
Abstract
Lung cancer represents the leading cause of cancer-related deaths in men and women worldwide. Targeted therapeutics, including the epidermal growth factor receptor (EGFR) inhibitor erlotinib, have recently emerged as clinical alternatives for the treatment of non-small cell lung cancer (NSCLC). However, the development of therapeutic resistance is a major challenge, resulting in low 5-year survival rates. Due to their ability to act as tumor suppressors, microRNAs (miRNAs) are attractive candidates as adjuvant therapeutics for the treatment of NSCLC. In this study, we examine the ability of 2 tumor suppressor miRNAs, let-7b and miR-34a to sensitize KRAS;TP53 mutant non-small cell lung cancer cells to the action of erlotinib. Treatment with these miRNAs, individually or in combination, resulted in synergistic potentiation of the anti-proliferative effects of erlotinib. This effect was observed over a wide range of miRNA and erlotinib interactions, suggesting that let-7b and miR-34a target oncogenic pathways beyond those inhibited by EGFR. Combinatorial treatment with let-7b and miR-34a resulted in the strongest synergy with erlotinib, indicating that these miRNAs can effectively target multiple cellular pathways involved in cancer cell proliferation and resistance to erlotinib. Together, our findings indicate that NSCLC cells can be effectively sensitized to erlotinib by supplementation with tumor suppressor miRNAs, and suggest that the use of combinations of miRNAs as adjuvant therapeutics for the treatment of lung cancer is a viable clinical strategy.
Introduction
Lung cancer is responsible for the most cancer-related deaths worldwide, making it a major medical issue.Citation1 Current therapeutic alternatives are largely ineffective, resulting in an average 5-year survival rate of only 16.8%.Citation1-3 Non-small cell lung cancer (NSCLC), which represents approximately 85% of lung cancer diagnoses, presents a particularly challenging cancer type due to its late detection and low sensitivity to chemotherapy and radiotherapy.Citation2,4,5 The dysregulation of potent signaling pathways through alterations in key oncogenes and tumor suppressors is one of the hallmarks of NSCLC, leading to uncontrolled cell proliferation and survival.Citation2,Citation5 The epidermal growth factor receptor (EGFR) pathway plays an important role in cell proliferation by activating major downstream signaling pathways, most importantly the RAS and phosphoinositide 3-kinase (PI3K) pathways; consequently, EGFR signaling can regulate cell proliferation, adhesion and migration, proliferation, apoptosis, and angiogenesis, thereby playing a crucial role in tumor initiation and progression.Citation2,6-8 Indeed, EGFR overexpression is correlated with disease progression, resistance to chemotherapy, and poor prognosis.Citation9
Activating mutations in the kinase domain of EGFR are believed to be early events in the development of NSCLC, and have been shown in vitro and in vivo to be able to transform cells and produce phenotypes resembling lung carcinomas.Citation10,11 EGFR mutations are present in 10–40% of NSCLC patients, while receptor overexpression is observed in 15–30% of cases.Citation2 The EGFR tyrosine kinase inhibitor erlotinib is used as a first-line therapy in patients with activating EGFR mutations, and as a second- or third-line agent for patients with lesions resistant to other chemotherapeutics.Citation12,13
While erlotinib is initially effective, particularly in patients bearing EGFR point mutations and deletions, most patients will eventually develop resistance and relapse.Citation14-16 A number of molecular lesions have been associated with resistance to erlotinib, including activating mutations in KRAS, which are found exclusively in NSCLC and are associated with resistance to EGFR inhibitors and chemotherapy, and poor prognosis.Citation16-18 Interestingly, EGFR mutations and KRAS mutations are mutually exclusive in NSCLC patients,Citation16 suggesting that patients bearing KRAS mutations may become resistant to erlotinib by regulation of the signaling pathway downstream of the receptor. Similarly, overexpression of alternative receptor tyrosine kinases such as MET and AXL, which can signal through the RAS and PI3K pathways is associated with resistance to erlotinib.Citation19-21 The binding of erlotinib to the receptor also appears to be affected in some resistant patients: a mutation in exon 20 of EGFR (T790M) was recently discovered to affect the binding of erlotinib to the receptor and lower its efficacy.Citation15,22 In addition to these alterations directly affecting the EGFR pathway, deleterious mutations in the crucial tumor suppressor gene TP53 are present in 50–70% of NSCLC patients, leading to impaired apoptotic mechanisms and increased cancer cell survival.Citation2,23,24
The RAS and p53 pathways are regulated in part by the tumor suppressive microRNAs (miRNAs) let-7 and miR-34, respectively.Citation25-30 In addition, these miRNAs can target several important oncogenes including MYC, HMGA2, and LIN28 for let-7Citation31–35, and MET, BCL2, AXL, CDK4, and MYC for miR-34.Citation26,27,36,37 Notably, let-7 and miR-34 are often lost or poorly expressed in lung tumors.Citation38-40 The ability of miRNAs to target multiple oncogenes with redundancy and cooperativity makes them attractive therapeutic options for the treatment of cancer.Citation41,42 Indeed, treatment with exogenous let-7 and miR-34 has been shown capable of both preventing and treating lung tumors in NSCLC animal models, including the therapeutically resistant KrasG12D/+;p53flx/flx mouse model,Citation39,43-46 and mimics of let-7 and miR-34 are currently in preclinical and Phase 1 trials, respectively, as therapeutics for the treatment of cancer.Citation40,47,48 Because let-7 and miR-34 target oncogenes central to the EGFR signaling pathway and are often downregulated in NSCLC, replacement therapies using these miRNAs in combination with erlotinib constitutes a promising therapeutic strategy. Indeed, miR-34 has been shown to enhance the effects of miR-34 in some NSCLC cell lines and in xenograft models.Citation49,50
In this study, we analyze the ability of let-7b and miR-34a to potentiate the effect of erlotinib on human NSCLC lines bearing mutations in RAS and TP53, which are associated with high resistance to chemotherapy and poor prognosis in patients.Citation39,41,43,51 Our data indicate that let-7b and miR-34a can synergize with erlotinib over a wide range of erlotinib concentrations. We also find that combinatorial treatment with let-7b and miR-34a shows the strongest synergy with erlotinib, resulting in greater potentiation of this agent than that induced by either miRNA individually. Our findings support a role for miRNA adjuvant therapy as a viable strategy for chemotherapeutic sensitization, and address the need for new combination treatments for the treatment of NSCLC.
