
ABSTRACT
While the emergence of immunotherapies has fundamentally altered the management of solid tumors, cancers exploit many complex biological mechanisms that result in resistance to these agents. These encompass a broad range of cellular activities – from modification of traditional paradigms of immunity via antigen presentation and immunoregulation to metabolic modifications and manipulation of the tumor microenvironment. Intervening on these intricate processes may provide clinical benefit in patients with solid tumors by overcoming resistance to immunotherapies, which is why it has become an area of tremendous research interest with practice-changing implications. This review details the major ways cancers avoid both natural immunity and immunotherapies through primary (innate) and secondary (acquired) mechanisms of resistance, and it considers available and emerging therapeutic approaches to overcoming immunotherapy resistance.
Introduction
Cancer immunotherapies have captured the attention of the scientific and lay communities alike, yet many obstacles continue to limit their use in solid tumors. Impressive clinical successes of immune checkpoint inhibitors (ICIs) have been notched in immunologically “hot” tumors like melanoma, non-small cell lung cancer (NSCLC), cutaneous squamous cell carcinoma, and renal cell carcinoma. Meanwhile, the efficacy of cancer vaccines and adoptive T cell treatments like tumor infiltrating lymphocytes (TILs) and chimeric antigen receptor (CAR) T cells have been much more tempered, noting the latter has enjoyed particular success in hematologic malignancies.Citation1 T cell engagers (TCEs), and specifically T cell engaging bispecific antibodies (e.g., BiTEⓇ), have recently become a much more active area of investigation in solid tumors, as well, though the large-scale translation of this technology to clinical practice is limited and is most notably characterized by tabentafusp-tebn for uveal melanoma.Citation2,Citation3
The majority of presently available (and investigational) cancer immunotherapies highlight the overarching importance of anti-tumor T cell activity. This is because CD8-expressing cytotoxic T lymphocytes (CTLs) are ultimately responsible for the final step in the process of immune regulation of cancers: killing tumor cells. They perform this task through two main processes. The most well-described is the secretion of the pore-forming protein perforin, which allows co-secreted granzymes to enter the target cell and induce apoptosis.Citation4 Additionally, CTL-induced Fas signaling results in Fas clustering on target cell membranes and the subsequent recruitment of the Fas-associated death domain (FADD), inducing apoptosis through caspases.Citation5
Although accurate, this characterization oversimplifies immunologic tumor death. In reality, the machinations that set the stage for the interaction between tumors and CTLs are carefully coordinated. These are summarized in and explored more in-depth in the following review. Recognition of cancer-related antigens is key to identifying tumors as “non-self” and thus immunogenic, involving antigen-presenting cells (APCs) that express antigenic peptides on major histocompatibility complexes (MHCs). CTLs also rely on supportive and regulatory network of immune cells that signal each other through complex cytokine signaling. Stimulatory and inhibitory immune checkpoints mediate the balance of immune activity to maximize anti-tumor effect without jeopardizing self-tolerance. Other immune cells like T regulatory cells (Tregs) and myeloid-derived suppressor cells (MDSCs) contribute immunosuppressive effects that are important to control excessive inflammatory responses. Tempering of immunity is also mediated by cancer-associated fibroblasts (CAFs) in the tumor microenvironment (TME).
Figure 1. Select tumor functions, anti-tumor immune components, and their interactions. (a) tumor-associated and tumor-specific antigens (TAA/TSA) are processed by antigen-presenting cells (APC), which then (b) communicate with both CD4+ and CD8+ T cells (CTL) through major histocompatibility class I and II (MHC-I, MHC-II), along with co-signals CD28/B7 and CD40/CD40L. (c) a complex interplay of cytokines including interleukin-2 (IL-2), IL-12, IL-15, IFNγ, TGF-β that are released by immune cells and from the tumor microenvironment tightly control the activation of the immune system through these steps (blue arrows/text). (d) this is further influenced by an individual’s microbiome, influencing immune responses. (e) Chemokines such as CXCL12, CCL5, and others (red arrows/text) mediate recruitment of immune cells to the tumor. Meanwhile, immune regulation is achieved through cellular components such as (f) regulatory T cells (Treg), as well as (g) myeloid-derived suppressor cells (MDSC) and a balance of M1 and M2 tumor-associated macrophages (TAM) in the tumor microenvironment (TME). (h) cancer associated fibroblasts (CAF) express fibroblast associated protein (FAP) and decrease responses to immunotherapies through generation of cytokines and chemokines, as well as causing a desmoplastic reaction in the TME. (i) specific tumor mutations also can provoke pro- or anti-immune functions through various mechanisms. (j) immune evasion is further achieved through depletion of metabolites, decreased glucose, oxygen (O2), tryptophan, arginine, and glutamine. (k) based on these factors, CD8+ CTLs perform direct killing of tumor cells after activation of the T cell receptor (TCR) and a balance of costimulatory (e.g., PD-1, LAG-3) and coinhibitory (e.g., 4-1BB, OX40) signals. Created with BioRender.
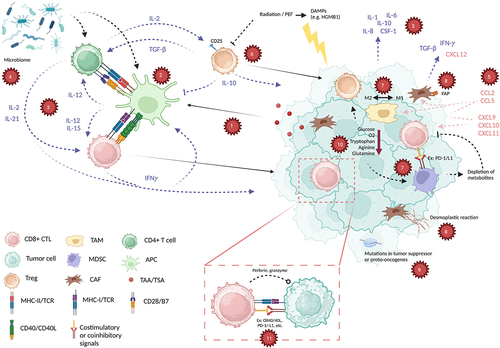
For some tumors, there is primary resistance to immunotherapies owing to baseline characteristics that impact the aforementioned factors and many others. In addition to methods of primary resistance that affect the traditional immune paradigm (i.e., antigen presentation, cytokine signaling, checkpoints, and immunoregulation), these include the presence of driver mutations as well as mutations that confer resistance to immunotherapies, a decreased tendency for immunogenic tumor cell death, metabolic derangements, and individual differences in microbiota. In tumors that are initially sensitive to immune-directed treatments, adaptations to immune surveillance allow them to secondarily resist currently available treatments. For example, primary (innate) and secondary (acquired) resistance to ICIs can be broken down into categories such as insufficient generation of anti-tumor T cells, inadequate function of those T cells, and impaired T cell memory.Citation6
In cases of both primary and secondary resistance, patients may be exposed to more toxic alternatives as a result of the lack of efficacy of immunotherapies for their tumors, driving the need for novel solutions to this problem. Therefore, overcoming this resistance has become an extraordinarily active field of research – with profound clinical implications. Still, the complexity of the interrelated processes involved highlights the need for better understanding of the immune landscape of cancer.This review provides an overview of the major ways that cancers avoid usual immune functions and immune-directed therapies. It also explores how these strategies can be overcome with available and investigational therapeutic approaches, while highlighting existing gaps in research and areas of future interest. Key ongoing clinical trials discussed in the appropriate sections are further summarized in for review.
Table 1. Selected ongoing trials addressing immunotherapy resistance in solid tumors.
Cancer antigens and antigen presentation
Before carrying out the functions of direct tumor killing, CTLs must first be able to recognize their targets through cancer-related antigens. These come in two primary forms: tumor-associated antigens (TAAs) and tumor-specific antigens (TSAs).Citation7 TAAs are more weakly immunogenic because they are encoded in germline tissues (and thus “self”) but preferentially expressed in tumors. These include familiar examples such as PSA, HER2, CEA, mesothelin, EGFR, and MAGE. Conversely, TSAs are unique neoantigens that arise from oncogenic driver mutations like single nucleotide variants (SNVs), insertion-deletions and fusions, and so they more strongly induce immune responses as they are seen as “non-self.” For this reason, though many targeted treatments have targeted on TAAs in the past, it may be prudent to focus on TSAs in the development of immunotherapies.
TAAs and TSAs are taken up by APCs like dendritic cells (DCs), macrophages, and B cells. These cells express MHC class I (MHC-I), which is subsequently loaded with relevant epitopes and prime CD8+ CTLs to kill tumor cells which express those epitopes through their own MHC-I. As such, one strategy employed by cancers to avoid immune killing is the downregulation of MHC-I on tumor cells. This is accomplished by somatic genetic defects (such as those in TAP1/TAP2Citation8–10 or MHC subunits like β2-microglobulin),Citation11 transcriptional silencing through epigenetic processes among other methods,Citation12 and post-transcriptional microRNA (miRNA) silencing.Citation13–16 Regardless of the cause, a decrease in MHC-I antigen presentation has been associated with resistance to ICI therapy as well as poorer clinical outcomes in solid tumors.Citation17,Citation18 Tumors may also interfere with the APCs themselves, through methods such as inhibition of DC recruitment, differentiation, and maturation.Citation7
Overcoming downregulation of antigen presentation through these means is an active area of research. Conventional chemotherapy has the potential to increase MHC-I expression, as has been shown with gemcitabine in colon, breast, and lung cancer cell lines.Citation19,Citation20 Similar findings were described by Messaoudene et al. in breast cancer patients treated with neoadjuvant chemotherapy, but differential immune responses were seen in primary tumors versus metastatic lymph nodes.Citation21 There are also indications that radiation similarly increases MHC-I density on tumor cells.Citation22 Taken together, these findings imply that traditional anti-cancer treatments have been modulating immune responses even without the explicit intention of doing so. It is possible that the intentional use of traditional chemotherapeutics for these effects (and not necessarily pure cytotoxicity) can be combined with immunotherapies for synergistic activity.
However, more targeted mechanisms for increasing MHC-I expression have also been identified. Transcriptional induction of MHC-I expression and subsequent antigen presentation was accomplished pre-clinically through the modulation of the JAK-STAT pathway (through NF-κ)Citation23 via administration of interferons (especially IFNγ at low doses),Citation24,Citation25 reduction in TRAF3,Citation26 and the use of CDK4/6 inhibitors.Citation27 Increases in MHC-I have also been seen with other changes in IFNγ and NF-κB pathways, involving application of retinoids, modulating expression of NLRC5, and agonism of Stimulator of Interferon Genes (STING).Citation13,Citation28 Therapeutic targeting of these mechanisms is under investigation, with multiple trials combining ICIs with treatments such as pegylated interferons (NCT04943679), STING agonists (NCT04609579), and CDK4/6 inhibitors (NCT05139082) in both the upfront and ICI-resistant settings. These trials, along with other active investigations into overcoming immune resistance, are summarized in . Overall, changes to transcriptional regulation of MHC-I and other methods of increasing its expression may serve a crucial purpose in overcoming both primary and adaptive resistance to immunotherapies.
Meanwhile, adoptive T cell therapies remain a hopeful method of circumventing the need for antigen presentation on MHC-I altogether. CAR T cells and TCEs are not reliant on MHC-I for anti-tumor activity, as they rely on manufactured mechanisms of immune activation. However, tumors present adaptive challenges to these treatments as well. They may either downregulate expression of cell surface antigen- or change-specific immunogenic epitopes, and this has been demonstrated in both hematologic malignancies (such as the therapeutic target CD19) and glioblastoma (IL13R⍺2).Citation29,Citation30 This, along with the antigen heterogeneity and low antigen density in solid tumors, is the reason that there is enthusiasm for developing such therapies to recognize multiple antigens.Citation2,Citation31
Cytokines
As previously noted, IFNγ signaling is crucial in the presentation of antigens to CTLs to identify tumors for killing. IFNγ is produced by T cells and NK cells through signaling by interleukin-12 (IL-12) and IL-18, inducing effects on both malignant and immune cells. IFNγ (type II IFN), along with types I (e.g. IFN⍺) and III (e.g. IFNγ) interferons, mediate upregulation of MHCs, modulation of apoptosis and differentiation, activation of the immune response, and inhibition of tumor growth.Citation32 However, multiple other cytokines are integral to anti-cancer immunity. IL-2 is produced primarily by CD4+ T cells and controls and supports the expansion of CD8+ CTLs, but it can also contrarily induce immunosuppression by inducing T regulatory cells (Tregs).Citation33 Cytokines with direct effects on the proliferation, activation, or support of anti-tumor CTLs also include IL-12, IL-15, and IL-21, among others.Citation34
Exploitation and hijacking of these interconnected signals by tumors can impair normal immune functions. For instance, it has been shown that persistent IL-2 production in the tumor microenvironment (TME) can surpass CD8+ T cell activation and induce T cell exhaustion by way of a pathway involving STAT5 and aryl hydrocarbon receptor (AhR).Citation35 In fact, this is one rationale for combining AhR inhibitors with immunotherapies in ICI-resistant tumors, as some clinical trials are now investigating (e.g. NCT05472506). Deletion of the genes encoding IL-2, IL-21, and especially IL-15 in colorectal cancers have also been shown to be associated with higher risk of recurrence, metastatic disease, and poorer patient survival, indicating both the importance of these signals in the TME and possible role for therapeutic (exogenous) administration to overcome this immune avoidance.Citation36
Unfortunately, the simple systemic administration of interleukins and interferons at anti-neoplastic doses has historically come with hard-to-tolerate or unacceptable toxicities, and short half-lives can limit their utility.Citation37 Additionally, because of the paradoxical pro-tumor effects that these signals may induce under different conditions, they do not represent the refined tools necessary to elicit the desired response. Still, modifications to these cytokines may allow their administration, inducing an anti-tumor effect while minimizing on-target, off-tumor toxicities.Citation38 One such example is a tumor-conditional pro-IL-15, which uses IL-15 Rβ fused to IL-15/IL-15 R⍺ to overcome ICI resistance, increase intratumoral CD8+ T cells, and improve the toxicity profile in mice.Citation39 Similarly, when a pro-IL-2 (IL-2 prodrug preferentially activated in tumors) was combined with checkpoint blockade in a model of anti-PD-L1 resistant mice, anti-tumor responses were seen with relatively minimal toxicity.Citation40 Comparable attempts at overcoming ICI resistance are being undertaken using variations of IL-21.Citation41 Modifications to interleukins – along with combination strategies – may very well make them viable cancer therapeutics despite their limited roles thus far.
