
ABSTRACT
Chaperonin containing TCP1 subunit 6A (CCT6A) was recently discovered to be involved in cancer pathogenesis and stemness; however, its role in oral squamous cell carcinoma (OSCC) has not been reported. The current study aimed to investigate the impact of CCT6A on OSCC cell malignant behaviors and stemness and to explore its potentially interreacted pathways. SCC-15 and HSC-3 cells were transfected with the plasmid loading control overexpression, CCT6A overexpression, control knockout, or CCT6A knockout. Wnt4 overexpression or Notch1 overexpression plasmids were transfected into CCT6A-knockout SCC-15 cells. Cell proliferation, apoptosis, invasion, stemness, Notch, and Wnt pathways were detected in both cell lines, whereas RNA sequencing was only performed in SCC-15 cells. CCT6A was upregulated in five OSCC cell lines, including SCC-15, HSC-3, SAT, SCC-9, and KON, compared to that in the control cell line. In SCC-15 and HSC-3 cells, CCT6A overexpression increased cell proliferation, invasion, sphere formation, CD133, and Sox2 expression, but decreased cell apoptosis; on the contrary, CCT6A knockout exhibited an opposite effect on the above indexes. RNA-sequencing data revealed that the Wnt and Notch pathways were involved in the CCT6A’effect on SCC-15 cell functions. CCT6A positively regulates the Wnt and Notch pathways in SCC-15 and HSC-3 cells. Importantly, it was shown that activation of the Wnt or Notch pathways attenuated the effect of CCT6A knockout on SCC-15 cell survival, invasion, and stemness. CCT6A may promote OSCC malignant behavior and stemness by activating the Wnt and Notch pathways.
Introduction
Oral carcinoma accounts for 2.0% of new cancer cases and 1.8% of new cancer-related deaths globally, with 377,713 and 177,757 deaths, respectively, among which oral squamous cell carcinoma (OSCC) accounts for the majority proportion.Citation1–3 Generally, the outcomes of OSCC are relatively acceptable compared to other cancers, profiting from early screening, surgical technical improvement and strategy, and novel targeted and immunochemotherapeutic drugs.Citation4–7 However, as mentioned above, the annual mortality rate is approximately equal to the annual new case rate regarding OSCC;Citation1 therefore, for the sake of further improving the prognostic consequences, further excavating the in-depth molecular mechanism of OSCC is needed.
Chaperonin containing TCP1 subunit 6A (CCT6A), belonging to the chaperonin containing TCP1 (CCT) family, participates in intracellular protein folding and aggregation.Citation8,Citation9 Recently, CCT6A has been uncovered as a new oncogene that participates in the initiation of various cancers.Citation10–17 For instance, CCT6A activates the transforming growth factor β (TGF-β) pathway and promotes non-small cell lung cancer cell proliferation, invasion, and migration.Citation10 Meanwhile, CCT6A upregulates cell cycle-related proteins (such as cyclin D) to enhance cell proliferation in hepatocellular carcinoma.Citation11 In the aspect of cancers of digestive tract, a previous study reveals that CCT6A enhances the invasive ability and epithelial-mesenchymal transition of esophageal squamous cell carcinoma in a TGF-β dependent way.Citation13 Moreover, CCT6A induces cell growth and invasion,Citation14 accelerates cancer-stem-cell phenotype transformation,Citation15 and relates to CD8+ T cell exhaust in colon cancer.Citation18 However, as for the role of CCT6A in OSCC, no previous reports are revealed.
Hence, this study was designed to explore the effect of CCT6A on OSCC cell survival, invasion, and stemness, as well as to discover the potentially mediated signaling pathways implicated in OSCC pathogenesis.
Materials and methods
Cell culture and CCT6A detection
Human Oral Keratinocytes (HOK, catalog number 2610; Sciencell, USA) and human OSCC cell lines, including SCC-15 (catalog number CRL-1623; ATCC, USA), HSC-3 (catalog number JCRB0623; JCRB, Japan), SCC-9 (catalog number CRL-1629; ATCC, USA), BHY (catalog number ACC-404; DSMZ, Germany), and KON (catalog number JCRB0194; JCRB, Japan), were cultured in DMEM/F12 (catalog number D8062; Sigma, USA), EMEM (catalog number M4655; Sigma, USA), or DMEM (catalog number D0822; Sigma, USA). All culture media were supplemented with 10% fetal bovine serum (FBS) (catalog number SH30088.03; Hyclone, USA). CCT6A expression in OSCC cell lines was analyzed (HOK served as a control).
Plasmid transfection
Negative control (NC) and CCT6A overexpression (OE-NC and OE-CCT6A) plasmids were constructed using the pEX-3 vector (catalog number C05003; GenePharma, China). The pGPH1 (catalog number C02004; GenePharma, China) vector was used to construct the NC and CCT6A knockout (KO-NC and KO-CCT6A) plasmids. When the SCC-15 or HSC-3 cells reached 70% confluence, the OE-NC, OE-CCT6A, KO-NC, and KO-CCT6A plasmids were transfected using the FuGENEⓇ 6 Transfection Reagent (catalog number E2692; Promega, USA). Transfection was performed according to the manufacturer’s instructions. The cells were marked as OE-NC, OE-CCT6A, KO-NC, and KO-CCT6A after transfection. SCC-15 and HSC-3 cells without transfection were used as normal controls.