Results
let-7 and miR-34 are downregulated in non-small cell lung cancer cell lines
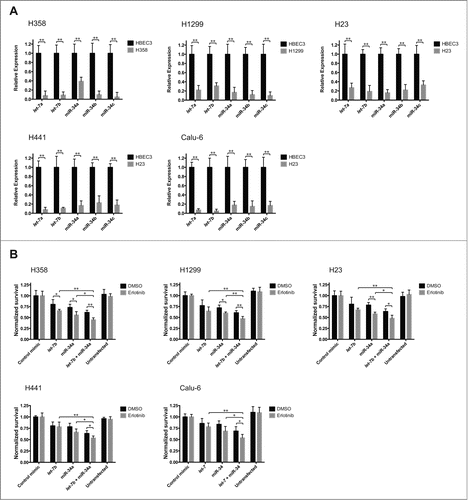
The miRNAs let-7 and miR-34 are often downregulated in non-small cell lung cancer (NSCLC) patients.Citation38-40 To determine whether the expression of let-7 and miR-34 is also altered in lung cancer cell lines, we selected NSCLC cell lines bearing open reading frame mutations in both KRAS and TP53 (H358, H23, H441, Calu-6) or NRAS and TP53 (H1299), collectively referred hereafter as KP lines. We profiled the levels of let-7a, let-7b, miR-34a, miR-34b, and miR-34c in these cell lines using quantitative reverse transcription PCR (RT-qPCR) (). Our analysis shows that these miRNAs are significantly downregulated (p < 0.01; Student's t-test) in the NSCLC lines tested, relative to the non-cancerous human bronchial epithelial cell line HBEC3.
Figure 1. A. The expression of let-7 and miR-34 variants is reduced in lung cancer cell lines. Total RNA from lung cancer cell lines was isolated, and the levels of let-7a, let-7b, miR-34a, miR-34b and miR-34c were determined using quantitative reverse transcription PCR (RT-qPCR). Expression levels are normalized to Rnu6B and plotted relative to the expression levels in a non-cancerous human bronchial epithelial cell line (HBEC3). Plotted: mean ± s.d.; n = 4 ; **P < 0.01, Student's t-test. B. let-7 and miR-34 potentiate the cytotoxic effect of erlotinib. Lung cancer cell lines were transfected with 25 nM of a control miRNA mimic, 25 nM of mimics of let-7b, miR-34a, or a half dose of each let-7b + miR-34a, or left untreated. 48 hours after transfection, the cells were treated with erlotinib at the experimentally determined EC50 for each line (Fig. S1). 72 hours after drug exposure, cell survival was determined via the sulforhodamine-B (SRB) assay. The absorbance measurements for each erlotinib + miRNA mimic treatment were normalized relative to the group treated with erlotinib + control miRNA mimic, while the measurements for the DMSO + miRNA mimic treatments were normalized to the DMSO + control mimic group. Plotted: mean ± s.d.; n = 4 ; *P < 0.05, **P < 0.01, Student's t-test.
let-7b and miR-34a supplementation sensitizes NSCLC cells to erlotinib treatment
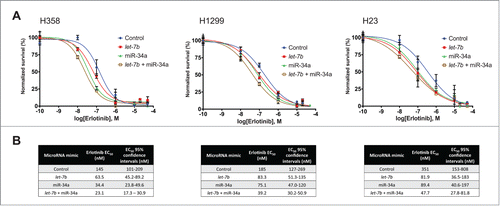
In order to establish whether let-7 and miR-34 supplementation could potentiate the effect of erlotinib on KP lines, we first evaluated their sensitivity to the drug by constructing erlotinib response profiles for each cell line. We determined the effect of serially increasing concentrations of erlotinib on the proliferation of H358, H1299, H23, H441, and Calu-6 cells, and calculated EC50 values for erlotinib in these cells ranging from 474.8 nM to 4.60 μM (Fig. S1).Having established the response baseline to erlotinib in KP cell lines, and to determine whether let-7 and miR-34 can affect their sensitivity to erlotinib, we transfected H358, H1299, H23, H441, and Calu-6 cells with low nanomolar concentrations (25 nM) of a control miRNA mimic, let-7b or miR-34a individually, or half the dose of each miRNA in combination. 48 hours after miRNA transfection, cells were treated with erlotinib at the calculated EC50 for each line (Fig. S1) or DMSO as a vehicle control. Treatment with erlotinib and let-7b, miR-34a, or let-7b + miR-34a resulted in a greater decrease in cell proliferation than that observed when cells were treated with erlotinib and a control miRNA mimic (). In addition, the decrease in proliferation was greater for cells treated with let-7b + miR-34a than for cells treated with either miRNA individually, in agreement with recent observations by Kasinski et al.Citation43 indicating that these miRNAs can synergize in NSCLC cells. To further explore the miRNA-driven sensitization of NSCLC cells to erlotinib, we transfected H358, H1299, H23, H441, and Calu-6 cells with 25 nM of a control miRNA mimic, let-7b or miR-34a individually, or half the dose of each miRNA provided in combination, followed by treatment with erlotinib in a dilution series. Treatment with let-7b, miR-34a, or the combination of these miRNAs resulted in a shift in the erlotinib response curve (), with a corresponding decrease in the erlotinib EC50 value (). This result confirmed the potentiating effect of let-7b and miR-34a, indicating that supplementation of these miRNAs can sensitize KP cells to the effects of erlotinib to further decrease cell proliferation. In addition, this analysis shows that the sensitizing effect is present at a wide range of erlotinib concentrations, and that the combination of the 2 miRNAs produced the greatest effect.