Meanwhile, tumor and TME production of IL-1, IL-6, IL-8, IL-10, colony stimulating factor-1 (CSF-1), and transforming growth factor-beta (TGF-β) can contribute directly to immunosuppression.Citation42,Citation43 Attempts to combine targets of these cytokines with existing immunotherapies are ongoing. For instance, the anti-IL-6 antibody tocilizumab is currently under investigation in combination with atezolizumab in patients with lung cancer who progressed on ICIs (NCT04691817). Tocilizumab has been shown to decrease the growth of oral squamous cell carcinoma in a murine model,Citation44 and its combination with anti-PD-L1 in mice is shown to enhance Th1 response and induce a synergistic therapeutic response, even in the setting of ICI resistance.Citation45–47 Still, clinical applications of IL-6 blockade have been limited to managing immunotherapy toxicities rather than potentiating their anti-cancer effects, as efficacy of this approach has not been established. Significant limitations in efficacy have also been seen with the anti-IL-1β agent canakinumab in lung cancer when combined with pembrolizumab (NCT03631199), but this is another potential strategy for overcoming tumor resistance to immunotherapy. In order to overcome the immunosuppressive aspects of the TME and drive more robust immune responses, co-administration of CAR T cells with low-dose IL-2 has been attempted, while others have modified (“armored”) CAR T cells to express a dominant negative receptor designed to inhibit TGF-β signaling.Citation48,Citation49
TGF-β is a highly pleiotropic cytokine that plays an important role in wound healing, angiogenesis, immunoregulation, and cancer. There are three isoforms (TGF-β1, TGF-β2, and TGF-β3), all of which signal through the same TGF-β receptor complex. All three isoforms are expressed as inactive protein complexes, and release of the active growth factor is required to allow signaling via the receptor complex. Signaling of the TGF-β pathway involves binding of TGF-β ligands to a type II receptor on the cell membrane, which recruits and phosphorylates a type I receptor. Phosphorylation of the type I receptor, in turn, phosphorylates the signaling factor Smad2 or Smad3, and trimerizes with Smad4 to enter the nucleus and bind to transcription factors to regulate the expression of specific genes.Citation50 Recent studies have revealed enhanced TGF-β signaling in some cancer patients who do not respond to ICIs, suggesting immunosuppressive changes in TME due to TGF-β signaling in the stroma that restrains anti-tumor immunity; some studies further indicate there is enhanced anti-tumor activity by ICIs when used in combination with a TGF-β inhibitor.Citation51–55
While multiple agents targeting TGF-β have been developed with a focus on the physiological effects in the TME, TGF-β inhibitors that inhibit multiple isoforms have been shown to have serious cardiac toxicity and bleeding in non-clinical studies.Citation56–58 In the clinical development of a pan-TGF-β inhibitor, a variety of on-target toxicities have been observed related to disruption of physiologic TGF-β signaling, including clinically significant bleeding.Citation59–62 Mouse and human genetic data suggest that some of the toxicity, including cardiotoxicity, may be related to inhibition of TGF-β isoforms 2 and 3.Citation63,Citation64 Recent analysis of RNA sequencing data from The Cancer Genome Atlas identified TGF-β1 as the most prevalent TGF-β isoform in solid tumors, which suggest TGF-β1 may be important in tumors.Citation54 Therefore, selectively inhibiting TGF-β1, while sparing isoforms 2 and 3, may lead to a potentially safer and more effective approach to targeting the TGF-β pathway in tumors and may result in potentiation of ICIs, and this approach is currently under investigation.
Finally, a subset of cytokines called chemokines can affect both pro- and anti-tumor responses based on their roles in chemotaxis and cell adhesion. The entire cohort of these signals is outside the scope of this review but is explored extensively elsewhere.Citation65 However, it is important to note that tumors sensitive to immune checkpoint blockade are generally rich in CXCL9, CXCL10, and CXCL11. Meanwhile, tumor and stromal cell production of CCL2 is associated with immunosuppression through recruitment of MDSCs and monocytes, and inhibition of its interaction with CCR2 has the potential to sensitize tumors to anti-PD-1 treatment in vitro.Citation66 Cancers that exhibit loss of the phosphatase and tensin homolog gene (PTEN) may be particularly effective at immune evasion through this mechanism, as they exhibit significant upregulation of CCL2.Citation67 Meanwhile, co-targeting of CCL5 with maraviroc and PD-1 with pembrolizumab has been investigated in mismatch repair-proficient colorectal cancer in the phase I PICASSO trial.Citation68 Although objective responses were underwhelming, the patient population had a longer survival than would be expected. Finally, developers of CAR T cells have also sought to more accurately target tumors by incorporating chemokine receptors into their constructs, including CXCR1, CXCR2, CCR4, CCR2b.Citation69
In summary, although cytokines have so far had limited success in cancer due to both middling effectiveness as monotherapies and unacceptable toxicity profiles, recent advancements may allow them to be incorporated into cancer care. They have shown most notable promise in boosting the effects of existing checkpoint inhibitors and CAR T cell therapies, and these parallel immunotherapy approaches may prove to be how cytokine treatments ultimately reach patients.
Costimulatory signals and immune checkpoints
Once T cells have been primed for antigen recognition, have been activated, and are recruited to the proximity of tumor cells by chemokines, they rely on a balance of stimulatory and inhibitory signals to direct their killing. This is accomplished through what are called immune checkpoints, the arbiters of these signals, beyond the interaction between MHC and the T cell receptor (TCR). In general, cancer therapeutics either block inhibitory checkpoint signaling (including PD-1, CTLA-4, LAG-3, TIM-3, TIGIT, BTLA, and VISTA) or induce stimulatory checkpoints (including CD27, CD28, 4-1BB, CD40, OX40, GITR, and ICOS) in order to generate anti-tumor responses.Citation70
The first checkpoint inhibitor was approved by the FDA in 2011, and since then the dominant immunotherapies both in clinical use and under active scientific investigation have been ICIs. Unfortunately, well-established monoclonal antibodies directed against CTLA-4 and PD-1/PD-L1 have shown activity only in certain solid tumors and limited to certain conditions such as high tumor mutational burden and microsatellite instability-high (MSI-H). Moreover, while responses can be durable in some cases of advanced/metastatic disease, resistance ultimately develops in the majority.
The mechanisms by which tumors develop resistance to ICIs are manifold. Resistance may be primary or acquired, and it generally occurs because of insufficient generation of effector T cells, dysfunction of those T cells, or lack of memory T cell development.Citation6 While these steps involve the aforementioned biological machinery (i.e. antigen presentation, cytokines, and chemokines), there is ongoing interest in determining what modifies surface expression of PD-1/-L1 and other checkpoints, as this dictates responses to ICIs (albeit not always reliably).Citation71–74 After transcription and translation, regulation of surface inhibitory immune checkpoints relies on some combination of delivery to the lipid bilayer of cells, glycosylation, internalization, and recycling or ubiquitination.Citation70 However, these are yet untargetable by clinically available drugs. Therefore, overcoming inhibitory checkpoint resistance largely relies on increasing antigen presentation/recognition, modulating the balance of cytokines, targeting multiple checkpoints simultaneously, and reducing the effect of immunosuppressive tumor-associated macrophages (TAMs) and MDSCs.
The efficacy of co-blockade of inhibitory checkpoints was first demonstrated through the use of ipilimumab (anti-CTLA-4) and nivolumab (anti-PD-1) in melanoma, and this combination has been expanded to other tumors including NSCLC and renal cell carcinoma.Citation75–77 More recently, relatlimab (anti-LAG-3) plus nivolumab was shown to have improved clinical efficacy compared with PD-1 blockade alone in patients with advanced melanoma with a 12-month progression-free survival (PFS) 47.7% vs. 36%.Citation78 This is a logical approach to overcoming the resistance to single checkpoint blockade because of the compensatory surface upregulation of other inhibitory checkpoints.Citation79 Attendant to these findings, combinations of anti-PD-1/-L1 treatments with ICIs targeting other inhibitory checkpoints have been under investigation in both pre-clinical and clinical trials, including those that target VISTA (e.g. NCT05082610),Citation80 TIM-3 (e.g. NCT02608268),Citation81 BTLA (e.g. NCT04137900),Citation82 and LAG-3 (e.g. NCT04080804).Citation83 Bispecific antibodies targeting multiple inhibitory checkpoints may also provide the opportunity to administer one agent with effects on two checkpoint targets such as TIM-3 and PD-1.Citation84,Citation85 Finally, some CAR T cell products have begun to incorporate dominant negative receptors (DNRs). A PD-1 DNR, for instance, provides a nonfunctional “decoy” receptor for tumors expressing PD-L1, allowing T cells to achieve better cytotoxicity.Citation86 Although the most common of these approaches has focused on the PD-1/PD-L1 checkpoint, others have incorporated DNRs against Fas and TGF-β.Citation87,Citation88
Targeting stimulatory checkpoints is also a viable way to overcome resistance to existing ICIs and adoptive T cell therapies. In fact, the rescue of T cell exhaustion during PD-1 therapy may be dependent on stimulatory factors such as CD28.Citation89 CD28 and 4-1BB are used as costimulatory domains in CAR T cell products, with neither demonstrating convincing benefit over the other.Citation90 Although agonists of such checkpoints are biologically promising as immune activators, they have historically been limited by toxicity in the form of cytokine release syndrome.Citation91 However, there are still models of combined 4-1BB stimulation and PD-1 inhibition that have some promise due to anti-tumor effect.Citation92 Similar models of CD27 checkpoint stimulation with PD-1 blockade have been established, and this includes synergism with adoptive T cell therapy.Citation93 In fact, many recent investigations have focused on multi-targeting through the use of bispecific antibodies. These new approaches may be able to potentiate anti-PD-1/-L1 therapies when given together at the outset, or else allow for improved and continued efficacy of established ICIs even after progression on those agents. Currently, they are being investigated in trials such as NCT05263180 (anti-PD-L1×OX40), NCT05442996 (anti-EGFRx4-1BB plus anti-PD-1), and NCT03809624 (anti-PD-L1×4-1BB). As compared with blocking inhibitory checkpoints, the stimulatory checkpoint approach has not advanced as quickly to clinical uses despite viability in the lab setting. These investigations provide hope that there is still a role for it, as long as their anti-tumor activity can be balanced against inflammatory adverse effects.
Cell-dependent immunosuppressive processes
Several immunosuppressive processes exist in order to keep the immune system from becoming so activated that it damages its host. Some of these have been reviewed already, as they are natural processes – inhibitory checkpoints, for instance – that are co-opted by cancers and their microenvironments. However, certain cells that mediate the dampening of an immune response deserve mention, as reducing their function may reinvigorate immune responses in tumors that have become resistant to immunotherapies. These include myeloid derived suppressor cells (MDSCs), tumor associated macrophages (TAMs), tumor associated neutrophils (TANs), and regulatory T cells (Tregs).