RNA sequencing (RNA-seq) and bioinformatics
Transfected SCC-15 cells (OE-NC, OE-CCT6A, KO-NC, and KO-CCT6A groups) were harvested 48 h after transfection. Total RNA was isolated with TRIzol and quantified using Agilent 2100 (Agilent, USA). Subsequently, according to the methods described in a previous study, an RNA-seq library was constructed and sequenced.Citation19 The RNA-seq data were analyzed using R packages (Version 3.6.3) (https://cran.r-project.org/bin/macosx/). To complete the calculation of the raw data, a feature count was applied. Expression normalization and differential expression analyses were performed using DESeq2. Principal component analysis (PCA) plots and heatmaps of mRNA were plotted using the Factoextra and pheatmap packages, respectively. When mRNAs had a fold change (FC) ≥2.0, and adjusted P value (BH multiple test correction) <0.05, the mRNAs were considered as differentially expressed genes (DEGs) and displayed as volcano plots. Cross-analysis was completed using the VennDiagram package. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were conducted using the DAVID web server.
Pathway screening and validation
KEGG enrichment of accordant DEGs was performed to identify potential signaling pathways. The accordant DEGs were defined as DEGs which were upregulated in overexpression term (OE-CCT6A vs. OE-NC) and downregulated in knockout term (KO-CCT6A vs. KO-NC), or downregulated in overexpression term and upregulated in knockout term. Using the rank of p values, the top five signaling pathways were screened out. The Notch and Wnt signaling pathways have been confirmed to be closely involved in the malignant progression of OSCC.Citation20,Citation21 Therefore, the expression of Notch1, Hes1, Wnt4, and β-catenin in SCC-15 and HSC-3 cells (Normal, OE-NC, OE-CCT6A, KO-NC and KO-CCT6A groups) was evaluated.
Compensation experiment
Notch1 and Wnt4 overexpression (OE-Notch1 and OE-Wnt4) plasmids were constructed with pEX-3 vector and transfected into SCC-15 cells according to the methods described in “Plasmid transfection” subsection. After transfection, the cells were divided into seven groups: (a) normal group, cells without transfection; (b) NC group, cells were transfected with KO-NC and OE-NC plasmids; (c) KO-CCT6A group, cells were transfected with OE-NC and KO-CCT6A plasmids; (d) OE-Notch1 group, cells were transfected with OE-Notch1 and KO-NC plasmids; (e) OE-Notch1 and KO-CCT6A group, cells were transfected with OE-Notch1 and KO-CCT6A plasmids; (f) OE-Wnt4 group, cells were transfected with OE-Wnt4 and KO-NC plasmids; (g) OE-Wnt4&KO-CCT6A group, cells were transfected with OE-Wnt4 and KO-CCT6A group.
Reverse transcription quantitative polymerase chain reaction (RT-qPCR)
Total RNA in cells (HOK cells, OSCC cells without transfection, OSCC cells that were harvested 48 h after transfection) was extracted with Trizol (catalog number 15,596,018; Invitrogen, USA) according to the manufacturer’s instructions. The concentration of total RNA was analyzed using Nanodrop 2000 (Invitrogen, USA). One microgram of total RNA was transcribed to cDNA using the RT Master Mix (catalog number LS2052; Promega, USA). Next, qPCR was conducted using the qPCR Master Mix (catalog number LS2062; Promega, USA). The results were analyzed by the 2−ΔΔCt method, with GAPDH serving as an internal reference. Primer sequences used are listed in Supplementary Table S1.
Western blot
The cells (HOK cells, OSCC cells without transfection, and OSCC cells that were harvested 48 h after transfection) were lysed using RIPA lysis buffer (catalog number P0013B; Beyotime, China). Twenty micrograms of protein, which was quantified using the BCA Protein Assay Kit (catalog number P0010; Beyotime, China), was separated on a 4–20% precast gel (catalog number P0057B; Beyotime, China). The proteins were then transferred to a nitrocellulose membrane (catalog number FFN08; Beyotime, China). The membrane was blocked with 5% BSA (catalog number ST023; Beyotime, China) and incubated with primary antibodies at 4°C overnight. After incubation with the secondary antibody, the membrane was visualized using an ECL luminescence reagent (catalog number C510045; Sangon, China). The antibodies used are listed in Supplementary Table S2.
Cell proliferation
At 0, 24, 48, and 72 h post-transfection, Cell Counting Kit-8 (CCK-8) reagent (catalog number 96,992; Sigma, USA) was incubated with cells for 3 h at 37°C. Optical density (OD) was then measured using a microplate reader (BioTek, USA).