Figure 2. Potentiation of the effect of erlotinib by let-7b and miR-34a over a wide range of erlotinib concentrations. (A) H358, H1299, and H23 cells were transfected with 25 nM of a control mimic, mimics of let-7b, miR-34a, or a half dose of each let-7b + miR-34a. 48 hours after transfection, cells were treated with erlotinib in a serial dilution series. Cell survival was determined following incubation for 72 hours via the sulforhodamine-B (SRB) assay, and normalized survival was calculated for each miRNA treatment. 100% normalized survival is defined as the cell survival of miRNA-transfected cells treated with DMSO as a vehicle control, and 0% normalized survival is defined as the cell survival of untransfected cells treated with 50 μM erlotinib. Plotted: mean ± s.d.; n = 3 . (B) Erlotinib EC50 values and their 95% confidence intervals were calculated for each for each miRNA mimic treatment.
let-7b and miR-34a synergize with erlotinib in NSCLC cells
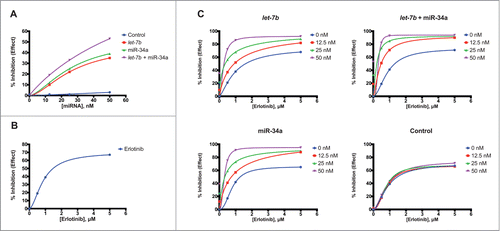
Our data indicates that treatment of KP cells with let-7b, miR-34a, and particularly the combination of let-7b and miR-34a is able to potentiate the anti-proliferative effect of erlotinib. However, these experiments did not allow us to determine whether this effect was additive or synergistic in nature. To assess synergy between let-7b, miR-34a, and erlotinib in KP cells, we first treated H358, H1299, and H23 cells with a control miRNA, let-7b, miR-34a, or a half-dose of each let-7b and miR-34a at concentrations ranging from 12.5 nM to 50 nM ( and S2A). This analysis yielded a response curve for each miRNA over a wide range of concentrations, and allowed us to build response profiles for each miRNA. For all cell lines, the greatest inhibition in proliferation was induced by transfection with let-7b and miR-34a in combination. No miRNA treatment induced greater than 55% inhibition of cell proliferation relative to untreated controls. H358 cells showed the greatest sensitivity to these miRNAs (), while H23 cells were the least affected (Fig S2A). Similarly, we constructed a new response curve for erlotinib, treating H358, H1299, and H23 cells with erlotinib concentrations from 0.5 μM to 5 μM ( and S2B). The maximal inhibition of proliferation induced by the drug was greatest in H358 and H1299 cells, ∼65% relative to controls. H23 cells were slightly less responsive, with maximal inhibition of 62%.To evaluate the ability of erlotinib to synergize with let-7b and miR-34a, we treated H358, H1299, and H23 NSCLC cells with varying concentrations of miRNA mimics and erlotinib. Specifically, cells were transfected with a control mimic, let-7b, miR-34a, or a half-dose of each let-7b and miR-34a at 12.5 nM, 25nM, or 50 nM. 48 hours after transfection, cells were treated with erlotinib at 0.5 μM, 1 μM, or 5 μM. We evaluated cell proliferation for each miRNA-erlotinib combination 48 hours after erlotinib treatment, and constructed effect curves for each miRNA ( and S3). These analyses illustrated the ability of let-7b and miR-34a to potentiate the action of erlotinib, as the observed effect on NSCLC cell proliferation increased markedly with the concentration of miRNA at lower doses of erlotinib. In addition, in H358 cells and H1299 cells, the combination treatment with a half-dose of each let-7b and miR-34a resulted in a lower concentration of erlotinib required to reach the maximal effect for every miRNA concentration, relative to treatments with let-7b and miR-34a individually. Similarly, treatment with 25 nM of the let-7b + miR-34a combination was able to induce largely the same effect as the 50 nM treatment of this combination at both 0.5 μM erlotinib (74% effect for 25 nM let-7b + miR-34a vs. 82% for 50 nM let-7b + miR-34a; H358 cells) and 1 μM erlotinib treatments (85% effect let-7b + miR-34a vs. 93% for 50 nM let-7b + miR-34a; H358 cells).
Figure 3. Dose-response analysis of let-7b and miR-34a in combination with erlotinib in H358 cells. (A) H358 cells were transfected with a control mimic, mimics of let-7b, miR-34a, or a half dose of each let-7b + miR-34a over a range of concentrations (12.5 nM to 50 nM). Five days after transfection, cell survival was determined using the sulforhodamine-B (SRB) assay. Curves representing % inhibition of cell proliferation relative to untransfected cells for each miRNA mimic were constructed using the Hill slope equation. Plotted data points represent the mean of 3 replicates. (B) H358 cells were treated with erlotinib over a range of concentrations (0.5 μM to 5 μM) 4 d after seeding. 72 hours after erlotinib treatment, cell survival was determined using the SRB assay. A curve representing % inhibition of cell proliferation relative to cells treated with DMSO as a vehicle control was plotted using the Hill slope equation. Plotted data points represent the mean of 3 replicates. (C) H358 cells were transfected with control mimic, mimics of let-7b, miR-34a, or a half dose of each let-7b + miR-34a at 3 concentrations (12.5 nM, 25 nM, 50 nM), or left untransfected. Two days after transfection, cells were treated with erlotinib in a serial dilution (0.5 μM to 5 μM). Cell survival was determined using the SRB assay 72 hours after erlotinib treatment. Curves representing % inhibition of cell proliferation relative to untransfected cells treated with DMSO as a vehicle control were constructed for each mimic treatment using the Hill slope equation. Plotted data points represent the mean of 3 replicates.