Historically, both characterization of MDSCs and their distinction from the myeloid lineage TAMs/TANs have been difficult.Citation94 Broadly, it is held that MDSCs have a pro-tumor effect whereas TAMs and TANs can either aid or inhibit cancer growth. Further, MDSCs represent a spectrum of cells and can be divided into polymorphonuclear (PMN-MDSC) and monocytic (M-MDSC) phenotypes. They support other immunosuppressive elements of the tumor and immune system like Tregs through arginase and indoleamine 1,2-dioxygenase (IDO)-dependent pathways; directly interfere with T cell functioning by producing reactive oxygen species, nitric oxide, and cytokines; modulate checkpoint expression; act as a source of chemokines; and mediate carcinogenesis, cancer progression, and metastasis.Citation95 Meanwhile, TAMs and TANs also have roles in tumor initiation, angiogenesis, and metastasis, inspiring treatments focus on these myeloid lineage cells as a strategy to induce or reinvigorate anti-tumor immune responses.Citation96
Since MDSCs are regulators of immunosuppression, they have been targeted as ways to overcome both primary and acquired resistance to immunotherapies. Platinum drugs have shown some activity against MDSCs, with evidence that cisplatin reduces PMN-MDSCs and oxaliplatin reduces M-MDSCs in vitro, indicating that different cytotoxic therapies may be able to target specific populations.Citation97–99 Bevacizumab has also demonstrated an ability to decrease PMN-MDSCs in lung cancer because of the reliance of these cells on VEGF signaling.Citation100 Other attempts at decreasing the number and/or function of MDSCs in tumors – or else forcing their differentiation into more mature cells – have used agents such as those targeting calgranulin A and B, anti-IL-1γ drugs, all-trans retinoic acid (ATRA), cytokine signaling inhibitors (CCL2/CCR2, CXCR1/2), histone deacetylase (HDAC) inhibitors, COX2 inhibitors, PDE5 inhibitors, and toll-like receptor (TLR) agonists.Citation101 Much of the evidence is currently pre-clinical, but some early phase data has been promising. For instance, adding an HDAC inhibitor (entinostat) to pembrolizumab after progression on ICI was associated with an overall response rate of 9.2% and median duration of response of 10.1 months in patients with lung cancer, and more benefit was seen in patients with high levels of circulating monocytes.Citation102
Meanwhile, targeting of TAMs mainly has focused on the tendency toward M2 polarization, which establishes an overall immunosuppressive state – though this characterization is an oversimplification of dynamic processes. The balance of M2 over M1 polarization may be at least partially related to hypoxia in the TME that occurs along with abnormal angiogenesis; this hypoxia then induces the triggering receptor expressed on myeloid cells-1 (TREM-1).Citation103,Citation104 In fact, a clinical trial is investigating a TREM-1 inhibitor in combination with pembrolizumab in patients with advanced cancers, including those who progressed on ICI (NCT04682431). TREM-2 is being targeted for similar reasons (NCT04691375). Furthermore, signaling between CSF-1 and its receptor (CSF-1 R), expressed on macrophages, aids in the survival and proliferation of TAMs.Citation105 Unfortunately, a phase I trial of pexidartinib (a CSF-1 R inhibitor) with durvalumab did not have significant clinical activity in patients with advanced pancreatic and colorectal cancers.Citation106 Zoledronic acid (ZA) may be another way to inhibit M2 polarization of TAMs, and a small study of ZA + ICI in patients with lung cancer showed improved clinical responses when compared with ICI therapy alone.Citation107 Trials of interventions targeting TANs (i.e. with PDE5 inhibitors, COX2 inhibitors, etc.) are even more limited,Citation96 especially in the setting of immunotherapy resistance, although there is active interest in better characterizing in vivo effects of these cells, as is being investigated in bone sarcomas (NCT04867421).
Regulatory T cells (Tregs) are a subset of CD4+ T cells that aid in immunosuppression through a number of mechanisms, and their activity is especially increased in the setting of PD-L1 targeting by ICIs.Citation108 Tregs have been difficult to specifically target with treatments, as anti-Treg therapies can have more broad effects on other immune cells populations, as well. However, Treg depletion with anti-CD25 agents like daclizumab may be able to selectively reduce the effects of Tregs in the TME, and this approach has been used to augment the effects of a dendritic cell vaccine in a phase I/II trial in patients with melanoma.Citation109 Inhibiting CCR4 with mogamulizumab, in combination with nivolumab, has also been shown to reduce populations of Treg cells while simultaneously increasing tumor infiltrating lymphocytes (TILs).Citation110 However, while a similar approach with mogamulizumab plus other ICIs (durvalumab or tremelimumab) also showed decreases in peripheral and tumoral Tregs, this did not correlate with clinical outcomes.Citation111 Similar to other approaches mentioned above, cytotoxic chemotherapy (especially cyclophosphamide),Citation112 checkpoint blockade, use of small molecules, and agents acting on angiogenesis have demonstrated effects on Tregs as they have with various other immune cells. However, narrowing the effects of these therapies to a small subpopulation of T cells remains a challenge in the clinical setting.
Cancer associated fibroblasts
Cancer-associated fibroblasts (CAFs) are a functional part of the TME that are derived from mesenchymal tissues. They have a number of roles that are distinct from normal fibroblasts, and some of these relate to the immune nature of tumors.Citation113 CAFs participate in complex interactions with T cells, MDSCs, TAMs, and TANs, and they provide a primary source of pro-tumor cytokines and chemokines (most notably TGF-β, IFNγ, and CXCL12).Citation114 Among other mechanisms, CAFs have also been shown to cause resistance to traditional cancer treatments like chemotherapy and radiation by mediating the desmoplastic reaction, as is seen in gastrointestinal cancers – most notably in pancreatic cancer, a quintessential example of an immunologically “cold” tumor.Citation115 Therefore, targeting CAFs has more recently garnered interest as a method to improve the efficacy of cancer therapeutics, especially with regards to immune processes.
One strategy to decrease the immunosuppressive effects of CAFs in tumors has been to prevent differentiation of resident fibroblasts into CAFs by blocking signaling of the TGF-β family. In a model of gastrointestinal stromal tumor (GIST), administering an anti-TGF-β1 antibody inhibited the transition of resident fibroblasts to CAFs.Citation116 Approached a different way, agonism of liver X receptors (LXR) inhibits signaling of TGF- and thus CAF differentiation.Citation117 A unique approach was taken by Freedman et al., who used an oncolytic virus that expressed a TCE targeting fibroblast activation protein (FAP) on CAFs. This caused T cell dependent killing of CAFs with an attendant increase in gene expression for checkpoint markers like CTLA-4 and LAG-3, decreases in fibroblast-associated gene expression, and repolarization of M2 macrophages.Citation118 Although these approaches have promise in reducing the immunosuppressive effects of CAFs, it remains to be seen whether they can aid in overcoming adaptive resistance to immunotherapies like ICIs and adoptive T cell therapies in the clinic. However, there is at least some pre-clinical evidence that interfering with functions of CAFs – such as blocking the interaction of CXCL12-CXCR4 and inhibiting the NOX4 enzyme – can enhance or restore responses to immunotherapies.Citation119,Citation120 Still, targeting CAFs is a more nascent area of research, meaning that any understanding of their roles is incomplete. Since they clearly affect the immune TME they remain a potential target for novel drug development – especially for tumors with well-defined desmoplastic reactions like those of the pancreas and breast.
Specific tumoral mutations with immunologic implications
Tumor-specific mutations play a critical role in the regulation of the TME, often generating a hostile local environment and characteristic chemical changes. This provides insight into how these mutations impart resistance to immunotherapies. Some of the most important specific mutations that affect tumor immunityacross solid malignanciesare those in tumor suppressor genes, proto-oncogenes, and primary driver mutations.
Several tumor suppressor genes have been characterized for their role in oncogenesis and their effect on immunotherapy response. PTEN mutations are implicated in the proliferation of numerous cancers and result in enhanced MAPK signaling, increased spatial clustering of tumor cells, and decreased CD8+ CTL tumor infiltration.Citation121 The release of immunosuppressive cytokines and modulation of the TME is correlated with decreased response to anti-PD-1 therapy in PTEN-mutated melanoma and glioblastoma patients, suggesting that combined MAPK inhibition with anti-PD-1 therapy may present an opportunity for future therapies and an ongoing area of needed research.Citation67,Citation121 Additionally, F-box/WD repeat-containing protein 7 (FBXW7) acts upon cellular proliferation targets including NOTCH1 and c-MYC, and mutations can be seen in a variety of malignancies including breast, colorectal, gastric, hepatocellular, and NSCLC.Citation122 Loss of FBXW7 impairs cytokine signaling, decreases CD8+ CTL infiltration, and decreases dsRNA sensing, corresponding with a diminished response to anti-PD-1 therapy. As such, restoration of dsRNA sensing results in a restored response.Citation123 Additionally, serine/threonine kinase 11 (STK11), also known as liver kinase B1 (LKB1), activates AMPK to inhibit growth via the mTOR pathway, and inactivation or mutation of STK11 has a profound effect on the TME via increased neutrophil recruitment through cytokine (IL-1) and chemokine (CXCL7, G-CSF, STAT3) production, increased IL-6 expression, and decreased CD8+ CTL infiltration.Citation124,Citation125 In fact, STK11-mutated NSCLC displays decreased PD-L1 expression and poor response to anti-PD-1 therapy, though this response was reversed with the administration of IL-6 blockade.Citation124
Proto-oncogenes have also been shown to modulate the TME, with both beneficial and adverse effects noted from an immunotherapeutic perspective. One notable proto-oncogene with pro-inflammatory TME modulation and induction of immunotherapy resistance is pescadillo ribosomal biogenesis factor 1 (PES1). PES1 is over-expressed in a variety of tumors and regulates the PI3K cell signaling pathway, enhancing malignant cellular proliferation.Citation126 PES1 expression also decreases IL-15 production, reduces CD8+ CTL esophageal tumor cell infiltration, and diminishes response to anti-PD-1 therapy; murine models suggest targeted knockdown of PES1 with co-administration of anti-PD-1 treatment resulted in improved CTL infiltration and increased pathologic response rates, and investigation of combined treatment with anti-PD-1 therapy and PES1 inhibition with CDK inhibitors represents an exciting area of future clinical inquiry.Citation127–129 Gain of function mutations and overexpression of TP53 are shown to correlate with overexpression of cytokines promoting angiogenesis, inflammation and cellular migration; this includes CCL2, which is associated with IL-6 upregulation and may confer resistance to immunotherapy through increased recruitment of Tregs.Citation130 On the other hand, other TP53 mutations are associated with increased tumor mutational burden (TMB), increased PD-1 expression, and increased CD8+ CTL tumor infiltration, which conversely may enhance the efficacy of immunotherapy; in fact, preclinical murine models have suggested targeted therapies to TP53 given in combination with anti-PD-1 therapies may enhance immunotherapy response, though this treatment approach requires further clinical trial investigation.Citation131–133
Finally, driver mutations have reshaped the classification and treatment of numerous malignancies, further supported by the emergence of targeted therapies. One such cancer is NSCLC, for which medical management is greatly influenced by the presence of EGFR, ALK, and other aberrations, as available targeted treatments have become first-line options. The presence of these mutations is shown to have a significant impact on the TME, as oncogene-associated lung adenocarcinoma has been shown to display low TMB, decreased expression of PD-1, and decreased CD8+ CTL tumor infiltration, which may contribute to their immunotherapy resistance.Citation134–136 Although many NSCLC immunotherapy clinical trials have excluded patients with EGFR or ALK mutations, several large trials included patients with these mutations which were then assessed via subgroup analyses. EGFR-mutated patients in CheckMate-057 (nivolumab versus docetaxel), CheckMate-010 (pembrolizumab versus docetaxel), and the OAK trial (atezolizumab versus docetaxel) did not experience improved OS with immunotherapy, and those in the BIRCH trial (atezolizumab) showed a 19% response rate in the first line setting (versus 23% in EGFR-wild type patients) and almost no response in the second line and beyond.Citation137–140 Conversely, EGFR- and ALK-mutated patients in the IMpower150 trial demonstrated an improvement in PFS with the addition of atezolizumab to bevacizumab and chemotherapy.Citation141 In the ATLANTIC trial, 12.2% of pre-treated patients with either mutation and PD-L1 expression more than 25% had encouraging responses to durvalumab, but this was less than counterparts who were EGFR- or ALK-wild type.Citation142
Nonetheless, attempts have been made to combine immunotherapy with mutation-targeted therapy or chemotherapy to overcome resistance, but these have encountered significant setbacks.Citation143 The TATTON trial, a phase Ib trial of osimertinib with durvalumab in EGFR-mutated NSCLC, found the combination not feasible due to increased incidence of interstitial lung disease (22%); this finding also resulted in the early closure of the phase III CAURAL trial, in which 1/12 patients reported development of interstitial lung disease.Citation144,Citation145 KEYNOTE-789, a phase III trial of platinum chemotherapy and pemetrexed with or without pembrolizumab in EGFR-mutated NSCLC patients who previously progressed on EGFR-targeted therapy, was closed early after no difference in OS was observed compared to placebo.Citation146 Similarly, disappointing results were noted in CheckMate-722, a phase III randomized trial which failed to show difference in PFS with the addition of nivolumab to platinum chemotherapy and pemetrexed in EGFR-mutated NSCLC patients who previously progressed on EGFR-targeted therapy.Citation147
Metabolic characteristics of tumors
Since the description of the Warburg effect in the 1920s, there has been considerable attention to the unique metabolic signature of cancer cells and how these mechanisms can be leveraged to improve cancer therapies. Enhanced glycolysis and lactic acid formation outside the mitochondria by tumor cells – a major source of energy production for increased cellular proliferation – results in glucose deprivation. This subsequently suppresses tumor-infiltrative CD4+ and CD8+ T cells, decreases CTL tumor infiltration, and profoundly alters the TME.Citation148–150 Acidification due to increased production of lactic acid directly decreases the cytotoxic activity of T cells while increasing recruitment of MDSCs to promote an immunosuppressive environment.Citation151 Nutrient depletion also results in a hypoxic environment with increased expression of hypoxia inducible factors (HIFs), which increase recruitment of MDSCs and Tregs while upregulating tumor expression of PD-L1.Citation152 Normalization of pH has been shown to increase CTL infiltration into tumors and improve response to immunotherapy, and numerous therapeutic strategies to mitigate TME acidification are currently being evaluated including proton transport inhibitors and alkalinizing agents.Citation153,Citation154
Critical attention has also been given to the utilization of specific ammino acids in TME modification. Tryptophan, whose degradation is mediated by the enzyme indoleamine 2,3-dioxygenase (IDO), is critical for T cell activation and proliferation.Citation155 IDO expression is a key feature of many tumors and serves as an intrinsic mechanism for immune resistance, further supported by IDO-mediated increased T cell PD-1 expression.Citation156,Citation157 While preclinical models have shown the potential of IDO1 inhibitors in combination with immunotherapy, the phase III trial KEYNOTE-252/ECHO-301 failed to show efficacy of epacadostat (an IDO1 inhibitor) with pembrolizumab compared to pembrolizumab alone in metastatic melanoma; nonetheless, epacadostat and alternative agents targeting tryptophan degradation, paired with ICIs, have been assessed via clinical trial in the treatment of various solid tumors (e.g. NCT03414229, NCT03459222).Citation158,Citation159
Arginine and glutamine have also been implicated in TME modification and immunotherapy resistance. Arginine depletion occurs in cancer cells through a variety of mechanisms, including decreased arginine synthesis via downregulation or silencing of arginosuccinate synthetase (ASS1) and increased arginine metabolism via HIF-mediated activation of arginase 1 (Arg1).Citation144,Citation152 Elevated arginase levels are associated with quiescence of CD8+ CTLs and Treg survival, promoting an immunosuppressive environment.Citation160,Citation161 Additionally, Arg1-mediated arginine metabolism produces urea and ornithine, the latter of which is used in the synthesis of polyamines which are necessary for cellular proliferation.Citation162
Glutamine is a critical amino acid for rapid cellular division, and many tumor cells express glutamine-uptake transporters to competitively harvest glutamine from the local environment to support their glutamine addiction.Citation163 Glutamine-deficient tumor-infiltrative CTLs have a diminished anti-tumor immune response, and glutamine restriction is associated with increased CD4+ Treg proliferation.Citation164,Citation165 Studies of both arginase inhibitors and glutaminase inhibitors with immunotherapy are ongoing, but none have achieved FDA approval yet, with studies such as the CANTANA trial (telanglenastat, glutaminase inhibitor) failing to show clinical benefit.Citation166
Nonetheless, manipulation of amino acid metabolism in cancer therapy remains both an area of needed research and potentially exciting future therapeutic option.