Cell apoptosis
Cells were collected for the detection of apoptosis 48 h after transfection. Detection was performed using the TUNEL Apoptosis Assay Kit (catalog number KGA1408; KeyGEN Biotech, China). Detection was performed according to the kit’s instructions. Images were taken using an inverted fluorescence microscope (Olympus, Tokyo, Japan).
Cell invasion
To complete cell invasion, cells were harvested 48 h post-transfection. The Transwell insert was coated with Matrigel matrix (catalog number 354,234; Corning, USA) at 37°C for 1 h prior to the experiment. The cells in 200 μL of FBS-free medium were seeded into the insert, and the chamber was filled with 500 μL of 10% FBS-containing medium. After incubation for another 24 h, the insert was collected and the Matrigel matrix was removed. The insert was fixed with 4% paraformaldehyde (catalog number E672002; Sangon, China) and stained with 0.1% crystal violet (catalog number A600331; Sangon, China). Images were taken using an inverted microscope (Olympus, Japan), and the invasive cells were counted.
Sphere formation
The cells were collected 48 h post-transfection. After counting, 300 cells were inoculated onto ultra-low attachment plates for 14 days. Sphere formation medium composed of DMEM/F12 (catalog number D8062; Sigma, USA), B-27 (catalog number 17,504,044; Thermo, USA), bFGF (catalog number 100-18B; Perprotech, USA), and EGF (catalog number GMP100–15; Perprotech, USA) was used for the incubation. Spheres with diameters larger than 50 μm were counted and photographed using a microscope (Olympus, Japan).
Statistical analysis
Statistical analyses were performed using GraphPad Prism 7.02 (GraphPad Software Inc., San Diego, CA, USA). The data are presented as the mean ± standard deviation. Comparisons among three or more groups were analyzed using Dunnett’s test or Tukey’s test. The P > .05, <0.05, <0.01, <0.001 were labeled as “NS”, “*”, “**”, “***”, respectively.
Results
Aberrant CCT6A expression in OSCC cell lines
Relative CCT6A mRNA expression increased in multiple OSCC cell lines compared to that in the control cell line (all P < .05) (). Moreover, relative CCT6A protein expression was also increased in various OSCC cell lines compared to that in the control cell line (all P < .05) (). Among all tested OSCC cells, SCC-15 and HSC-3 cell lines exhibited higher CCT6A expression and were therefore chosen for subsequent experiments.
Figure 1. CCT6A expression. CCT6A mRNA expression (a) and protein expression (b, c) were increased in OSCC cells compared to control cells.
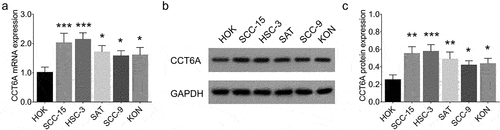
Effect of CCT6A on OSCC cell survival and invasion
In SCC-15 cells, the CCT6A overexpression plasmid increased CCT6A expression, while the CCT6A knockout plasmid reduced CCT6A expression (all P < .01) (). Moreover, the CCT6A overexpression plasmid increased cell proliferation (P < .05) and invasive cell count (P < .01), while suppressing cell apoptosis (P < .05), CCT6A knockout plasmid reduced cell proliferation (P < .05) and invasive cell count (P < .05), while increasing cell apoptosis (P < .001) ().
Figure 2. CCT6A induced OSCC malignant behaviors. CCT6A mRNA expression (a) and protein expression (b, c) after transfection into SCC-15 cells. Effect of CCT6A on cell proliferation (d), cell apoptosis rate (e, g) and invasive cell count (f, h) in SCC-15 cells. CCT6A mRNA expression (i) and protein expression (j, k) after transfection into HSC-3 cells. Effect of CCT6A on cell proliferation (i), cell apoptosis rate (m, o) and invasive cell count (n, p) in HSC-3 cells.
Furthermore, in HSC-3 cells, CCT6A expression was upregulated by the CCT6A overexpression plasmid and downregulated by the CCT6A knockout plasmid as well (all P < .001) (). Besides, the CCT6A overexpression plasmid elevated cell proliferation (P < .05) and invasive cell count (P < .01), but decreasing cell apoptosis rate (P < .01) in HSC-3 cells (). Oppositely, the CCT6A knockout plasmid reduced cell proliferation (P < .01) and invasive cell count (P < .01), while enhancing cell apoptosis rate (P < .001) in HSC-3 cells.
Effect of CCT6A on OSCC cell stemness
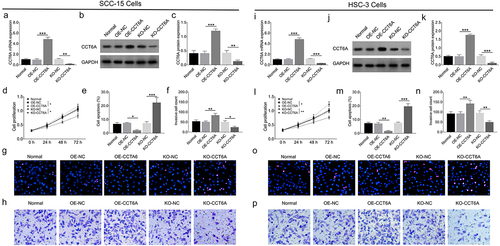
In SCC-15 cells, the CCT6A overexpression plasmid increased sphere formation ability (P < .05), CD133 (P < .05), and Sox2 (P < .01) expression, while the CCT6A knockout plasmid reduced these indices (all P < .01) ().