To directly determine whether the potentiation of erlotinib by let-7b and miR-34a is due to synergy, we used the inhibition of proliferation calculated for each co-treatment to calculate combination index (CI) values for each miRNA-drug combination in H358, H1299, and H23 KP NSCLC cells ( and S4).Citation52 CI values for various miRNA-erlotinib combinations were calculated to be below 0.5 in the 3 cell lines tested, indicating strong synergy between the treatments. In contrast a control miRNA showed no synergy (Fig. S4) at any of the erlotinib concentrations tested. Treatment with let-7b at low doses (12.5 nM) was able to induce strong synergy with erlotinib when the effect levels were above 50%. Transfection with let-7b at 50 nM resulted in extremely low CI values (≤0.22) even at low doses of erlotinib (0.5 μM) in the 3 NSCLC cell lines tested. Similarly, miR-34a transfection produced strong synergy with erlotinib even when both drugs were administered at low doses. Treatment with miR-34a at concentrations of 25 nM and higher resulted in CI values smaller than 0.25 at all erlotinib treatment concentrations in H358 and H1299 cells. Likewise, treatment with miR-34a at 50 nM in these 2 cell lines resulted in extremely low CI values (≤0.11) at all erlotinib concentrations. CI values for the combinatorial treatments tested in H23 cells also indicate strong synergy with erlotinib (Fig. S4B). However, synergy for let-7b and miR-34a with erlotinib was less strong in H23 cells than in H358 and H1299 cells, particularly for the miR-34a treatment.
Figure 4. let-7b and miR-34a synergize with erlotinib in H358 cells. (A) Tables showing combination index (CI) values and % inhibition of cell proliferation (relative to untransfected cells) for H358 cells transfected with let-7b, miR-34a, or half dose of each let-7b and miR-34a, and treated in combination with erlotinib at various concentrations. Reported CI values represent the average of 3 replicate experiments. Cells highlighted in green have CI values smaller than 0.5, indicating strong synergy. (B) Combination index plots of for H358 cells treated with miRNA mimics at 25 nM in combination with erlotinib at concentrations from 0 μM to 5 μM. (C) Bar graph indicating the CI values for H358 cells treated with a control miRNA mimic, or mimics of let-7b, miR-34a, or a half dose of each let-7b and miR-34a at 25 nM, and erlotinib at 1 μM. The dashed line indicates CI = 0.5 as a threshold indicating strong synergy. Plotted: mean ± s.d.; n = 3 ; *P < 0.05, **P < 0.01, Student's t-test.
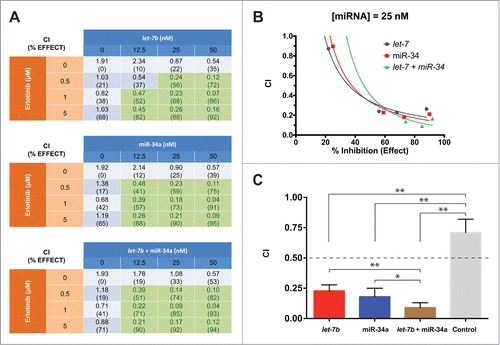
Strong synergy with erlotinib at various miRNA:drug ratios was also observed in NSCLC cells treated with a half-dose of each let-7b and miR-34a. Notably, the CI values when the miRNAs were transfected in combination at a total concentration of 50 nM are similar to those observed when the individual miRNAs are provided. However, when let-7b + miR-34a were provided at 12.5 nM or 25 nM, this combinatorial treatment resulted in lower CI values than the equivalent concentration of either individual miRNA in all 3 NSCLC cell lines, indicating stronger synergy at lower miRNA levels ( and S4B). In H1299 cells, the CI values for let-7b + miR-34a at 12.5 nM are lower than those for let-7b alone, but comparable or higher to those for the individual transfection of miR-34a (Fig. S4B).
The enhanced ability of the let-7b + miR-34a combination to synergize with erlotinib is illustrated in the combination index plots in and S5A,B. At miRNA treatments of 12.5 nM and 25 nM, there is a shift toward greater effect and lower CI value for the let-7b + miR-34a treatment, being most evident when cells were treated with 1 μM erlotinib ( and S5A,B). This shift is indicative of greater synergistic ability for this combinatorial treatment relative to the individual miRNA treatments. Similarly, and S5C, which compare CI values for the different miRNA mimic treatments at 25 nM, show graphically that while individual miRNA treatments result in strong potentiation of erlotinib, the strongest synergy in H358 and H1299 cells was achieved with the combination of let-7b + miR-34a ( and S5A,B).
let-7b and miR-34a fail to potentiate the effect of other tested chemotherapeutics
Our findings indicate that let-7b and miR-34a can synergize with erlotinib and potentiate its antiproliferative effect on NSCLC cells. To explore whether these miRNAs can also affect the sensitivity of NSCLC lines to other chemotherapeutics, we first evaluated their response profile to cisplatin, a broadly used platinum-based chemotherapeutic in the treatment of NSCLC. We transfected H358, H23, and H441 cells with 25 nM of a control miRNA mimic, let-7b or miR-34a individually, or a combination treatment consisting of half the dose of each miRNA. 48 hours after transfection, cell lines were treated with cisplatin in a dilution series. No miRNA treatment was able to significantly alter the cisplatin EC50 for these cell lines, compared to control mimic (Figs. S6A and S6B), suggesting that neither let-7b nor miR-34a supplementation at the concentration provided is able to sensitize the NSCLC lines tested to the effects of cisplatin. We also examined whether let-7b and miR-34a could potentiate the effect of 2 other chemotherapeutics of clinical importance for NSCLC treatmentCitation6,53: etoposide and paclitaxel.Citation2 We transfected H358 cells with 25 nM of a control miRNA mimic, let-7b or miR-34a individually, or half the dose of each miRNA in combination. 48 hours following miRNA transfection, cells were treated with either etoposide or paclitaxel in a dilution series. As with cisplatin, the miRNA treatments tested were unable to alter the etoposide or paclitaxel EC50 relative to control mimic (Figs. S6C–S6F). This result indicates that let-7b and miR-34a are unable to sensitize this NSCLC cell line to the antiproliferative effects of etoposide or paclitaxel.
Discussion
The development of secondary resistance is a major challenge in the treatment of lung cancer. Combinatorial treatment is a promising strategy to overcome resistance; however, it is often hindered by a lack of suitable adjuvant therapeutics. MiRNAs have recently emerged as promising candidates for the treatment of a variety of cancers, due largely to their ability to modulate multiple targets in key oncogenic pathways.