Cancer cell death
Programmed cell death (PCD), and the ways cancer cells evade and resist this process, is one of the hallmarks of cancer.Citation167 PCD via apoptosis, efferocytosis, and necroptosis result in a complex cascade of biochemical signals including chemokines, mitogens, and extracellular vesicles that promote chemotaxis and cellular proliferation in the TME.Citation168 The release of danger-associated molecular patterns (DAMPs) such as calreticulin (CRT), heat shock proteins (HSPs), and high mobility group box protein 1 (HMGB1) after PCD promote a pro-inflammatory state and increased activity of dendritic cells, NK cells, and CTLs.Citation169,Citation170 Through the process of immunoediting, immunogenic tumor cells associated with the aforementioned processes are selectively lost during tumor proliferation. Combined with a host of alterations to antigen presentation and lymphocyte proliferation and activation, this may serve as a significant mechanism of immunotherapy resistance.Citation171,Citation172
A variety of novel treatments are under active investigation to induce immunogenic cell death and enhance immunotherapy efficacy. Vaccine therapies promote tumor antigen uptake with APC activation and CTL tumor infiltration, which works synergistically with conventional immunotherapy.Citation173,Citation174 Vaccines utilizing both tumor-associated antigens (TAAs) and tumor-specific antigens (TSAs) have been studied in an array of primary tumor types including leukemia, NSCLC, gynecologic, genitourinary, and neurologic cancers.Citation174,Citation175 Radiation promotes immunogenic cell death via release of DAMPs such as HMGB1 which subsequently increase antigen presentation and CD8+ CTL proliferation.Citation176 Combination radiotherapy and immunotherapy has been utilized successfully across a variety of primary tumors, and the addition of radiotherapy may both stimulate immunogenically “hot” tumors and convert immunogenically “cold” tumors.Citation177–179 Meanwhile, utilization of pulsed electric fields (PEFs) is associated with the release of DAMPs such as HMGB1 and CRT which modulate the TME, resulting in decreased Tregs and MDSCs with increased CD8+ CTLs.Citation180–182 Preclinical data with combination PEF and immunotherapy shows elevated HMGB1, increased CD8+ CTL tumor infiltration, and improved survival compared to immunotherapy alone.Citation183–185
Microbiota
The role of the microbiome has been explored in a wide spectrum of diseases, and recent attention has been given to its contribution to cancer pathogenesis and immunotherapy responsiveness. Bacteria and bacterial metabolites act upon a variety of cells including DCs, stimulating T cell activation and cytokine release resulting in profound modulation of the local inflammatory environment; specific genera including Bifidobacterium have been shown to increase lymphocyte recruitment and T cell activation, while others including Enterococcus and Akkermansia increase central memory CD4+ T cells.Citation186,Citation187 Higher gut microbiome diversity is associated with an increased response to anti-PD-1 therapy in melanoma patients, and certain bacterial species including Bifidobacterium longum, Enterococcus faecium, and Collinsella aerofaciens were found in higher quantities in patients with metastatic melanoma who had clinical responses to anti-PD-1 therapy.Citation188,Citation189 Conversely, relatively low levels of these bacteria have been correlated with non-response to anti-PD-1 therapy, as with Akkermansia muciniphila in non-responders to anti-PD1 therapy with lung and kidney cancers.Citation190 As demonstrated by Routy et al., fecal microbiota transplantation (FMT) from non-responders to anti-PD-1 therapy to antibiotic-treated mice failed to improve anti-PD-1 response, but oral supplementation with Akkermansia muciniphila restored responsiveness to anti-PD1 therapy.Citation190 This observation has been further explored by evaluating cancer patients with suspected dysbiosis related to antibiotic use. A recent meta-analysis included 12 studies enrolling 6,010 cancer patients and analyzed concurrent use of antibiotic with ICIs. Results indicated significantly worse PFS and OS with concurrent use of ICIs and antibiotics compared to those without antibiotic use.Citation191 This observation provides further credence to the role of microbiota in cancer-immunity regulation and opens potential avenues for future research.
From these observations, the safety and efficacy of FMT as a strategy to overcome immunotherapy resistance has gained early clinical trial support. A phase I trial of 10 patients with anti-PD-1 refractory metastatic melanoma received FMT from 2 donors with documented complete response (CR) to anti-PD-1 therapy. Two patients achieved partial response (PR) at 90 days, while 1 achieved CR and none demonstrated grade 2–4 adverse events from FMT.Citation192 Similarly, a phase II trial of 16 patients with anti-PD-1 refractory melanoma received FMT from 7 donors with documented CR or PR to anti-PD-1 therapy, and 6 of 15 assessed patients experiencing clinical benefit.Citation193 Notably, response to anti-PD1 therapy after FMT was associated with increased CD8+ CTL activation, decreased IL-8 expression, increased TNF expression, and durable modification of microbiome composition with increased expression of bacteria such as Akkermansia muciniphilia. Ongoing studies seek to build upon these foundational findings, including a randomized double-blinded phase Ib/IIa trial of 24 metastatic melanoma patients to receive FMT from either anti-PD-1 responders or non-responders (NCT05251389).Citation194
Conclusion
The natural immune response to solid tumors is a carefully coordinated one. Intrinsically, it involves a host of cell–cell interactions through antigen presentation and immune checkpoints, direct cell killing, inflammatory signaling via cytokines and chemokines, and a balance with immunosuppressive components at each of these steps. However, it is also extrinsically influenced by other elements in the TME such as CAFs, driver and immunity-influencing mutations, methods of cancer cell death, and the host gut microbiome. Because of these observations, cancer immunity dysregulation has increasingly become recognized not only as a harbinger of carcinogenesis but also as a target for novel therapeutics. This review is unable to address every method by which tumors exhibit either primary or adaptive resistance to immunotherapies, especially as more mechanisms are being discovered through continued investigation.
However, with each one of these methods of resistance – both intrinsic and extrinsic – comes a possible way to overcome it. A preponderance of strategies is still being investigated in the pre-clinical setting, and some have made it into clinical trials with varying successes. These have mainly focused on how oncologists can overcome the resistance that tumors develop to ICIs, as these immunotherapies have enjoyed first-mover advantage over others. However, this limitation should not bar the investigation into other immunotherapies such as CAR T cell products, vaccines, and T cell engagers, especially because the immunologic methods by which tumors evade these treatments may differ significantly from the methods by which they resist ICIs.
In summary, understanding and overcoming resistance to immunotherapies is a nascent, but rich field of research. The community has much to learn, but small advances in the fundamental science and clinical trial settings give hope that more leaps in effective treatments are on the horizon.
Author contributions
All authors conceptualized the manuscript. DSL and SAB researched, prepared, and drafted the manuscript, and BB wrote, reviewed, and edited the manuscript.
Ethical disclosure
This review article did not include any individual patient information and did not require input from an Institutional Review Board. The authors have no ethical disclosures to report.
Data sharing statement
This review article did not include any individual patient information. Therefore, there is no process by which data sharing needs delineation.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: From t cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651–17. doi:10.1038/s41577-020-0306-5.
- de Miguel M, Umana P, Gomes de Morais AL, Moreno V, Calvo E. T-cell–engaging therapy for solid tumors. Clinical Cancer Research. 2021;27(6):1595–1603. doi:10.1158/1078-0432.CCR-20-2448.
- Nathan P, Hassel JC, Rutkowski P, Baurain J-F, Butler MO, Schlaak M, Sullivan RJ, Ochsenreither S, Dummer R, Kirkwood JM, et al. Overall survival benefit with tebentafusp in metastatic uveal melanoma. N Engl J Med. 2021;385(13):1196–1206. doi:10.1056/NEJMoa2103485.
- Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol. 2015;15(6):388–400. doi:10.1038/nri3839.
- Fu Q, Fu TM, Cruz AC, Sengupta P, Thomas SK, Wang S, Siegel RM, Wu H, Chou JJ. Structural basis and functional role of intramembrane trimerization of the fas/cd95 death receptor. Mol Cell. 2016;61(4):602–613. doi:10.1016/j.molcel.2016.01.009.
- Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. 2018;118(1):9–16. doi:10.1038/bjc.2017.434.
- Jhunjhunwala S, Hammer C, Delamarre L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat Rev Cancer. 2021;21(5):298–312. doi:10.1038/s41568-021-00339-z.
- Kaklamanis L, Townsend A, Doussis-Anagnostopoulou IA, Mortensen N, Harris AL, Gatter KC. Loss of major histocompatibility complex-encoded transporter associated with antigen presentation (tap) in colorectal cancer. Am J Pathol. 1994;145(3):505–509.
- Seliger B, Höhne A, Knuth A, Bernhard H, Ehring B, Tampé R, Huber C. Reduced membrane major histocompatibility complex class i density and stability in a subset of human renal cell carcinomas with low tap and lmp expression. Clin Cancer Res. 1996;2(8):1427–1433.
- Vitale M, Rezzani R, Rodella L, Zauli G, Grigolato P, Cadei M, Hicklin DJ, Ferrone S. Hla class i antigen and transporter associated with antigen processing (tap1 and tap2) down-regulation in high-grade primary breast carcinoma lesions. Cancer Res. 1998;58(4):737–742.
- Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, Bjorgaard SL, Hammond MR, Vitzthum H, Blackmon SM, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun. 2017;8(1):1136. doi:10.1038/s41467-017-01062-w.
- Nie Y, Yang G, Song Y, Zhao X, So C, Liao J, Wang LD, Yang CS. DNA hypermethylation is a mechanism for loss of expression of the hla class i genes in human esophageal squamous cell carcinomas. Carcinogenesis. 2001;22(10):1615–1623. doi:10.1093/carcin/22.10.1615.
- Taylor BC, Balko JM. Mechanisms of mhc-i downregulation and role in immunotherapy response. Front Immunol. 2022;13(844866). doi:10.3389/fimmu.2022.844866.
- Lazaridou MF, Gonschorek E, Massa C, Friedrich M, Handke D, Mueller A, Jasinski-Bergner S, Dummer R, Koelblinger P, Seliger B. Identification of mir-200a-5p targeting the peptide transporter tap1 and its association with the clinical outcome of melanoma patients. Oncoimmunology. 2020;9(1):1774323. doi:10.1080/2162402X.2020.1774323.
- Colangelo T, Polcaro G, Ziccardi P, Pucci B, Muccillo L, Galgani M, Fucci A, Milone MR, Budillon A, Santopaolo M, et al. Proteomic screening identifies calreticulin as a mir-27a direct target repressing mhc class i cell surface exposure in colorectal cancer. Cell Death Disease. 2016;7(2):e2120. doi:10.1038/cddis.2016.28.
- Yi M, Xu L, Jiao Y, Luo S, Li A, Wu K. The role of cancer-derived micrornas in cancer immune escape. J Hematol Oncol. 2020;13(1):25. doi:10.1186/s13045-020-00848-8.
- Park HS, Cho U, Im SY, Yoo CY, Jung JH, Suh YJ, Choi HJ. Loss of human leukocyte antigen class i expression is associated with poor prognosis in patients with advanced breast cancer. J Pathol Transl Med. 2019;53(2):75–85. doi:10.4132/jptm.2018.10.11.