Figure 3. CCT6A promoted OSCC stemness. Effect of CCT6A on sphere formatted number (a, b), CD133 and sox expressions (c, d) in SCC-15 cells. Effect of CCT6A on sphere formatted number (e, f), CD133 and sox expressions (g, h) in HSC-3 cells.
Furthermore, in HSC-3 cells, the CCT6A overexpression plasmid elevated sphere formation ability (P < .01), CD133 (P < .05), and Sox2 (P < .05) expression, but the CCT6A knockout plasmid suppressed the above indexes (all P < .05) ().
Determination of CCT6A mediated pathways in OSCC cells
to determine the CCT6A mediated pathways in OSCC, high-throughput sequencing was performed in SCC-15 cells between OE-CCT6A group vs. OE-NC group as well as KO-CCT6A group vs. KO-NC group, as displayed in Supplementary Figure S1a-j.
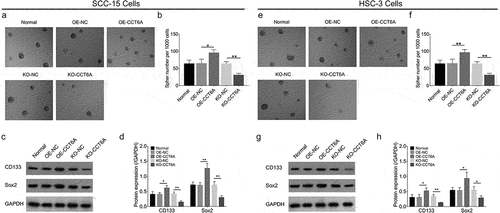
Further cross analysis exhibited that 40 accordant DEGs were in the intersection group of OE-CCT6A vs. OE-NC upregulated DEGs and KO-CCT6A vs. KO-NC downregulated DEGs; 89 accordant DEGs were in the intersection group of OE-CCT6A vs. OE-NC downregulated DEGs and KO-CCT6A vs. KO-NC upregulated DEGs (), with the detailed information of 50 top accordant DEGs shown in .
Figure 4. Cross analysis. Cross analysis (a) of accordant DEGs. Further GO enrichment (b) and KEGG enrichment (c) analyses for cross analysis.
Table 1. Top 50 accordant DEGs.
Further GO analysis revealed that accordant DEGs were enriched in gene sets involved in protein binding, regulation of cell proliferation, and the canonical Wnt signaling pathway (). Besides, KEGG analysis displayed that accordant DEGs were enriched in gene sets involved in pathways in cancer, PI3K-Akt signaling pathway and Notch signaling pathway () with the detailed information of the top 5 enriched pathway shown in . Among the top five pathways, Notch and Wnt signaling pathways have been reported to be highly involved in OSCC pathogenesis and stemness. Rescue experiments were performed to further determine whether CCT6A regulated Notch and Wnt pathways to induce cell malignant behaviors and stemness in OSCC, by co-transfecting the CCT6A knockout plasmid and Notch1 overexpression plasmid versus transfecting each one plasmid individually, and by co-transfecting the CCT6A knockout plasmid and Wnt4 overexpression plasmid versus transfecting each one plasmid individually.
Table 2. Top 5 pathways.
Effect of CCT6A on notch and wnt signaling in OSCC
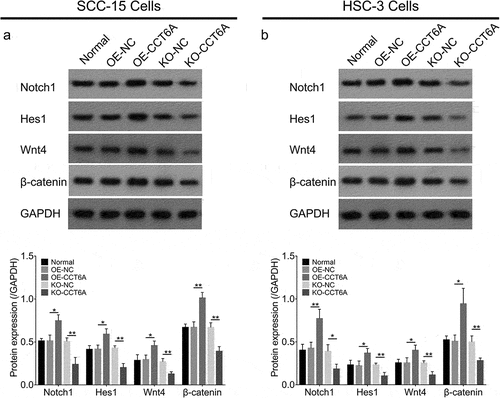
In SCC-15 cells, the CCT6A overexpression plasmid increased Notch1 (P < .05), Hes1 (P < .05), Wnt4 (P < .05), and β-catenin (P < .01) expression levels, while the CCT6A knockout plasmid reduced the expression of Notch1, Hes1, Wnt4, and β-catenin (all P < .01) (). Furthermore, the effect of CCT6A overexpression or knockout plasmid displayed the same effect on Notch and Wnt signaling in HSC-3 cells as in SCC-15 cells (all P < .05) ().
Figure 5. CCT6A activated Wnt and Notch pathways in OSCC. Effect of CCT6A on Wnt and Notch pathways in SCC-15 cells (a) and in HSC-3 cells (b).
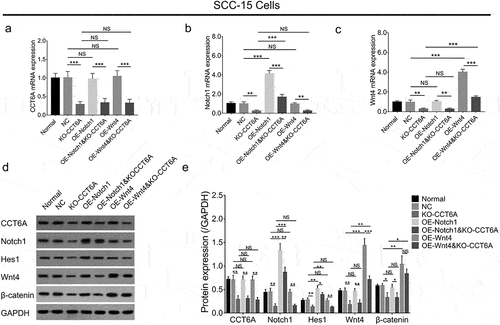
Notch and wnt pathway detection in rescue experiments
In SCC-15 cells, Notch1 and Wnt4 overexpression plasmids activated the Notch and Wnt signaling pathways, respectively (). Furthermore, Notch1 and Wnt4 overexpression plasmids could compensate for the effect of the CCT6A knockout plasmid on regulating Notch and Wnt pathways, respectively ().