In this study, we evaluated the potential for 2 tumor-suppressive miRNAs, let-7b and miR-34a, to potentiate the effect of the EGFR inhibitor erlotinib in NSCLC cells bearing clinically relevant mutations in KRAS or NRAS, and TP53 (KP cells). Our findings indicate that these miRNAs can synergize with erlotinib to induce a potent antiproliferative effect on NSCLC cells. Both let-7b and miR-34a were individually able to sensitize KP cells to erlotinib, indicating they target oncogenic pathways important for both cancer cell proliferation and the response to erlotinib. These results are in agreement with the recent finding that miR-34a can sensitize lung cancer and hepatocellular carcinoma cells to erlotnib.Citation54
Our results also indicate that administration of let-7b and miR-34a in combination resulted in the strongest sensitization to erlotinib, potentiating the effect of this drug more strongly than the equivalent dose of either miRNA individually. This observation supports previous findings from our group indicating that let-7 and miR-34 can synergize to inhibit cell proliferation and tumor growth.Citation43 This effect may be explained by the fact that let-7b and miR-34a can target distinct sets of potent oncogenes to modulate key pathways involved in tumor growth and survival, including KRAS and MYC for let-7, and BCL2, MET, MYC, and AXL for miR-34.Citation25-30,32,36,37
From a therapeutic perspective, our results support a potential role for let-7b and miR-34a supplementation as adjuvant therapeutics for the treatment of RAS, TP53-mutant NSCLC. Two findings in the present study are of particular translational interest: first, the sensitizing effect of let-7b and miR-34a, individually and in combination, is present even at concentrations of erlotinib well above the EC50, suggesting that these miRNAs target a subset of cellular pathways that erlotinib fails to inhibit. Based on this observation, we predict that miRNA adjuvant therapy would be a robust clinical approach, resistant to the appearance of mutations that might limit the response to erlotinib. Second, the sensitizing effect induced by let-7b and miR-34a was observed even when these miRNAs and erlotinib were provided at low concentrations, suggesting that delivery of these miRNAs could be a plausible strategy to lower the dose of erlotinib required for an effective therapeutic response. Together, these observations indicate that let-7b and miR-34a might represent viable co-therapeutics for the potentiation of current agents and the prevention of secondary resistance.
Our study also indicates, however, that the ability of let-7b and miR-34a to act as co-therapeutics is not universal, as they failed to potentiate the effect of 3 commonly used first-line chemotherapeutics.Citation6,53 Further studies are required to determine whether the sensitizing effect reported here is specific to erlotinib, or is also present with other targeted therapeutics, including other EGFR inhibitors and inhibitors of other receptor tyrosine kinases, such as ERBB2 (HER2) and MET.
While our findings ascertain the ability of these miRNAs to potentiate the in vitro response to erlotinib in NSCLC cells, it is unclear whether this response is also present in other types of cancer driven by receptor tyrosine kinase activation. In addition, animal studies will be essential to determine whether miRNAs can also sensitize tumors to erlotinib in vivo, and whether systemic delivery of miRNAs as co-therapeutics is a safe and effective strategy. The current study begins to set the foundation for the use of combinatorial let-7b + miR-34a supplementation as an adjuvant therapeutic for the treatment of lung cancer.
Materials and Methods
Cell culture
Cell lines were obtained from ATCC. Cells were cultured in RPMI media (Life Technologies, http://www.lifetechnologies.com/order/catalog/product/11875093) supplemented with fetal bovine serum (Sigma-Aldrich, http://www.sigmaaldrich.com/catalog/product/sigma/f0392) to a final concentration of 10% (v/v), and penicillin/streptomycin (Life Technologies, https://www.lifetechnologies.com/order/catalog/product/15070063) to a final concentration of 1% (v/v), following standard tissue culture procedure. Cells were maintained at 37°C in a humidified incubator at 5% CO2 concentration. Cells were passaged at a 1:10 dilution every 3–4 d.
RNA isolation
Total RNA was isolated from cell lines at 80% confluency using TRIzol reagent (Life Technologies, https://www.lifetechnologies.com/order/catalog/product/15596026), according to the manufacturer's instructions. Briefly, growth media was removed from the culture dish, and 8 mL of TRIzol was directly added to each 100 mm dish. Lysis was achieved by pipetting the cells up and down repeatedly. The sample was incubated at room temperature for 5 minutes, followed by the addition of 1.6 mL of chloroform to each sample. The samples were shaken and incubated for 3 minutes at room temperature, and subsequently centrifuged at 12,000 × g at 4°C for 15 minutes. The aqueous phase was recovered and 10 μg GlycoBlue (Life Technologies, http://www.lifetechnologies.com/order/catalog/product/AM9516) was added to each sample. Four mL of 100% isopropanol was added to each sample, followed by incubation at room temperature for 10 minutes, and centrifugation at 12,000 × g for 20 minutes at 4°C. The RNA pellet was gently washed with 5 mL of 75% ethanol, mixed, and centrifuged at 7500 x g for 10 minutes at 4°C. The RNA pellet was air dried at room temperature for 10 minutes, and then resuspended in 30 μL of nuclease-free water.
cDNA synthesis and qPCR
cDNA was synthesized from 1 μg of isolated RNA using the miScript II RT Kit (Qiagen, http://www.qiagen.com/products/catalog/assay-technologies/mirna/miscript-ii-rt-kit), according to the manufacturer's instructions. Following reverse transcription, the reaction mix was diluted by adding 200 μL of RNase-free water. qPCR was performed using 2 μL of the diluted cDNA on a Roche LightCycler 480 II (Roche, http://lifescience.roche.com/shop/products/lightcycler14301–480-instrument-ii), using the miScript SYBR Green PCR Kit (Qiagen, http://www.qiagen.com/products/catalog/assay-technologies/mirna/miscript-sybr-green-pcr-kit) in conjunction with miScript primer assays (Qiagen, http://www.qiagen.com/products/catalog/assay-technologies/mirna/miscript-primer-assays). RNU6–6P was used as a normalization control. Relative miRNA expression in cancer cell lines versus human bronchial epithelial cells (HBEC3) was calculated using the ΔΔCt method. Statistical significance was determined via the Student's t-test (2-tailed), using Prism 6 software (GraphPad, http://www.graphpad.com/scientific-software/prism/).