- Hanagiri T, Shigematsu Y, Kuroda K, Baba T, Shiota H, Ichiki Y, Nagata Y, Yasuda M, Uramoto H, So T, et al. Prognostic implications of human leukocyte antigen class i expression in patients who underwent surgical resection for non-small-cell lung cancer. J Surg Res. 2013;181(2):e57–63. doi:10.1016/j.jss.2012.07.029.
- Liu WM, Fowler DW, Smith P, Dalgleish AG. Pre-treatment with chemotherapy can enhance the antigenicity and immunogenicity of tumours by promoting adaptive immune responses. Br J Cancer. 2010;102(1):115–123. doi:10.1038/sj.bjc.6605465.
- Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell. 2015;28(6):690–714. doi:10.1016/j.ccell.2015.10.012.
- Messaoudene M, Mourikis TP, Michels J, Fu Y, Bonvalet M, Lacroix-Trikki M, Routy B, Fluckiger A, Rusakiewicz S, Roberti MP, et al. T-cell bispecific antibodies in node-positive breast cancer: novel therapeutic avenue for mhc class i loss variants. Ann Oncol. 2019;30(6):934–944. doi:10.1093/annonc/mdz112.
- Garnett CT, Palena C, Chakraborty M, Tsang KY, Schlom J, Hodge JW. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic t lymphocytes. Cancer Res. 2004;64(21):7985–7994. doi:10.1158/0008-5472.CAN-04-1525.
- Zhou F. Molecular mechanisms of ifn-gamma to up-regulate mhc class i antigen processing and presentation. Int Rev Immunol. 2009;28(3–4):239–260. doi:10.1080/08830180902978120.
- Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-γ. Annu Rev Immunol. 1997;15(1):15(749–795. doi:10.1146/annurev.immunol.15.1.749.
- Propper DJ, Chao D, Braybrooke JP, Bahl P, Thavasu P, Balkwill F, Turley H, Dobbs N, Gatter K, Talbot DC, et al. Low-dose ifn-gamma induces tumor mhc expression in metastatic malignant melanoma. Clin Cancer Res. 2003;9(1):84–92.
- Gu SS, Zhang W, Wang X, Jiang P, Traugh N, Li Z, Meyer C, Stewig B, Xie Y, Bu X, et al. Therapeutically increasing mhc-i expression potentiates immune checkpoint blockade. Cancer Discov. 2021;11(6):1524–1541. doi:10.1158/2159-8290.CD-20-0812.
- Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, Khan N, Ubellacker JM, Xie S, Metzger-Filho O, et al. Cdk4/6 inhibition triggers anti-tumour immunity. Nature. 2017;548(7668):471–475. doi:10.1038/nature23465.
- Kobayashi KS, van den Elsen PJ. Nlrc5: a key regulator of mhc class i-dependent immune responses. Nat Rev Immunol. 2012;12(12):813–820. doi:10.1038/nri3339.
- Krenciute G, Prinzing BL, Yi Z, Wu MF, Liu H, Dotti G, Balyasnikova IV, Gottschalk S. Transgenic expression of il15 improves antiglioma activity of il13rα2-car t cells but results in antigen loss variants. Cancer Immunol Res. 2017;5(7):571–581. doi:10.1158/2326-6066.CIR-16-0376.
- Ruella M, Maus MV. Catch me if you can: leukemia escape after cd19-directed t cell immunotherapies. Comput Struct Biotechnol J. 2016. 14:14(357–362. doi:10.1016/j.csbj.2016.09.003.
- Marofi F, Motavalli R, Safonov VA, Thangavelu L, Yumashev AV, Alexander M, Shomali N, Chartrand MS, Pathak Y, Jarahian M, et al. Car t cells in solid tumors: challenges and opportunities. Stem Cell Res Ther. 2021;12(1):81. doi:10.1186/s13287-020-02128-1.
- Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer. 2016;16(3):131–144. doi:10.1038/nrc.2016.14.
- Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol. 2012;12(3):180–190. doi:10.1038/nri3156.
- Conlon KC, Miljkovic MD, Waldmann TA. Cytokines in the treatment of cancer. J Interferon Cytokine Res. 2019;39(1):6–21. doi:10.1089/jir.2018.0019.
- Liu Y, Zhou N, Zhou L, Wang J, Zhou Y, Zhang T, Fang Y, Deng J, Gao Y, Liang X, et al. Il-2 regulates tumor-reactive cd8(+) t cell exhaustion by activating the aryl hydrocarbon receptor. Nat Immunol. 2021;22(3):358–369. doi:10.1038/s41590-020-00850-9.
- Mlecnik B, Bindea G, Angell HK, Sasso MS, Obenauf AC, Fredriksen T, Lafontaine L, Bilocq AM, Kirilovsky A, Tosolini M, et al. Functional network pipeline reveals genetic determinants associated with in situ lymphocyte proliferation and survival of cancer patients. Sci Transl Med. 2014;6(228):ra228237–ra228237. doi:10.1126/scitranslmed.3007240.
- Baldo BA. Side effects of cytokines approved for therapy. Drug Saf. 2014;37(11):921–943. doi:10.1007/s40264-014-0226-z.
- Deckers J, Anbergen T, Hokke AM, de Dreu A, Schrijver DP, de Bruin K, Toner YC, Beldman TJ, Spangler JB, de Greef TFA, et al. Engineering cytokine therapeutics. Nat Rev Bioengin. 2023;1(4):286–303. doi:10.1038/s44222-023-00030-y.
- Guo J, Liang Y, Xue D, Shen J, Cai Y, Zhu J, Fu YX, Peng H. Tumor-conditional il-15 pro-cytokine reactivates anti-tumor immunity with limited toxicity. Cell Res. 2021;31(11):1190–1198. doi:10.1038/s41422-021-00543-4.
- Hsu EJ, Cao X, Moon B, Bae J, Sun Z, Liu Z, Fu Y-X. A cytokine receptor-masked il2 prodrug selectively activates tumor-infiltrating lymphocytes for potent antitumor therapy. Nat Commun. 2021;12(1):2768. doi:10.1038/s41467-021-22980-w.
- Mortezaee K, Majidpoor J. Checkpoint inhibitor/interleukin-based combination therapy of cancer. Cancer Med. 2022;11(15):2934–2943. doi:10.1002/cam4.4659.
- Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, Lichtor T, Decker WK, Whelan RL, Kumara HMCS, et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Seminars In Cancer Biology. 2015;35:35(S185–S198. doi:10.1016/j.semcancer.2015.03.004.
- Briukhovetska D, Dörr J, Endres S, Libby P, Dinarello CA, Kobold S. Interleukins in cancer: from biology to therapy. Nat Rev Cancer. 2021;21(8):481–499. doi:10.1038/s41568-021-00363-z.
- Shinriki S, Jono H, Ota K, Ueda M, Kudo M, Ota T, Oike Y, Endo M, Ibusuki M, Hiraki A, et al. Humanized anti-interleukin-6 receptor antibody suppresses tumor angiogenesis and in vivo growth of human oral squamous cell carcinoma. Clin Cancer Res. 2009;15(17):5426–5434. doi:10.1158/1078-0432.CCR-09-0287.
- Tsukamoto H, Fujieda K, Miyashita A, Fukushima S, Ikeda T, Kubo Y, Senju S, Ihn H, Nishimura Y, Oshiumi H. Combined blockade of il6 and pd-1/pd-l1 signaling abrogates mutual regulation of their immunosuppressive effects in the tumor microenvironment. Cancer Res. 2018;78(17):5011–5022. doi:10.1158/0008-5472.CAN-18-0118.
- Liu H, Shen J, Lu K. Il-6 and pd-l1 blockade combination inhibits hepatocellular carcinoma cancer development in mouse model. Biochem Bioph Res Co. 2017;486(2):239–244. doi:10.1016/j.bbrc.2017.02.128.
- Weber R, Groth C, Lasser S, Arkhypov I, Petrova V, Altevogt P, Utikal J, Umansky V. Il-6 as a major regulator of mdsc activity and possible target for cancer immunotherapy. Cell Immunol. 2021;359(104254):104254. doi:10.1016/j.cellimm.2020.104254.
- Narayan V, Barber-Rotenberg JS, Jung I-Y, Lacey SF, Rech AJ, Davis MM, Hwang W-T, Lal P, Carpenter EL, Maude SL, et al. Psma-targeting tgfβ-insensitive armored car t cells in metastatic castration-resistant prostate cancer: a phase 1 trial. Nat Med. 2022;28(4):724–734. doi:10.1038/s41591-022-01726-1.
- Junghans RP, Ma Q, Rathore R, Gomes EM, Bais AJ, Lo AS, Abedi M, Davies RA, Cabral HJ, Al-Homsi AS, et al. Phase i trial of anti-psma designer car-t cells in prostate cancer: possible role for interacting interleukin 2-t cell pharmacodynamics as a determinant of clinical response. The Prostate. 2016;76(14):1257–1270. doi:10.1002/pros.23214.
- Massagué J, Wotton D. Transcriptional control by the tgf-beta/smad signaling system. EMBO J. 2000;19(8):1745–1754. doi:10.1093/emboj/19.8.1745.
- Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. Tgfβ attenuates tumour response to pd-l1 blockade by contributing to exclusion of t cells. Nature. 2018;554(7693):544–548. doi:10.1038/nature25501.
- Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, Sevillano M, Ibiza S, Cañellas A, Hernando-Momblona X, et al. Tgfβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554(7693):538–543. doi:10.1038/nature25492.
- Greco R, Qu H, Qu H, Theilhaber J, Shapiro G, Gregory R, Winter C, Malkova N, Sun F, Jaworski J, et al. Pan-tgfβ inhibition by sar439459 relieves immunosuppression and improves antitumor efficacy of pd-1 blockade. Oncoimmunology. 2020;9(1):1811605. doi:10.1080/2162402X.2020.1811605.
- Martin CJ, Datta A, Littlefield C, Kalra A, Chapron C, Wawersik S, Dagbay KB, Brueckner CT, Nikiforov A, Danehy FT Jr., et al. Selective inhibition of tgfβ1 activation overcomes primary resistance to checkpoint blockade therapy by altering tumor immune landscape. Sci Transl Med. 2020;12(536). doi:10.1126/scitranslmed.aay8456.
- Lan Y, Moustafa M, Knoll M, Xu C, Furkel J, Lazorchak A, Yeung TL, Hasheminasab SM, Jenkins MH, Meister S, et al. Simultaneous targeting of tgf-β/pd-l1 synergizes with radiotherapy by reprogramming the tumor microenvironment to overcome immune evasion. Cancer Cell. 2021;39(10):1388–1403.e1310. doi:10.1016/j.ccell.2021.08.008.
- Anderton MJ, Mellor HR, Bell A, Sadler C, Pass M, Powell S, Steele SJ, Roberts RR, Heier A. Induction of heart valve lesions by small-molecule alk5 inhibitors. Toxicol Pathol. 2011;39(6):916–924. doi:10.1177/0192623311416259.
- Mitra MS, Lancaster K, Adedeji AO, Palanisamy GS, Dave RA, Zhong F, Holdren MS, Turley SJ, Liang W-C, Wu Y, et al. A potent pan-tgfβ neutralizing monoclonal antibody elicits cardiovascular toxicity in mice and cynomolgus monkeys. Toxicol Sci. 2020;175(1):24–34. doi:10.1093/toxsci/kfaa024.
- Gueorguieva I, Cleverly AL, Stauber A, Sada Pillay N, Rodon JA, Miles CP, Yingling JM, Lahn MM. Defining a therapeutic window for the novel tgf-β inhibitor ly2157299 monohydrate based on a pharmacokinetic/pharmacodynamic model. Br J Clin Pharmacol. 2014;77(5):796–807. doi:10.1111/bcp.12256.
- Brandes AA, Carpentier AF, Kesari S, Sepulveda-Sanchez JM, Wheeler HR, Chinot O, Cher L, Steinbach JP, Capper D, Specenier P, et al. A phase ii randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro Oncol. 2016;18(8):1146–1156. doi:10.1093/neuonc/now009.
- Haque S, Morris JC. Transforming growth factor-β: a therapeutic target for cancer. Hum Vaccin Immunother. 2017;13(8):1741–1750. doi:10.1080/21645515.2017.1327107.
- Melisi D, Garcia-Carbonero R, Macarulla T, Pezet D, Deplanque G, Fuchs M, Trojan J, Oettle H, Kozloff M, Cleverly A, et al. Galunisertib plus gemcitabine vs. Gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br J Cancer. 2018;119(10):1208–1214. doi:10.1038/s41416-018-0246-z.
- Robbrecht D, Doger B, Grob J-J, Bechter OE, de Miguel MJ, Vieito M, Schadendorf D, Curigliano G, Borbath I, Butler MO, et al. Safety and efficacy results from the expansion phase of the first-in-human study evaluating tgfβ inhibitor sar439459 alone and combined with cemiplimab in adults with advanced solid tumors. J Clin Oncol. 2022;40(16_suppl):2524–2524. doi:10.1200/JCO.2022.40.16_suppl.2524.
- Lindsay ME, Schepers D, Bolar NA, Doyle JJ, Gallo E, Fert-Bober J, Kempers MJ, Fishman EK, Chen Y, Myers L, et al. Loss-of-function mutations in tgfb2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet. 2012;44(8):922–927. doi:10.1038/ng.2349.