Figure 6. Notch1 and Wnt4 compensated the effect of CCT6A on regulating Notch and Wnt pathways. CCT6A mRNA expression (a) Notch1 mRNA expression (b), Wnt4 mRNA expression (c) and their protein expressions (d, e) in rescue experiments.
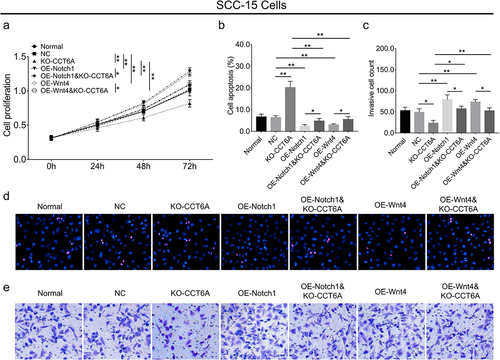
OSCC cell proliferation, apoptosis and invasion in rescue experiments
In SCC-15 cells, Notch1 overexpression plasmid increased cell proliferation (P < .01) and invasive cell count (P < .01), while suppressing cell apoptosis (P < .01), Wnt4 overexpression plasmid elevated cell proliferation (P < .01) and invasive cell count (P < .01), and inhibited cell apoptosis (P < .01) (). Interestingly, it was noteworthy that Notch1 and Wnt4 overexpression plasmids attenuated the effect of the CCT6A knockout plasmid on regulating the above-mentioned cell malignant behaviors (all P < .05) ().
Figure 7. Notch1 and Wnt4 compensated the effect of CCT6A knockout on regulating OSCC malignant behaviors. Notch1 and Wnt4 attenuated the effect of CCT6A knockout on regulating cell proliferation (a), cell apoptosis rate (b, d) and invasive cell count (c, e) in SCC-15 cells.
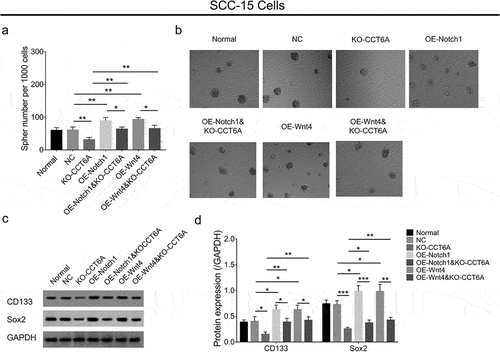
OSCC cell stemness in rescue experiments
In SCC-15 cells, Notch1 overexpression plasmid increased sphere formation ability (P < .01), CD133 (P < .05), and Sox2 (P < .05) expression; meanwhile, the Wnt4 overexpression plasmid exhibited a similar impact on the above indices as Notch1 overexpression plasmid did (all P < .05) (). Importantly, Notch1 and Wnt4 overexpression plasmids could compensate for the effect of CCT6A knockout on regulating OSCC cell stemness, reflected by sphere formation ability, CD133, and Sox2 expression (all P < .05) ().
Figure 8. Notch1 and Wnt4 compensated the effect of CCT6A knockout on regulating OSCC stemness. Notch1 and Wnt4 attenuated the effect of CCT6A knockout on regulating sphere formatted number (a, b), CD133 and sox expressions (c, d) in SCC-15 cells.
Discussion
CCTs, also known as tailless complex polypeptide 1 ring complex (TRiC), are a collection of proteins that are critical for cellular protein homeostasis by regulating cytoskeletal protein folding (such as actin and tubulins).Citation22 Besides, CCTs are also responsible for cancer cell proliferation via regulation of the autophagy process.Citation23–25 CCT6A, as a recently discovered subunit of CCT, has displayed oncogenic properties in several cancers.Citation10–15 However, no molecular study has investigated the effect of CCT6A on OSCC cell malignant phenotypes and stemness. Therefore, we conducted this study and discovered that CCT6A overexpression induced cell malignancy and stemness, whereas its knockout displayed an opposite trend in OSCC. The possible reasons to explain these findings were: (a) CCT6A upregulated cell cycle-related pathways, which further enhanced the cell proliferation rate and eventually resulted in OSCC cell malignant behaviors.Citation11,Citation26 (b) As a family of CCT, upregulation of CCT6A may lead to aberrant chaperone-mediated autophagy and altered cellular metabolic processes (changing from oxidative phosphorylation to aerobic glycolysis), which further led to a malignant phenotype in OSCC.Citation27 (c) CCT6A might directly interact with several tumor suppressors, leading to dysregulation and loss of their suppressive function, thereby resulting in OSCC cell malignant behaviors.Citation22,Citation28 Taken together, CCT6A displayed oncogenic properties in OSCC pathogenesis.