Transfection
48 hours before transfection, 4000–5000 cells per well were seeded in 96-well, flat-bottom cell culture dishes in RPMI media supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Three wells were seeded for each data point. Twenty-four hours before transfection, the cells were gently washed twice using 100 μL of sterile PBS (Life Technologies, http://www.lifetechnologies.com/order/catalog/product/10010023), and cultured in RPMI media supplemented with 10% fetal bovine serum. Cells were transfected with miRvana miRNA mimics (Life Technologies, https://www.lifetechnologies.com/us/en/home/life-science/epigenetics-noncoding-rna-research/mirna-profiling-/mirvana-mimics-inhibitors.html.html) using DharmaFECT1 (GE Healthcare, http://dharmacon.gelifesciences.com/transfection/dharmafect-1-transfection-reagent/), according to the manufacturer's instructions. Briefly, the miRNA mimics were complexed with DharmaFECT1 in Opti-MEM reduced serum media (Life Technologies, http://www.lifetechnologies.com/order/catalog/product/31985070), and added to the cells in a final volume of 100 μL/well. Four hours after transfection, the transfection media was replaced with RPMI media supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin.
Erlotinib treatment Erlotinib hydrochloride (AbCam, www.abcam.com/Erlotinib-CP-358774-ab120881.html) was dissolved in sterile DMSO (Sigma Aldrich, http://www.sigmaaldrich.com/catalog/product/sigma/d2650?lang=en andregion=) to obtain a 10 mM stock solution. For the determination of EC50 values, 4000–5000 cells were seeded in 96-well, flat-bottom cell culture dishes in RPMI media supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin, and incubated for 48 hours. Three wells were seeded for each data point. Following this incubation, the cells were rinsed with 2 gentle washes of 100 μL of sterile PBS (Life Technologies, http://www.lifetechnologies.com/order/catalog/product/10010023), and cultured in RPMI media supplemented with 10% fetal bovine serum. After a further 48-hour incubation, cells were rinsed with 2 gentle washes of 100 μL of sterile PBS. Erlotinib dilutions were prepared serially in RPMI supplemented with 10% FBS, and added to the cells to a final concentration of 1 nM – 50 μM in a final volume of 100 μL/well. Cells were incubated for 72 hours, and cell survival was determined using the SRB assay, and normalized to cells treated with DMSO to a final concentration of 0.5% as a vehicle control.
For fixed-concentration miRNA mimic + erlotinib treatments and combination index analysis, cells were plated and transfected 48 hours after plating as detailed above. 48 hours after miRNA mimic transfection, cells were washed with PBS, treated with the indicated concentrations of erlotinib, diluted from a 10 mM stock using RPMI media supplemented with 10% FBS, and added to the cells in a final volume of 100 μL/well. 72 hours after erlotinib addition, cell survival was determined using the SRB assay.
For curve shift analyses, cells were plated as above, and transfected with miRNA mimics 48 hours after plating. Following a 48-hour incubation, cells were washed with PBS, and erlotinib dilutions were prepared serially and added to the cells to a final concentration of 1 nM – 50 μM, in a final volume of 100 μL/well. In addition, for each miRNA transfection group, 3 wells were treated with DMSO at a final concentration of 0.5% as a vehicle control. Cell survival was determined using the SRB assay 72 hours after erlotinib addition, and normalized survival was calculated for each treatment as follows: 100% normalized survival is defined as the cell survival of miRNA-transfected cells treated with DMSO as a vehicle control; 0% normalized survival is defined as the cell survival of untransfected cells treated with 50 μM erlotinib.
Cisplatin treatment
Cisplatin was kindly provided by Dr. J. Weidhaas as a 1 mg/mL solution. For curve shift analyses, 4000–5000 cells were seeded in 96-well, flat-bottom cell culture dishes in RPMI media supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin, and incubated for 24 hours. Three wells were seeded for each data point. Following this incubation, the cells were rinsed with 2 gentle washes of 100 μL of PBS, and cultured in RPMI media supplemented with 10% fetal bovine serum. Twenty-four hours later, cells were transfected with miRNA mimics as detailed above. Following a 48-hour incubation, cells were rinsed with PBS, and cisplatin dilutions in RPMI supplemented with 10% FBS were prepared serially and added to the cells to a final concentration of 0.1 nM – 1 mM, in a final volume of 100 μL/well. Cell survival was determined using the SRB assay 72 hours after cisplatin addition.
Etoposide treatment
Etoposide was kindly provided by Dr. J. Weidhaas as a 20 mg/mL solution. For curve shift analysis, 4000–5000 cells were seeded in 96-well, flat-bottom cell culture dishes in RPMI media supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin, and incubated for 24 hours. Three wells were seeded for each data point. Following this incubation, the cells were rinsed with 2 gentle washes of 100 μL of PBS, and cultured in RPMI media supplemented with 10% fetal bovine serum. Twenty-four hours later, cells were transfected with miRNA mimics as detailed above. Following a further 48-hour incubation, cells were rinsed with PBS, and etoposide dilutions in RPMI supplemented with 10% FBS were prepared serially and added to the cells to a final concentration of 0.1 nM – 1 mM, in a final volume of 100 μL/well. Cell survival was determined using the SRB assay 72 hours after etoposide addition.
Paclitaxel treatment
Paclitaxel was kindly provided by Dr. J. Weidhaas as a 6 mg/mL solution. For curve shift analysis, 4000–5000 cells were seeded in 96-well, flat-bottom cell culture dishes in RPMI media supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin, and incubated for 24 hours. Three wells were seeded for each data point. Following this incubation, the cells were rinsed with 2 gentle washes of 100 μL PBS, and cultured in RPMI media supplemented with 10% fetal bovine serum. Cells were transfected with miRNA mimics 24 hours later, as detailed above. Following a 48-hour incubation, cells were rinsed with PBS, and paclitaxel dilutions were prepared serially in RPMI supplemented with 10% FBS and added to the cells to a final concentration of 1 pM – 10 nM, in a final volume of 100 μL/well. Cell survival was determined using the SRB assay 72 hours after paclitaxel addition.