- Bertoli-Avella AM, Gillis E, Morisaki H, Verhagen JMA, de Graaf BM, van de Beek G, Gallo E, Kruithof BPT, Venselaar H, Myers LA, et al. Mutations in a tgf-β ligand, tgfb3, cause syndromic aortic aneurysms and dissections. J Am Coll Cardiol. 2015;65(13):1324–1336. doi:10.1016/j.jacc.2015.01.040.
- Ozga AJ, Chow MT, Luster AD. Chemokines and the immune response to cancer. Immunity. 2021;54(5):859–874. doi:10.1016/j.immuni.2021.01.012.
- Tu MM, Abdel-Hafiz HA, Jones RT, Jean A, Hoff KJ, Duex JE, Chauca-Diaz A, Costello JC, Dancik GM, Tamburini BAJ, et al. Inhibition of the ccl2 receptor, ccr2, enhances tumor response to immune checkpoint therapy. Commun Biol. 2020;3(1):720. doi:10.1038/s42003-020-01441-y.
- Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, et al. Loss of pten promotes resistance to t cell–mediated immunotherapy. Cancer Discov. 2016;6(2):202–216. doi:10.1158/2159-8290.CD-15-0283.
- Haag GM, Springfeld C, Grün B, Apostolidis L, Zschäbitz S, Dietrich M, Berger A-K, Weber TF, Zoernig I, Schaaf M, et al. Pembrolizumab and maraviroc in refractory mismatch repair proficient/microsatellite-stable metastatic colorectal cancer – the piccasso phase i trial. Eur J Cancer. 2022;167:167(112–122. doi:10.1016/j.ejca.2022.03.017.
- Liu G, Rui W, Zhao X, Lin X. Enhancing car-t cell efficacy in solid tumors by targeting the tumor microenvironment. Cell Mol Immunol. 2021;18(5):1085–1095. doi:10.1038/s41423-021-00655-2.
- He X, Xu C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020;30(8):660–669. doi:10.1038/s41422-020-0343-4.
- Ngiow SF, Young A, Jacquelot N, Yamazaki T, Enot D, Zitvogel L, Smyth MJ. A threshold level of intratumor cd8+ t-cell pd1 expression dictates therapeutic response to anti-pd1. Cancer Res. 2015;75(18):3800–3811. doi:10.1158/0008-5472.CAN-15-1082.
- Lefler DS, Snook AE, Bashir B. Immune checkpoint inhibitors in luminal gastrointestinal malignancies: going beyond msi-h/dmmr, tmb and pd-l1. Immunotherapy. 2022;14(11):885–902. doi:10.2217/imt-2022-0012.
- Gandini S, Massi D, Mandalà M. Pd-l1 expression in cancer patients receiving anti pd-1/pd-l1 antibodies: a systematic review and meta-analysis. Crit Rev Oncol Hematol. 2016. 100:100(88–98. doi:10.1016/j.critrevonc.2016.02.001.
- Doroshow DB, Bhalla S, Beasley MB, Sholl LM, Kerr KM, Gnjatic S, Wistuba II, Rimm DL, Tsao MS, Hirsch FR. Pd-l1 as a biomarker of response to immune-checkpoint inhibitors. Nat Rev Clin Oncol. 2021;18(6):345–362. doi:10.1038/s41571-021-00473-5.
- Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon R-A, Reed K, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–133. doi:10.1056/NEJMoa1302369.
- Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim S-W, Carcereny Costa E, Park K, Alexandru A, Lupinacci L, de la Mora Jimenez E, et al. Nivolumab plus ipilimumab in advanced non–small-cell lung cancer. N Engl J Med. 2019;381(21):2020–2031. doi:10.1056/NEJMoa1910231.
- Motzer RJ, Tannir NM, McDermott DF, Arén Frontera O, Melichar B, Choueiri TK, Plimack ER, Barthélémy P, Porta C, George S, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med. 2018;378(14):1277–1290. doi:10.1056/NEJMoa1712126.
- Tawbi HA, Schadendorf D, Lipson EJ, Ascierto PA, Matamala L, Castillo Gutiérrez E, Rutkowski P, Gogas HJ, Lao CD, De Menezes JJ, et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N Engl J Med. 2022;386(1):24–34. doi:10.1056/NEJMoa2109970.
- Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H, et al. Adaptive resistance to therapeutic pd-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7(10501). doi:10.1038/ncomms10501.
- Sasikumar PG, Sudarshan NS, Adurthi S, Ramachandra RK, Samiulla DS, Lakshminarasimhan A, Ramanathan A, Chandrasekhar T, Dhudashiya AA, Talapati SR, et al. Pd-1 derived ca-170 is an oral immune checkpoint inhibitor that exhibits preclinical anti-tumor efficacy. Commun Biol. 2021;4(1):699. doi:10.1038/s42003-021-02191-1.
- Curigliano G, Gelderblom H, Mach N, Doi T, Tai D, Forde PM, Sarantopoulos J, Bedard PL, Lin CC, Hodi FS, et al. Phase i/ib clinical trial of sabatolimab, an anti-tim-3 antibody, alone and in combination with spartalizumab, an anti-pd-1 antibody, in advanced solid tumors. Clin Cancer Res. 2021;27(13):3620–3629. doi:10.1158/1078-0432.CCR-20-4746.
- Choi J, Medikonda R, Saleh L, Kim T, Pant A, Srivastava S, Kim YH, Jackson C, Tong L, Routkevitch D, et al. Combination checkpoint therapy with anti-pd-1 and anti-btla results in a synergistic therapeutic effect against murine glioblastoma. Oncoimmunology. 2021;10(1):1956142. doi:10.1080/2162402X.2021.1956142.
- Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL, et al. Immune inhibitory molecules lag-3 and pd-1 synergistically regulate t-cell function to promote tumoral immune escape. Cancer Res. 2012;72(4):917–927. doi:10.1158/0008-5472.CAN-11-1620.
- Sung E, Ko M, Won JY, Jo Y, Park E, Kim H, Choi E, Jung UJ, Jeon J, Kim Y, et al. Lag-3xpd-l1 bispecific antibody potentiates antitumor responses of t cells through dendritic cell activation. Mol Ther. 2022;30(8):2800–2816. doi:10.1016/j.ymthe.2022.05.003.
- Liu J, Luan Y, Deng H, Wang F, Wang C, Zhang Z. A bivalent tim-3/pd-1 bispecific antibody for the treatment of pd-1 antibody resistant or refractory nsclc. J Clin Oncol. 2022;40(16_suppl):e14597–e14597. doi:10.1200/JCO.2022.40.16_suppl.e14597.
- Chen N, Morello A, Tano Z, Adusumilli PS. Car t-cell intrinsic pd-1 checkpoint blockade: a two-in-one approach for solid tumor immunotherapy. Oncoimmunology. 2017;6(2):e1273302. doi:10.1080/2162402X.2016.1273302.
- Tian Y, Li Y, Shao Y, Zhang Y. Gene modification strategies for next-generation car t cells against solid cancers. J Hematol Oncol. 2020;13(1):54. doi:10.1186/s13045-020-00890-6.
- Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, Maus MV, Fraietta JA, Zhao Y, June CH. Dominant-negative tgf-β receptor enhances psma-targeted human car t cell proliferation and augments prostate cancer eradication. Mol Ther. 2018;26(7):1855–1866. doi:10.1016/j.ymthe.2018.05.003.
- Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, Konieczny BT, Daugherty CZ, Koenig L, Yu K, et al. Rescue of exhausted cd8 t cells by pd-1-targeted therapies is cd28-dependent. Science. 2017;355(6332):1423–1427. doi:10.1126/science.aaf0683.
- Cappell KM, Kochenderfer JN. A comparison of chimeric antigen receptors containing cd28 versus 4-1bb costimulatory domains. Nat Rev Clin Oncol. 2021;18(11):715–727. doi:10.1038/s41571-021-00530-z.
- Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine storm in a phase 1 trial of the anti-cd28 monoclonal antibody tgn1412. N Engl J Med. 2006;355(10):1018–1028. doi:10.1056/NEJMoa063842.
- Qiao Y, Qiu Y, Ding J, Luo N, Wang H, Ling X, Sun J, Wu Z, Wang Y, Liu Y, et al. Cancer immune therapy with pd-1-dependent cd137 co-stimulation provides localized tumour killing without systemic toxicity. Nat Commun. 2021;12(1):6360. doi:10.1038/s41467-021-26645-6.
- Buchan SL, Fallatah M, Thirdborough SM, Taraban VY, Rogel A, Thomas LJ, Penfold CA, He LZ, Curran MA, Keler T, et al. Pd-1 blockade and cd27 stimulation activate distinct transcriptional programs that synergize for cd8(+) t-cell-driven antitumor immunity. Clin Cancer Res. 2018;24(10):2383–2394. doi:10.1158/1078-0432.CCR-17-3057.
- Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7(1):12150. doi:10.1038/ncomms12150.
- Hao Z, Li R, Wang Y, Li S, Hong Z, Han Z. Landscape of myeloid-derived suppressor cell in tumor immunotherapy. Biomarker Res. 2021;9(1):77. doi:10.1186/s40364-021-00333-5.
- Wu L, Zhang XH. Tumor-associated neutrophils and macrophages-heterogenous but not chaotic. Front Immunol. 2020;11(553967). doi:10.3389/fimmu.2020.553967.
- Wu K, Tan MY, Jiang JT, Mu XY, Wang JR, Zhou WJ, Wang X, Li MQ, He YY, Liu ZH. Cisplatin inhibits the progression of bladder cancer by selectively depleting g-mdscs: a novel chemoimmunomodulating strategy. Clin Immunol. 2018. 193:193(60–69. doi:10.1016/j.clim.2018.01.012.
- Li T, Liu T, Zhu W, Xie S, Zhao Z, Feng B, Guo H, Yang R. Targeting mdsc for immune-checkpoint blockade in cancer immunotherapy: Current progress and new prospects. Clin Med Insights Oncol. 2021. 15:11795549211035540. doi:10.1177/11795549211035540.
- Kim NR, Kim YJ. Oxaliplatin regulates myeloid-derived suppressor cell-mediated immunosuppression via downregulation of nuclear factor-κb signaling. Cancer Med. 2019;8(1):276–288. doi:10.1002/cam4.1878.
- Koinis F, Vetsika EK, Aggouraki D, Skalidaki E, Koutoulaki A, Gkioulmpasani M, Georgoulias V, Kotsakis A. Effect of first-line treatment on myeloid-derived suppressor cells’ subpopulations in the peripheral blood of patients with non-small cell lung cancer. J Thorac Oncol. 2016;11(8):1263–1272. doi:10.1016/j.jtho.2016.04.026.
- Li K, Shi H, Zhang B, Ou X, Ma Q, Chen Y, Shu P, Li D, Wang Y. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct Target Ther. 2021;6(1):362. doi:10.1038/s41392-021-00670-9.
- Hellmann MD, Jänne PA, Opyrchal M, Hafez N, Raez LE, Gabrilovich DI, Wang F, Trepel JB, Lee M-J, Yuno A, et al. Entinostat plus pembrolizumab in patients with metastatic nsclc previously treated with anti–pd-(l)1 therapy. Clin Cancer Res. 2021;27(4):1019–1028. doi:10.1158/1078-0432.CCR-20-3305.
- Raggi F, Pelassa S, Pierobon D, Penco F, Gattorno M, Novelli F, Eva A, Varesio L, Giovarelli M, Bosco MC. Regulation of human macrophage m1-m2 polarization balance by hypoxia and the triggering receptor expressed on myeloid cells-1. Front Immunol. 2017;8(1097). doi:10.3389/fimmu.2017.01097.
- Wu Q, Zhou W, Yin S, Zhou Y, Chen T, Qian J, Su R, Hong L, Lu H, Zhang F, et al. Blocking triggering receptor expressed on myeloid cells-1-positive tumor-associated macrophages induced by hypoxia reverses immunosuppression and anti-programmed cell death ligand 1 resistance in liver cancer. Hepatology. 2019;70(1):198–214. doi:10.1002/hep.30593.
- Laoui D, Van Overmeire E, De Baetselier P, Van Ginderachter JA, Raes G. Functional relationship between tumor-associated macrophages and macrophage colony-stimulating factor as contributors to cancer progression. Front Immunol. 2014;5(489). doi:10.3389/fimmu.2014.00489.
- Voissière A, Gomez-Roca C, Chabaud S, Rodriguez C, Nkodia A, Berthet J, Montane L, Bidaux A-S, Treilleux I, Eberst L, et al. The csf-1r inhibitor pexidartinib impacts dendritic cell differentiation through inhibition of flt3 signaling and may antagonize the effect of durvalumab in patients with advanced cancer – results from a phase 1 study. medRxiv. 2023; 2023.2002.2015.23285939. doi: 10.1101/2023.02.15.23285939
- Zheng Y, Wang PP, Fu Y, Chen Y-y, Ding Z-Y. Zoledronic acid enhances the efficacy of immunotherapy in non-small cell lung cancer. Int Immunopharmacol. 2022;110(109030). doi:10.1016/j.intimp.2022.109030.