Apart from the direct cell malignant behavior induced by CCT6A in several cancers, the underlying molecular mechanism is also of great interest. One study discovered that CCT6A targets the TGF-β signaling pathway to induce cell malignancy in lung cancer, while another study showed that CCT6A regulates G1-to-S phase protein to accelerate cell proliferation in hepatocellular carcinoma.Citation10,Citation11 These findings are not consistent, suggesting that CCT6A may target multiple oncogenic pathways in cancer development. In our high-throughput sequencing analysis, we discovered that a large number of DEGs and multiple pathways (such as Notch, Wnt, and other pathways modifying stem cell pluripotency) were activated by the overexpression or knockout of CCT6A in OSCC. Meanwhile, Wnt and Notch pathways are two well-characterized oncogenic pathways in OSCC development.Citation20,Citation21 Therefore, we chose these pathways in the present experiment to determine whether CCT6A regulated these two pathways to induce OSCC cell malignant behaviors.
In the aspect of the Notch pathway, it is proposed to be a very complex participant closely involved in cancers, whose complexity is revealed by its affluent oncogenic or anti-oncogenic characteristics in diverse malignancies.Citation29,Citation30 Regarding OSCC, the Notch pathway not only promotes its growth, invasion, and epithelial-mesenchymal transition, but also facilitates cancer stemness and drug resistance.Citation31–33 In line with these previous discoveries, we discovered that knockout of CCT6A inhibited the Notch pathway, and this inhibition could be compensated by overexpression of Notch1. These data suggest that CCT6A activates the Notch pathway to induce malignancy in OSCC cells.
The Wnt signaling pathway consists of Wnt ligands (such as Wnt3, Wnt4, and Wnt10a), Wnt receptors (such as Frizzled and LRP5/6), and downstream signaling molecules (such as GSK-3β, β-catenin, and c-Myc), which regulate cellular proliferation, survival, and stem cell self-renewal via a complex intracellular signaling cascade.Citation34 As for the role of Wnt pathway in OSCC pathogenesis, canonical Wnt family members activate TCF receptor gene and prevent β-catenin from degradation, which further leads to transactivation of target oncogenes (such as c-MYC) and OSCC cell growth and invasion.Citation20,Citation35 Besides, aberrant Wnt singling also involves in the inhibition of chemotherapy-induced apoptosis and results in cancer stemness.Citation36,Citation37 In the present study, we found that CCT6A knockout suppressed the Wnt pathway, and that these effects could be attenuated by Wnt4 overexpression in OSCC cells. These data suggest that CCT6A activates the Wnt pathway to induce malignancy in OSCC cells. Further in vivo experiments are required to confirm the effect of CCT6A on OSCC pathogenesis.
In conclusion, this study showed that CCT6A may be implicated in the malignant behavior and stemness of OSCC via activation of the Notch and Wnt pathways.
Supplementary Figure 1.tif
Download TIFF Image (28.9 MB)Supplementary Table 1.docx
Download MS Word (15.2 KB)Supplementary Table 2 clean.docx
Download MS Word (15.7 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data used to support the findings of this study have been included in this article. The datasets and materials used in this study are available from the corresponding author upon reasonable request.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15384047.2023.2287122
Additional information
Funding
Notes on contributors
Yangyi Chen
Yangyi Chen received His B.S. degree in oral medicine from Central South University in 2019. Currently, he is a professional master student in the China Medical University. His research focuses on oral squamous cell carcinoma and cytokines.
Yongge Chen
Yongge Chen received His B.S. degree in clinical medicine from Hebei University of Engineering in 2016. Now, he is a chief physician in Handan Central Hospital of Hebei Province. His research focuses on chemoradiotherapy of head and neck cancer and minimally invasive therapy.
Weixian Liu
Weixian Liu received His B.S. degree in oral medicine from Jiamusi University in 1983. Later, he received his Master degree and PhD degree in oral and maxillofacial surgery from the China Medical University in 1987 and 1996. Now, he is a chief physician, doctoral supervisor in Shengjing Hospital of China Medical University. His research focuses on tissue injury and repair, clinical study of dental implant, and early detection of oral and maxillofacial malignancy, et al.
References
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–10. doi:10.3322/caac.21660.
- Fritz C, Ng JJ, Harris J, Romeo DJ, Prasad A, Moreira A, Rajasekaran K. Clinical practice guidelines for management of head and neck squamous cell carcinoma of unknown primary: an AGREE II appraisal. Eur Arch Otorhinolaryngol. 2023;280(9):4195–4204. doi:10.1007/s00405-023-07997-9.
- Chamoli A, Gosavi AS, Shirwadkar UP, Wangdale KV, Behera SK, Kurrey NK, Kalia K, Mandoli A. 2021. Overview of oral cavity squamous cell carcinoma: risk factors, mechanisms, and diagnostics. Oral Oncol. 121:105451. doi:10.1016/j.oraloncology.2021.105451.