Sulforhodamine B (SRB) assays
72 hours after erlotinib addition, cells in 96-well plates were fixed by adding tricholoroacetic acid (Sigma-Aldrich, http://www.sigmaaldrich.com/catalog/product/sial/t6399) to the media to a final concentration of 10% (v/v). After fixation, cells were rinsed 5 times with distilled water, and air-dried at room temperature for ∼5 hours. The fixed cells were stained for 30 minutes with sulforhodamine-B (SRB) (Sigma-Aldrich, http://www.sigmaaldrich.com/catalog/product/aldrich/230162) dye dissolved in 1% acetic acid (v/v) to a final concentration of 0.4% (w/v). Excess dye was removed by washing with 1% acetic acid, and the plates were then allowed to air-dry at room temperature. Protein-bound dye was extracted by adding 100 μL/well of 10 mM unbuffered Tris base with gentle agitation. Following extraction, absorbance at 490 nm was determined using a Wallac VICTORCitation2 spectrophotometer (PerkinElmer, http://www.perkinelmer.com/catalog/product/id/1420–832).
EC50 values, dose-response curves, and combination index values and plots
EC50 values were calculated from the SRB absorbance readings using non-linear regression (curve fit) analysis in Prism 6 software. Dose-response curves were calculated from the SRB absorbance readings using the Hill slope equation in Prism 6 software. Combination index (CI) values were determined using CompuSyn (http://www.combosyn.com) software to analyze the effect on cell survival derived from SRB absorbance readings at different miRNA mimic + erlotinib concentrations, based on the linear form transformation method developed by Chou et al. Combination index plots were constructed using Prism 6 software, based on the CI analysis performed in CompuSyn. Statistical significance was determined via Student's t-test (one-tailed), using Prism 6 software (GraphPad, http://www.graphpad.com/scientific-software/prism/).
Disclosure of Potential Conflicts of Interest
CS declares no conflicts of interest. FJS is a consultant and a shareholder for Mirna Therapeutics (Austin, TX), which is developing miRNA therapeutics.
1003008_Supplementary_Materials.zip
Download Zip (3 MB)Acknowledgments
The authors thank Dr. Joanne Weidhaas and Sunitha Nallur at the Yale School of Medicine for providing us with cisplatin, etoposide and erlotinib. We thank Dr. Andrea Kasinski and Eliot Shimer at the Department of MCDB, Yale University for their technical assistance, and for useful discussions of our results.
Funding
This work was supported by pilot funds from the Yale Cancer Center and NIH CA131301.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Related Research Data
References
- American Cancer Society: Cancer Facts & Figures 2014. Available: www.cancer.org, 2014
- Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med 2008, 359:1367-1380; PMID:18815398; http://dx.doi.org/10.1056/NEJMra0802714
- Herbst RS, Johnson DH, Mininberg E, Carbone DP, Henderson T, Kim ES, Blumenschein G, Jr., Lee JJ, Liu DD, Truong MT, et al. Phase I/II trial evaluating the anti-vascular endothelial growth factor monoclonal antibody bevacizumab in combination with the HER-1/epidermal growth factor receptor tyrosine kinase inhibitor erlotinib for patients with recurrent non-small-cell lung cancer. J Clin Oncol 2005, 23:2544-2555; PMID:15753462; http://dx.doi.org/10.1200/JCO.2005.02.477
- Sekido Y, Fong KM, Minna JD. Molecular genetics of lung cancer. Ann Rev Med 2003, 54:73-87; PMID:12471176; http://dx.doi.org/10.1146/annurev.med.54.101601.152202
- Minna JD, Roth JA, Gazdar AF. Focus on lung cancer. Cancer Cell 2002, 1:49-52; PMID:12086887; http://dx.doi.org/10.1016/S1535-6108(02)00027-2
- Herbst RS, Lynch TJ, Sandler AB. Beyond doublet chemotherapy for advanced non-small-cell lung cancer: combination of targeted agents with first-line chemotherapy. Clin Lung Cancer 2009, 10:20-27; PMID:19289368; http://dx.doi.org/10.3816/CLC.2009.n.003
- Miller VA, Riely GJ, Zakowski MF, Li AR, Patel JD, Heelan RT, Kris MG, Sandler AB, Carbone DP, Tsao A, et al. Molecular characteristics of bronchioloalveolar carcinoma and adenocarcinoma, bronchioloalveolar carcinoma subtype, predict response to erlotinib. J Clin Oncol 2008, 26:1472-1478; PMID:18349398; http://dx.doi.org/10.1200/JCO.2007.13.0062
- Woodburn JR. The epidermal growth factor receptor and its inhibition in cancer therapy. Pharmacol Ther 1999, 82:241-250; PMID:10454201; http://dx.doi.org/10.1016/S0163-7258(98)00045-X
- Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer 2001, 37 Suppl 4:S9-15; http://dx.doi.org/10.1016/S0959-8049(01)00231-3
- Westra WH. Early glandular neoplasia of the lung. Respir Res 2000, 1:163-169; PMID:11667981; http://dx.doi.org/10.1186/rr28
- Tang X, Shigematsu H, Bekele BN, Roth JA, Minna JD, Hong WK, Gazdar AF, Wistuba II. EGFR tyrosine kinase domain mutations are detected in histologically normal respiratory epithelium in lung cancer patients. Cancer Res 2005, 65:7568-7572; PMID:16140919
- Herbst RS, Sandler A. Bevacizumab and erlotinib: a promising new approach to the treatment of advanced NSCLC. Oncologist 2008, 13:1166-1176; PMID:18997180; http://dx.doi.org/10.1634/theoncologist.2008-0108
- Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer 2007, 7:169-181; PMID:17318210; http://dx.doi.org/10.1038/nrc2088
- Sequist LV, Bell DW, Lynch TJ, Haber DA. Molecular predictors of response to epidermal growth factor receptor antagonists in non-small-cell lung cancer. J Clin Oncol 2007, 25:587-595; PMID:17290067; http://dx.doi.org/10.1200/JCO.2006.07.3585