- Togashi Y, Shitara K, Nishikawa H. Regulatory t cells in cancer immunosuppression — implications for anticancer therapy. Nat Rev Clin Oncol. 2019;16(6):356–371. doi:10.1038/s41571-019-0175-7.
- Jacobs JF, Punt CJ, Lesterhuis WJ, Sutmuller RP, Brouwer HM, Scharenborg NM, Klasen IS, Hilbrands LB, Figdor CG, de Vries IJ, et al. Dendritic cell vaccination in combination with anti-cd25 monoclonal antibody treatment: a phase i/ii study in metastatic melanoma patients. Clin Cancer Res. 2010;16(20):5067–5078. doi:10.1158/1078-0432.CCR-10-1757.
- Doi T, Muro K, Ishii H, Kato T, Tsushima T, Takenoyama M, Oizumi S, Gemmoto K, Suna H, Enokitani K, et al. A phase i study of the anti-cc chemokine receptor 4 antibody, mogamulizumab, in combination with nivolumab in patients with advanced or metastatic solid tumors. Clin Cancer Res. 2019;25(22):6614–6622. doi:10.1158/1078-0432.CCR-19-1090.
- Zamarin D, Hamid O, Nayak-Kapoor A, Sahebjam S, Sznol M, Collaku A, Fox FE, Marshall MA, Hong DS. Mogamulizumab in combination with durvalumab or tremelimumab in patients with advanced solid tumors: A phase i study. Clin Cancer Res. 2020;26(17):4531–4541. doi:10.1158/1078-0432.CCR-20-0328.
- Madondo MT, Quinn M, Plebanski M. Low dose cyclophosphamide: mechanisms of t cell modulation. Cancer Treat Rev. 2016. 42:42(3–9. doi:10.1016/j.ctrv.2015.11.005.
- Ping Q, Yan R, Cheng X, Wang W, Zhong Y, Hou Z, Shi Y, Wang C, Li R. Cancer-associated fibroblasts: overview, progress, challenges, and directions. Cancer Gene Ther. 2021;28(9):984–999. doi:10.1038/s41417-021-00318-4.
- Glabman RA, Choyke PL, Sato N. Cancer-associated fibroblasts: tumorigenicity and targeting for cancer therapy. Cancers Basel. 2022;14(16):3906. doi:10.3390/cancers14163906.
- Ham IH, Lee D, Hur H. Cancer-associated fibroblast-induced resistance to chemotherapy and radiotherapy in gastrointestinal cancers. Cancers Basel. 2021;13(5):1172. doi:10.3390/cancers13051172.
- Yoon H, Tang C-M, Banerjee S, Delgado AL, Yebra M, Davis J, Sicklick JK. Tgf-β1-mediated transition of resident fibroblasts to cancer-associated fibroblasts promotes cancer metastasis in gastrointestinal stromal tumor. Oncogenesis. 2021;10(2):13. doi:10.1038/s41389-021-00302-5.
- Morén A, Bellomo C, Tsubakihara Y, Kardassis D, Mikulits W, Heldin CH, Moustakas A. Lxrα limits tgfβ-dependent hepatocellular carcinoma associated fibroblast differentiation. Oncogenesis. 2019;8(6):36. doi:10.1038/s41389-019-0140-4.
- Freedman JD, Duffy MR, Lei-Rossmann J, Muntzer A, Scott EM, Hagel J, Campo L, Bryant RJ, Verrill C, Lambert A, et al. An oncolytic virus expressing a t-cell engager simultaneously targets cancer and immunosuppressive stromal cells. Cancer Res. 2018;78(24):6852–6865. doi:10.1158/0008-5472.CAN-18-1750.
- Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, et al. Targeting cxcl12 from fap-expressing carcinoma-associated fibroblasts synergizes with anti-pd-l1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110(50):20212–20217. doi:10.1073/pnas.1320318110.
- Ford K, Hanley CJ, Mellone M, Szyndralewiez C, Heitz F, Wiesel P, Wood O, Machado M, Lopez MA, Ganesan AP, et al. Nox4 inhibition potentiates immunotherapy by overcoming cancer-associated fibroblast-mediated cd8 t-cell exclusion from tumors. Cancer Res. 2020;80(9):1846–1860. doi:10.1158/0008-5472.CAN-19-3158.
- Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, Bordbar D, Shan D, Samanamud J, Mahajan A, et al. Immune and genomic correlates of response to anti-pd-1 immunotherapy in glioblastoma. Nat Med. 2019;25(3):462–469. doi:10.1038/s41591-019-0349-y.
- Yeh C-H, Bellon M, Nicot C. Fbxw7: a critical tumor suppressor of human cancers. Mol Cancer. 2018;17(1):115. doi:10.1186/s12943-018-0857-2.
- Gstalder C, Liu D, Miao D, Lutterbach B, DeVine AL, Lin C, Shettigar M, Pancholi P, Buchbinder EI, Carter SL, et al. Inactivation of fbxw7 impairs dsrna sensing and confers resistance to pd-1 blockade. Cancer Discov. 2020;10(9):1296–1311. doi:10.1158/2159-8290.CD-19-1416.
- Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, Buczkowski KA, Liu Y, Awad MM, Denning WL, et al. Stk11/lkb1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress t-cell activity in the lung tumor microenvironment. Cancer Res. 2016;76(5):999–1008. doi:10.1158/0008-5472.CAN-15-1439.
- Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, Schrock AB, Hartmaier RJ, Trabucco SE, Gay L, et al. Stk11/lkb1 mutations and pd-1 inhibitor resistance in kras-mutant lung adenocarcinoma. Cancer Discov. 2018;8(7):822–835. doi:10.1158/2159-8290.CD-18-0099.
- Wang J, Sun J, Zhang N, Yang R, Li H, Zhang Y, Chen K, Kong D. Pes1 enhances proliferation and tumorigenesis in hepatocellular carcinoma via the pi3k/akt pathway. Life Sci. 2019. 219:219(182–189. doi:10.1016/j.lfs.2018.12.054.
- Ma N, Hua R, Yang Y, Liu Z-C, Pan J, Yu B-Y, Sun Y-F, Xie D, Wang Y, Li Z-G. Pes1 reduces cd8+ t cell infiltration and immunotherapy sensitivity via interrupting ilf3-il15 complex in esophageal squamous cell carcinoma. J Biomed Sci. 2023;30(1):20. doi:10.1186/s12929-023-00912-8.
- Ala M. Target c-myc to treat pancreatic cancer. Cancer Biol Ther. 2022;23(1):34–50. doi:10.1080/15384047.2021.2017223.
- Jin X, Fang R, Fan P, Zeng L, Zhang B, Lu X, Liu T. Pes1 promotes bet inhibitors resistance and cells proliferation through increasing c-myc expression in pancreatic cancer. J Exp Clin Cancer Res. 2019;38(1):463. doi:10.1186/s13046-019-1466-7.
- Ham SW, Jeon H-Y, Jin X, Kim E-J, Kim J-K, Shin YJ, Lee Y, Kim SH, Lee SY, Seo S, et al. Tp53 gain-of-function mutation promotes inflammation in glioblastoma. Cell Death Differ. 2019;26(3):409–425. doi:10.1038/s41418-018-0126-3.
- Dong Z-Y, Zhong W-Z, Zhang X-C, Su J, Xie Z, Liu S-Y, Tu H-Y, Chen H-J, Sun Y-L, Zhou Q, et al. Potential predictive value of tp53 and kras mutation status for response to pd-1 blockade immunotherapy in lung adenocarcinoma. Clin Cancer Res. 2017;23(12):3012–3024. doi:10.1158/1078-0432.CCR-16-2554.
- Chen X, Zhang T, Su W, Dou Z, Zhao D, Jin X, Lei H, Wang J, Xie X, Cheng B, et al. Mutant p53 in cancer: from molecular mechanism to therapeutic modulation. Cell Death Disease. 2022;13(11):974. doi:10.1038/s41419-022-05408-1.
- Xiao Y, Chen J, Zhou H, Zeng X, Ruan Z, Pu Z, Jiang X, Matsui A, Zhu L, Amoozgar Z, et al. Combining p53 mrna nanotherapy with immune checkpoint blockade reprograms the immune microenvironment for effective cancer therapy. Nat Commun. 2022;13(1):758. doi:10.1038/s41467-022-28279-8.
- Nagahashi M, Sato S, Yuza K, Shimada Y, Ichikawa H, Watanabe S, Takada K, Okamoto T, Okuda S, Lyle S, et al. Common driver mutations and smoking history affect tumor mutation burden in lung adenocarcinoma. J Surg Res. 2018;230:230(181–185. doi:10.1016/j.jss.2018.07.007.
- Gainor JF, Shaw AT, Sequist LV, Fu X, Azzoli CG, Piotrowska Z, Huynh TG, Zhao L, Fulton L, Schultz KR, et al. Egfr mutations and alk rearrangements are associated with low response rates to pd-1 pathway blockade in non–small cell lung cancer: a retrospective analysis. Clin Cancer Res. 2016;22(18):4585–4593. doi:10.1158/1078-0432.CCR-15-3101.
- Addeo A, Passaro A, Malapelle U, Banna GL, Subbiah V, Friedlaender A. Immunotherapy in non-small cell lung cancer harbouring driver mutations. Cancer Treat Rev. 2021. 96:96(. doi:10.1016/j.ctrv.2021.102179.
- Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, et al. Nivolumab versus docetaxel in advanced nonsquamous non–small-cell lung cancer. N Engl J Med. 2015;373(17):1627–1639. doi:10.1056/NEJMoa1507643.
- Herbst RS, Baas P, Kim D-W, Felip E, Pérez-Gracia JL, Han J-Y, Molina J, Kim J-H, Arvis CD, Ahn M-J, et al. Pembrolizumab versus docetaxel for previously treated, pd-l1-positive, advanced non-small-cell lung cancer (keynote-010): a randomised controlled trial. Lancet. 2016;387(10027):1540–1550. doi:10.1016/S0140-6736(15)01281-7.
- Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, Gadgeel SM, Hida T, Kowalski DM, Dols MC, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (oak): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389(10066):255–265. doi:10.1016/S0140-6736(16)32517-X.
- Peters S, Gettinger S, Johnson ML, Jänne PA, Garassino MC, Christoph D, Toh CK, Rizvi NA, Chaft JE, Carcereny Costa E, et al. Phase ii trial of atezolizumab as first-line or subsequent therapy for patients with programmed death-ligand 1–selected advanced non–small-cell lung cancer (birch). J Clin Oncol. 2017;35(24):2781–2789. doi:10.1200/JCO.2016.71.9476.
- Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, Rodríguez-Abreu D, Moro-Sibilot D, Thomas CA, Barlesi F, et al. Atezolizumab for first-line treatment of metastatic nonsquamous nsclc. N Engl J Med. 2018;378(24):2288–2301. doi:10.1056/NEJMoa1716948.
- Garassino MC, Cho B-C, Kim J-H, Mazières J, Vansteenkiste J, Lena H, Corral Jaime J, Gray JE, Powderly J, Chouaid C, et al. Durvalumab as third-line or later treatment for advanced non-small-cell lung cancer (atlantic): an open-label, single-arm, phase 2 study. Lancet Oncol. 2018;19(4):521–536. doi:10.1016/S1470-2045(18)30144-X.
- Passaro A, Jänne PA, Mok T, Peters S. Overcoming therapy resistance in egfr-mutant lung cancer. Nat Cancer. 2021;2(4):377–391. doi:10.1038/s43018-021-00195-8.
- Oxnard GR, Yang JCH, Yu H, Kim SW, Saka H, Horn L, Goto K, Ohe Y, Mann H, Thress KS, et al. Tatton: A multi-arm, phase ib trial of osimertinib combined with selumetinib, savolitinib, or durvalumab in egfr-mutant lung cancer. Ann Oncol. 2020;31(4):507–516. doi:10.1016/j.annonc.2020.01.013.
- Yang J-H, Shepherd FA, Kim D-W, Lee G-W, Lee JS, Chang G-C, Lee SS, Wei Y-F, Lee YG, Laus G, et al. Osimertinib plus durvalumab versus osimertinib monotherapy in egfr t790m–positive nsclc following previous egfr tki therapy: caural brief report. J Thorac Oncol. 2019;14(5):933–939. doi:10.1016/j.jtho.2019.02.001.
- Yang J-H, Lee DH, Lee J-S, Fan Y, de Marinis F, Okamoto I, Inoue T, Rodriguez Cid JR, Zhang L, Yang C-T, et al. Pemetrexed and platinum with or without pembrolizumab for tyrosine kinase inhibitor (tki)-resistant, egfr-mutant, metastatic nonsquamous nsclc: phase 3 keynote-789 study. J Clin Oncol. 2023;41(17_suppl):LBA9000–LBA9000. doi:10.1200/JCO.2023.41.17_suppl.LBA9000.
- Mok TSK, Nakagawa K, Park K, Ohe Y, Girard N, Kim HR, Wu YL, Gainor J, Lee SH, Chiu CH, et al. Lba8 nivolumab (nivo) + chemotherapy (chemo) vs chemo in patients (pts) with egfr-mutated metastatic non-small cell lung cancer (mnsclc) with disease progression after egfr tyrosine kinase inhibitors (tkis) in checkmate 722. Ann Oncol. 2022;33:33(S1561–S1562. doi:10.1016/j.annonc.2022.10.350.
- Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8(6):519–530. doi:10.1085/jgp.8.6.519.
- Singer K, Kastenberger M, Gottfried E, Hammerschmied CG, Büttner M, Aigner M, Seliger B, Walter B, Schlösser H, Hartmann A, et al. Warburg phenotype in renal cell carcinoma: high expression of glucose-transporter 1 (glut-1) correlates with low cd8+ t-cell infiltration in the tumor. Int J Cancer. 2011;128(9):2085–2095. doi:10.1002/ijc.25543.
- Ho P-C, Bihuniak Jessica D, Macintyre Andrew N, Staron M, Liu X, Amezquita R, Tsui Y-C, Cui G, Micevic G, Perales Jose C, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor t cell responses. Cell. 2015;162(6):1217–1228. doi:10.1016/j.cell.2015.08.012.
- Marchiq I, Pouysségur J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/h(+) symporters. J Mol Med (Berl). 2016;94(2):155–171. doi:10.1007/s00109-015-1307-x.
- Ramapriyan R, Caetano MS, Barsoumian HB, Mafra ACP, Zambalde EP, Menon H, Tsouko E, Welsh JW, Cortez MA. Altered cancer metabolism in mechanisms of immunotherapy resistance. Pharmacology & Therapeutics. 2019. 195:162–171. doi:10.1016/j.pharmthera.2018.11.004.
- Pilon-Thomas S, Kodumudi KN, El-Kenawi AE, Russell S, Weber AM, Luddy K, Damaghi M, Wojtkowiak JW, Mulé JJ, Ibrahim-Hashim A, et al. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res. 2016;76(6):1381–1390. doi:10.1158/0008-5472.CAN-15-1743.
- Hamaguchi R, Isowa M, Narui R, Morikawa H, Wada H. Clinical review of alkalization therapy in cancer treatment. Front Oncol. 2022. 12:12(. doi:10.3389/fonc.2022.1003588.
- Hwu P, Du MX, Lapointe R, Do M, Taylor MW, Young HA. Indoleamine 2,3-dioxygenase production by human dendritic cells results in the inhibition of t cell proliferation. J Immunol. 2000;164(7):3596–3599. doi:10.4049/jimmunol.164.7.3596.
- Uyttenhove C, Pilotte L, Théate I, Stroobant V, Colau D, Parmentier N, Boon T, Van den Eynde BJ. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9(10):1269–1274. doi:10.1038/nm934.
- Rui Q, Chen Z, Chen-Ji W, Wei X, Jian-Yuan Z, Yan L, Yi-Yuan Y, Peng-Cheng L, Yao L, Shimin Z, et al. Tryptophan potentiates cd8+ t cells against cancer cells by trip12 tryptophanylation and surface pd-1 downregulation. J Immunother Cancer. 2021;9(7):e002840. doi:10.1136/jitc-2021-002840.
- Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, Arance A, Carlino MS, Grob JJ, Kim TM, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (echo-301/keynote-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019;20(8):1083–1097. doi:10.1016/S1470-2045(19)30274-8.
- Chen S, Tan J, Zhang A. The ups, downs and new trends of ido1 inhibitors. Bioorg Chem. 2021;110(104815):104815. doi:10.1016/j.bioorg.2021.104815.
- Liu Y, Van Ginderachter JA, Brys L, De Baetselier P, Raes G, Geldhof AB. Nitric oxide-independent ctl suppression during tumor progression: association with arginase-producing (m2) myeloid cells1. J Immunol. 2003;170(10):5064–5074. doi:10.4049/jimmunol.170.10.5064.
- Lowe MM, Boothby I, Clancy S, Ahn RS, Liao W, Nguyen DN, Schumann K, Marson A, Mahuron KM, Kingsbury GA, et al. Regulatory t cells use arginase 2 to enhance their metabolic fitness in tissues. JCI Insight. 2019;4(24). doi:10.1172/jci.insight.129756.
- Cheng C-T, Qi Y, Wang Y-C, Chi KK, Chung Y, Ouyang C, Chen Y-R, Oh ME, Sheng X, Tang Y, et al. Arginine starvation kills tumor cells through aspartate exhaustion and mitochondrial dysfunction. Commun Biol. 2018;1(1):178. doi:10.1038/s42003-018-0178-4.
- Pallett LJ, Dimeloe S, Sinclair LV, Byrne AJ, Schurich A. A glutamine ‘tug-of-war’: Targets to manipulate glutamine metabolism for cancer immunotherapy. Immunother Adv. 2021;1(1):ltab010. doi:10.1093/immadv/ltab010.
- Ma G, Zhang Z, Li P, Zhang Z, Zeng M, Liang Z, Li D, Wang L, Chen Y, Liang Y, et al. Reprogramming of glutamine metabolism and its impact on immune response in the tumor microenvironment. Cell Commun Signal. 2022;20(1):114. doi:10.1186/s12964-022-00909-0.
- Metzler B, Gfeller P, Guinet E. Restricting glutamine or glutamine-dependent purine and pyrimidine syntheses promotes human t cells with high foxp3 expression and regulatory properties. J Immunol. 2016;196(9):3618–3630. doi:10.4049/jimmunol.1501756.
- Tannir NM, Agarwal N, Porta C, Lawrence NJ, Motzer R, McGregor B, Lee RJ, Jain RK, Davis N, Appleman LJ, et al. Efficacy and safety of telaglenastat plus cabozantinib vs placebo plus cabozantinib in patients with advanced renal cell carcinoma: the cantata randomized clinical trial. JAMA Oncol. 2022;8(10):1411–1418. doi:10.1001/jamaoncol.2022.3511.
- Hanahan D, Weinberg Robert A. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013.
- Gadiyar V, Lahey KC, Calianese D, Devoe C, Mehta D, Bono K, Desind S, Davra V, Birge RB. Cell death in the tumor microenvironment: implications for cancer immunotherapy. Cells. 2020;9(10):2207. doi:10.3390/cells9102207.
- Showalter A, Limaye A, Oyer JL, Igarashi R, Kittipatarin C, Copik AJ, Khaled AR. Cytokines in immunogenic cell death: applications for cancer immunotherapy. Cytokine. 2017. 97:97(123–132. doi:10.1016/j.cyto.2017.05.024.
- Li Z, Lai X, Fu S, Ren L, Cai H, Zhang H, Gu Z, Ma X, Luo K. Immunogenic cell death activates the tumor immune microenvironment to boost the immunotherapy efficiency. Adv Sci. 2022;9(22):2201734. doi:10.1002/advs.202201734.
- Tang S, Ning Q, Yang L, Mo Z, Tang S. Mechanisms of immune escape in the cancer immune cycle. Int Immunopharmacol. 2020;86(106700):106700. doi:10.1016/j.intimp.2020.106700.
- Lu T, Wang S, Xu L, Zhou Q, Singla N, Gao J, Manna S, Pop L, Xie Z, Chen M, et al. Tumor neoantigenicity assessment with csin score incorporates clonality and immunogenicity to predict immunotherapy outcomes. Sci Immunol. 2020;5(44):eaaz3199. doi:10.1126/sciimmunol.aaz3199.
- Aaes TL, Vandenabeele P. The intrinsic immunogenic properties of cancer cell lines, immunogenic cell death, and how these influence host antitumor immune responses. Cell Death Differ. 2021;28(3):843–860. doi:10.1038/s41418-020-00658-y.
- Liu J, Fu M, Wang M, Wan D, Wei Y, Wei X. Cancer vaccines as promising immuno-therapeutics: platforms and current progress. J Hematol Oncol. 2022;15(1):28. doi:10.1186/s13045-022-01247-x.
- Lin MJ, Svensson-Arvelund J, Lubitz GS, Marabelle A, Melero I, Brown BD, Brody JD. Cancer vaccines: the next immunotherapy frontier. Nat Cancer. 2022;3(8):911–926. doi:10.1038/s43018-022-00418-6.
- Herrera FG, Bourhis J, Coukos G. Radiotherapy combination opportunities leveraging immunity for the next oncology practice. CA Cancer J Clin. 2017;67(1):65–85. doi:10.3322/caac.21358.
- Formenti SC, Rudqvist N-P, Golden E, Cooper B, Wennerberg E, Lhuillier C, Vanpouille-Box C, Friedman K, Ferrari de Andrade L, Wucherpfennig KW, et al. Radiotherapy induces responses of lung cancer to ctla-4 blockade. Nat Med. 2018;24(12):1845–1851. doi:10.1038/s41591-018-0232-2.
- Demaria S, Coleman CN, Formenti SC. Radiotherapy: changing the game in immunotherapy. Trends Cancer. 2016;2(6):286–294. doi:10.1016/j.trecan.2016.05.002.
- Shaverdian N, Lisberg AE, Bornazyan K, Veruttipong D, Goldman JW, Formenti SC, Garon EB, Lee P. Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: a secondary analysis of the keynote-001 phase 1 trial. Lancet Oncol. 2017;18(7):895–903. doi:10.1016/S1470-2045(17)30380-7.
- Dai Z, Wang Z, Lei K, Liao J, Peng Z, Lin M, Liang P, Yu J, Peng S, Chen S, et al. Irreversible electroporation induces cd8+ t cell immune response against post-ablation hepatocellular carcinoma growth. Cancer Lett. 2021;503:503(1–10. doi:10.1016/j.canlet.2021.01.001.
- Zhang X, Zhang Y, Chen J, Wu Y, Zhang J, Wang J. Nanosecond pulsed electric field inhibits malignant melanoma growth by inducing the change of systemic immunity. Med Oral Patol Oral Cir Bucal. 2019;24(4):e555–e561. doi:10.4317/medoral.22976.
- Szlasa W, Janicka N, Sauer N, Michel O, Nowak B, Saczko J, Kulbacka J. Chemotherapy and physical therapeutics modulate antigens on cancer cells. Front Immunol. 2022. 13:13(. doi:10.3389/fimmu.2022.889950.
- Silvestrini M, Pastori C, Tamakloe S, O’Brien T, Allen C, Neal R. Ep02.04-002 synergy of local treatment with pulsed electric fields and anti-pd1 checkpoint blockade. J Thorac Oncol. 2022;17(9, Supplement):S232. doi:10.1016/j.jtho.2022.07.387.
- Pastori C, Wagh M, Nafie E, Murad F, Trikha M, Neal R. Abstract 6392: combination of pulsed electric field, immunotherapy and cisplatin significantly prolongs survival in an orthotopic breast cancer mouse model. Cancer Res. 2023;83(7_Supplement):6392–6392. doi:10.1158/1538-7445.AM2023-6392.
- Nafie E, Wagh M, Pastori C, Trikha M, Neal R. Abstract 6638: pulsed electric fields combined with anti-pd1 prolongs survival and triggers an adaptive immune response in an io-non-responsive orthotopic mouse model. Cancer Res. 2023;83(7_Supplement):6638–6638. doi:10.1158/1538-7445.AM2023-6638.
- Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, Wargo JA. The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell. 2018;33(4):570–580. doi:10.1016/j.ccell.2018.03.015.
- Yi M, Yu S, Qin S, Liu Q, Xu H, Zhao W, Chu Q, Wu K. Gut microbiome modulates efficacy of immune checkpoint inhibitors. J Hematol Oncol. 2018;11(1):47. doi:10.1186/s13045-018-0592-6.
- Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman K, Wei SC, et al. Gut microbiome modulates response to anti–pd-1 immunotherapy in melanoma patients. Science. 2018;359(6371):97–103. doi:10.1126/science.aan4236.
- Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre M-L, Luke JJ, Gajewski TF. The commensal microbiome is associated with anti–pd-1 efficacy in metastatic melanoma patients. Science. 2018;359(6371):104–108. doi:10.1126/science.aao3290.
- Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP, et al. Gut microbiome influences efficacy of pd-1–based immunotherapy against epithelial tumors. Science. 2018;359(6371):91–97. doi:10.1126/science.aan3706.
- Jiang S, Geng S, Chen Q, Zhang C, Cheng M, Yu Y, Zhang S, Shi N, Dong M. Effects of concomitant antibiotics use on immune checkpoint inhibitor efficacy in cancer patients. Front Oncol. 2022;12(823705). doi:10.3389/fonc.2022.823705.
- Baruch EN, Youngster I, Ben-Betzalel G, Ortenberg R, Lahat A, Katz L, Adler K, Dick-Necula D, Raskin S, Bloch N, et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science. 2021;2021371(6529):602–609. doi:10.1126/science.abb5920.
- Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin J-M, Morrison RM, Deblasio RN, Menna C, Ding Q, Pagliano O, et al. Fecal microbiota transplant overcomes resistance to anti–pd-1 therapy in melanoma patients. Science. 2021;2021371(6529):595–602. doi:10.1126/science.abf3363.
- Borgers JSW, Burgers FH, Terveer EM, van Leerdam ME, Korse CM, Kessels R, Flohil CC, Blank CU, Schumacher TN, van Dijk M, et al. Conversion of unresponsiveness to immune checkpoint inhibition by fecal microbiota transplantation in patients with metastatic melanoma: study protocol for a randomized phase ib/iia trial. BMC Cancer. 2022;22(1):1366. doi:10.1186/s12885-022-10457-y.