- Yang SY, Li SH, Liu JL, Sun XQ, Cen YY, Ren RY, Ying SC, Chen Y, Zhao ZH, Liao W. Histopathology-based diagnosis of oral squamous cell carcinoma using deep learning. J Dent Res. 2022;101(11):1321–1327. doi:10.1177/00220345221089858.
- Shetty KSR, Kurle V, Greeshma P, Ganga VB, Murthy SP, Thammaiah SK, Prasad PK, Chavan P, Halkud R, Krishnappa R. 2021. Salvage Surgery in recurrent oral squamous cell carcinoma. Front Oral Health. 2:815606. doi:10.3389/froh.2021.815606.
- Kain JJ, Birkeland AC, Udayakumar N, Morlandt AB, Stevens TM, Carroll WR, Rosenthal EL, Warram JM. Surgical margins in oral cavity squamous cell carcinoma: Current practices and future directions. Laryngoscope. 2020;130(1):128–138. doi:10.1002/lary.27943.
- Chakraborty R, Darido C, Liu F, Maselko M, Ranganathan S. Head and Neck Cancer Immunotherapy: Molecular Biological Aspects of Preclinical and Clinical Research. Cancers Basel. 2023;15(3). doi:10.3390/cancers15030852.
- Willison KR. The substrate specificity of eukaryotic cytosolic chaperonin CCT. Philos Trans R Soc Lond B Biol Sci. 2018;373(1749):20170192. doi:10.1098/rstb.2017.0192.
- Zheng L, Chen X, Zhang L, Qin N, An J, Zhu J, Jin H, Tuo B. A potential tumor marker: chaperonin containing TCP‑1 controls the development of malignant tumors (review). Int J Oncol. 2023;63(3). doi:10.3892/ijo.2023.5554.
- Ying Z, Tian H, Li Y, Lian R, Li W, Wu S, Zhang HZ, Wu J, Liu L, Song J, et al. CCT6A suppresses SMAD2 and promotes prometastatic TGF-beta signaling. J Clin Invest. 2017;127(5):1725–1740. doi:10.1172/JCI90439.
- Zeng G, Wang J, Huang Y, Lian Y, Chen D, Wei H, Lin C, Huang Y. 2019. Overexpressing CCT6A contributes to cancer cell growth by affecting the G1-to-S phase transition and predicts a negative prognosis in hepatocellular carcinoma. Onco Targets Ther. 12:10427–10439. doi:10.2147/OTT.S229231.
- Zeng W, Wu M, Cheng Y, Liu L, Han Y, Xie Q, Li J, Wei L, Fang Y, Chen Y, et al. CCT6A knockdown suppresses osteosarcoma cell growth and Akt pathway activation in vitro. PLoS ONE. 2022;17(12):e0279851. doi:10.1371/journal.pone.0279851.
- Xia X, Zhao S, Chen W, Xu C, Zhao D. CCT6A promotes esophageal squamous cell carcinoma cell proliferation, invasion and epithelial-mesenchymal transition by activating TGF-beta/Smad/c-myc pathway. Ir J Med Sci. 2023. doi:10.1007/s11845-023-03357-y.
- Yang X, Tong Y, Ye W, Chen L. HOXB2 increases the proliferation and invasiveness of colon cancer cells through the upregulation of CCT6A. Mol Med Rep. 2022;25(5). doi:10.3892/mmr.2022.12690.
- Peng X, Chen G, Lv B, Lv J. MicroRNA-148a/152 cluster restrains tumor stem cell phenotype of colon cancer via modulating CCT6A. Anticancer Drugs. 2022;33(1):e610–e21. doi:10.1097/CAD.0000000000001198.
- Peng S, Yu J, Wang Y. 2023. CCT6A dysregulation in surgical prostate cancer patients: association with disease features, treatment information, and prognosis. Ir J Med Sci. doi:10.1007/s11845-023-03461-z
- Wang H, Wang X, Xu L, Lin Y, Zhang J, Yang Y. 2022. CCT6A and CHCHD2 are coamplified with EGFR and associated with the unfavorable Clinical outcomes of lung adenocarcinoma. Dis Markers. 2022:1–16. doi:10.1155/2022/1560199.
- Hu J, Han C, Zhong J, Liu H, Liu R, Luo W, Chen P, Ling F. 2021. Dynamic network biomarker of pre-exhausted CD8(+) T cells contributed to T cell exhaustion in colorectal cancer. Front Immunol. 12:691142. doi:10.3389/fimmu.2021.691142.
- Head SR, Komori HK, LaMere SA, Whisenant T, Van Nieuwerburgh F, Salomon DR, Ordoukhanian P. 2014. Library construction for next-generation sequencing: overviews and challenges. Biotechniques. 56(2):61–64. 66, 68, passim. doi: 10.2144/000114133
- Xie J, Huang L, Lu YG, Zheng DL. 2020. Roles of the Wnt signaling pathway in head and neck squamous cell carcinoma. Front Mol Biosci. 7:590912. doi:10.3389/fmolb.2020.590912.
