selective and immediate dissociation of p21 from cyclin D-CDK4 to inhibit CDK2. Nat Commun. 2021;12(1):3356. doi:10.1038/s41467-021-23612-z.
- Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25(38):5220–5227. doi:10.1038/sj.onc.1209615.
- Mizrahi JD, Surana R, Valle JW, Shroff RT. Pancreatic cancer. Lancet. 2020;395(10242):2008–2020. doi:10.1016/S0140-6736(20)30974-0.
- Li Y, Xi Z, Chen X, Cai S, Liang C, Wang Z, Li Y, Tan H, Lao Y, Xu H, et al. Natural compound oblongifolin C confers gemcitabine resistance in pancreatic cancer by downregulating src/MAPK/ERK pathways. Cell Death Disease. 2018;9(5):538. doi:10.1038/s41419-018-0574-1.
- Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013 Oct 31;369(18):1691–1703. doi10.1056/NEJMoa1304369.
- Chen W, Mook RA Jr., Premont RT, Wang J. Niclosamide: beyond an antihelminthic drug. Cell Signal. 2018;41:89–96. doi:10.1016/j.cellsig.2017.04.001.
- Osada T, Chen M, Yang XY, Spasojevic I, Vandeusen JB, Hsu D, Clary BM, Clay TM, Chen W, Morse MA, et al. Antihelminth compound niclosamide downregulates Wnt signaling and elicits antitumor responses in tumors with activating APC mutations. Cancer Res. 2011;71(12):4172–4182. doi:10.1158/0008-5472.CAN-10-3978.
- Li R, Hu Z, Sun SY, Chen ZG, Owonikoko TK, Sica GL, Ramalingam SS, Curran WJ, Khuri FR, Deng X, et al. Niclosamide overcomes acquired resistance to erlotinib through suppression of STAT3 in non–small cell lung cancer. Mol Cancer Ther. 2013;12(10):2200–2212. doi:10.1158/1535-7163.MCT-13-0095.
- Li R, You S, Hu Z, Chen ZG, Sica GL, Khuri FR, Curran WJ, Shin DM, Deng X, et al. Inhibition of STAT3 by niclosamide synergizes with erlotinib against head and neck cancer. PloS One. 2013;8(9):e74670. doi:10.1371/journal.pone.0074670.
- Park SJ, Shin JH, Kang H, Hwang JJ, Cho DH. Niclosamide induces mitochondria fragmentation and promotes both apoptotic and autophagic cell death. BMB Rep. 2011;44(8):517–522. doi:10.5483/BMBRep.2011.44.8.517.
- Han Z, Li Q, Wang Y, Wang L, Li X, Ge N, Wang Y, Guo C. Niclosamide induces cell cycle arrest in G1 phase in head and neck squamous cell carcinoma through let-7d/CDC34 axis. Front Pharmacol. 2018;9:1544. doi:10.3389/fphar.2018.01544.
- Li Z, Yu Y, Sun S, Qi B, Wang W, Yu A. Niclosamide inhibits the proliferation of human osteosarcoma cell lines by inducing apoptosis and cell cycle arrest. Oncol Rep. 2015;33(4):1763–1768. doi:10.3892/or.2015.3766.
- Yu K, Wang T, Li Y, Wang C, Wang X, Zhang M, Xie Y, Li S, An Z, Ye T, et al. Niclosamide induces apoptosis through mitochondrial intrinsic pathway and inhibits migration and invasion in human thyroid cancer in vitro. Biomed Pharmacother. 2017;92:403–411. doi:10.1016/j.biopha.2017.05.097.
- Wang C, Zhou X, Xu H, Shi X, Zhao J, Yang M, Zhang L, Jin X, Hu Y, Li X, et al. Niclosamide inhibits cell growth and enhances drug sensitivity of hepatocellular carcinoma cells via STAT3 signaling pathway. J Cancer. 2018;9(22):4150–4155. doi:10.7150/jca.26948.
- Wang W, Li M, Ponnusamy S, Chi Y, Xue J, Fahmy B, Fan M, Miranda-Carboni GA, Narayanan R, Wu J, et al. ABL1-dependent OTULIN phosphorylation promotes genotoxic Wnt/β-catenin activation to enhance drug resistance in breast cancers. Nat Commun. 2020;11(1):3965. doi:10.1038/s41467-020-17770-9.