- Liu W, Shi X, Wang B. Retraction note: microRNA-133a exerts tumor suppressive role in oral squamous cell carcinoma through the Notch signaling pathway via downregulation of CTBP2. Cancer Gene Ther. 2023;30(8):1179–1179. doi:10.1038/s41417-023-00649-4.
- Roh SH, Kasembeli M, Bakthavatsalam D, Chiu W, Tweardy DJ. Contribution of the type II Chaperonin, TRiC/CCT, to Oncogenesis. Int J Mol Sci. 2015;16(11):26706–26720. doi:10.3390/ijms161125975.
- Andrade-Tomaz M, de Souza I, Rocha CRR, Gomes LR. The role of chaperone-mediated autophagy in cell cycle control and its implications in cancer. Cells. 2020;9(9):2140. doi:10.3390/cells9092140.
- Chen X, Chen X, Huang Y, Lin J, Wu Y, Chen Y. TCP1 increases drug resistance in acute myeloid leukemia by suppressing autophagy via activating AKT/mTOR signaling. Cell Death Disease. 2021;12(11):1058. doi:10.1038/s41419-021-04336-w.
- Ghozlan H, Showalter A, Lee E, Zhu X, Khaled AR. 2021. Chaperonin-containing TCP1 complex (CCT) promotes breast cancer growth through correlations with key cell cycle regulators. Front Oncol. 11:663877. doi:10.3389/fonc.2021.663877.
- Huang K, Zeng Y, Xie Y, Huang L, Wu Y. Bioinformatics analysis of the prognostic value of CCT6A and associated signalling pathways in breast cancer. Mol Med Rep. 2019;19(5):4344–4352. doi:10.3892/mmr.2019.10100.
- Kon M, Kiffin R, Koga H, Chapochnick J, Macian F, Varticovski L, Cuervo AM. Chaperone-mediated autophagy is required for tumor growth. Sci Transl Med. 2011;3(109):109ra17. doi:10.1126/scitranslmed.3003182.
- Trinidad AG, Muller PA, Cuellar J, Klejnot M, Nobis M, Valpuesta JM, Vousden KH. Interaction of p53 with the CCT complex promotes protein folding and wild-type p53 activity. Mol Cell. 2013;50(6):805–817. doi:10.1016/j.molcel.2013.05.002.
- Ferreira A, Aster JC. 2022. Notch signaling in cancer: complexity and challenges on the path to clinical translation. Semin Cancer Biol. 85:95–106. doi:10.1016/j.semcancer.2021.04.008.
- Porcheri C, Mitsiadis TA. Notch in head and neck cancer. Adv Exp Med Biol. 2021;1287:81–103. doi:10.1007/978-3-030-55031-8_7.
- Weaver AN, Burch MB, Cooper TS, Della Manna DL, Wei S, Ojesina AI, Rosenthal EL, Yang ES. Notch signaling activation is associated with patient mortality and increased FGF1-mediated invasion in squamous cell carcinoma of the oral cavity. Mol Cancer Res. 2016;14(9):883–891. doi:10.1158/1541-7786.MCR-16-0114.
- Kalafut J, Czerwonka A, Anameric A, Przybyszewska-Podstawka A, Misiorek JO, Rivero-Muller A, Nees M. Shooting at moving and hidden targets—tumour cell plasticity and the Notch signalling pathway in head and neck squamous cell carcinomas. Cancers Basel. 2021;13(24):6219. doi:10.3390/cancers13246219.
- Lee SH, Do SI, Lee HJ, Kang HJ, Koo BS, Lim YC. Notch1 signaling contributes to stemness in head and neck squamous cell carcinoma. Lab Invest. 2016;96(5):508–516. doi:10.1038/labinvest.2015.163.
- MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17(1):9–26. doi:10.1016/j.devcel.2009.06.016.
- Chen S, Guttridge DC, You Z, Zhang Z, Fribley A, Mayo MW, Kitajewski J, Wang CY. Wnt-1 signaling inhibits apoptosis by activating beta-catenin/T cell factor-mediated transcription. J Cell Biol. 2001;152(1):87–96. doi:10.1083/jcb.152.1.87.
- Liu SC, Huang CS, Huang CM, Hsieh MS, Huang MS, Fong IH, Yeh CT, Lin CC. 2021. Isoorientin inhibits epithelial-to-mesenchymal properties and cancer stem-cell-like features in oral squamous cell carcinoma by blocking Wnt/beta-catenin/STAT3 axis. Toxicol Appl Pharmacol. 424:115581. doi:10.1016/j.taap.2021.115581.
- Mohapatra P, Shriwas O, Mohanty S, Ghosh A, Smita S, Kaushik SR, Arya R, Rath R, Das Majumdar SK, Muduly DK, et al. CMTM6 drives cisplatin resistance by regulating Wnt signaling through the ENO-1/AKT/GSK3beta axis. JCI Insight. 2021. 6(4). doi:10.1172/jci.insight.143643.