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005, 2:e73; PMID:15737014; http://dx.doi.org/10.1371/journal.pmed.0020073
- Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, Zakowski MF, Heelan RT, Kris MG, Varmus HE. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med 2005, 2:e17; PMID:15696205; http://dx.doi.org/10.1371/journal.pmed.0020017
- Eberhard DA, Johnson BE, Amler LC, Goddard AD, Heldens SL, Herbst RS, Ince WL, Janne PA, Januario T, Johnson DH, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol 2005, 23:5900-5909; PMID:16043828; http://dx.doi.org/10.1200/JCO.2005.02.857
- Riely GJ, Kris MG, Rosenbaum D, Marks J, Li A, Chitale DA, Nafa K, Riedel ER, Hsu M, Pao W, et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin Cancer Res 2008, 14:5731-5734; PMID:18794081; http://dx.doi.org/10.1158/1078-0432.CCR-08-0646
- Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A 2007, 104:20932-20937; PMID:18093943; http://dx.doi.org/10.1073/pnas.0710370104
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316:1039-1043; PMID:17463250; http://dx.doi.org/10.1126/science.1141478
- Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nature Genet 2012, 44:852-860; PMID:22751098; http://dx.doi.org/10.1038/ng.2330
- Inukai M, Toyooka S, Ito S, Asano H, Ichihara S, Soh J, Suehisa H, Ouchida M, Aoe K, Aoe M, et al. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res 2006, 66:7854-7858; PMID:16912157; http://dx.doi.org/10.1158/0008-5472.CAN-06-1951
- Sun S, Schiller JH, Gazdar AF. Lung cancer in never smokers–a different disease. Nat Rev Cancer 2007, 7:778-790; PMID:17882278; http://dx.doi.org/10.1038/nrc2190
- Wistuba II, Mao L, Gazdar AF. Smoking molecular damage in bronchial epithelium. Oncogene 2002, 21:7298-7306; PMID:12379874; http://dx.doi.org/10.1038/sj.onc.1205806
- Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ. RAS is regulated by the let-7 microRNA family. Cell 2005, 120:635-647; PMID:15766527; http://dx.doi.org/10.1016/j.cell.2005.01.014
- Bommer GT, Gerin I, Feng Y, Kaczorowski AJ, Kuick R, Love RE, Zhai Y, Giordano TJ, Qin ZS, Moore BB, et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol 2007, 17:1298-1307; PMID:17656095; http://dx.doi.org/10.1016/j.cub.2007.06.068
- He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, et al. A microRNA component of the p53 tumour suppressor network. Nature 2007, 447:1130-1134; PMID:17554337; http://dx.doi.org/10.1038/nature05939
- Tarasov V, Jung P, Verdoodt B, Lodygin D, Epanchintsev A, Menssen A, Meister G, Hermeking H. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle 2007, 6:1586-1593; PMID:17554199; http://dx.doi.org/10.4161/cc.6.13.4436
- Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, Bentwich Z, Oren M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell 2007, 26:731-743; PMID:17540598; http://dx.doi.org/10.1016/j.molcel.2007.05.017
- Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 2007, 26:745-752; PMID:17540599; http://dx.doi.org/10.1016/j.molcel.2007.05.010
- Buechner J, Tomte E, Haug BH, Henriksen JR, Lokke C, Flaegstad T, Einvik C. Tumour-suppressor microRNAs let-7 and mir-101 target the proto-oncogene MYCN and inhibit cell proliferation in MYCN-amplified neuroblastoma. Br J Cancer 2011, 105:296-303; PMID:21654684; http://dx.doi.org/10.1038/bjc.2011.220
- Kim HH, Kuwano Y, Srikantan S, Lee EK, Martindale JL, Gorospe M. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev 2009, 23:1743-1748; PMID:19574298; http://dx.doi.org/10.1101/gad.1812509
- Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res 2004, 64:3753-3756; PMID:15172979; http://dx.doi.org/10.1158/0008-5472.CAN-04-0637
- Lee YS, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev 2007, 21:1025-1030; PMID:17437991; http://dx.doi.org/10.1101/gad.1540407
- Viswanathan SR, Daley GQ, Gregory RI. Selective blockade of microRNA processing by Lin28. Science 2008, 320:97-100; PMID:18292307; http://dx.doi.org/10.1126/science.1154040
- Kaller M, Liffers ST, Oeljeklaus S, Kuhlmann K, Roh S, Hoffmann R, Warscheid B, Hermeking H. Genome-wide characterization of miR-34a induced changes in protein and mRNA expression by a combined pulsed SILAC and microarray analysis. Mol Cell Proteomics 2011, 10:M111 010462; http://dx.doi.org/10.1074/mcp.M111.010462
- Wei JS, Song YK, Durinck S, Chen QR, Cheuk AT, Tsang P, Zhang Q, Thiele CJ, Slack A, Shohet J, Khan J. The MYCN oncogene is a direct target of miR-34a. Oncogene 2008, 27:5204-5213; PMID:18504438; http://dx.doi.org/10.1038/onc.2008.154
- Adams BD, Kasinski AL, Slack FJ. Aberrant regulation and function of microRNAs in cancer. Curr Biol 2014, 24:R762-776; PMID:25137592; http://dx.doi.org/10.1016/j.cub.2014.06.043
- Kasinski AL, Slack FJ. miRNA-34 prevents cancer initiation and progression in a therapeutically resistant K-ras and p53-induced mouse model of lung adenocarcinoma. Cancer Res 2012, 72:5576-5587; PMID:22964582; http://dx.doi.org/10.1158/0008-5472.CAN-12-2001
- Kasinski AL, Slack FJ. Epigenetics and genetics. MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nat Rev Cancer 2011, 11:849-864; PMID:22113163; http://dx.doi.org/10.1038/nrc3166
- Stahlhut C, Slack FJ. MicroRNAs and the cancer phenotype: profiling, signatures and clinical implications. Genome Med 2013, 5:111; PMID:24373327; http://dx.doi.org/10.1186/gm516
- Stahlhut Espinosa CE, Slack FJ. The role of microRNAs in cancer. Yale J Biol Med 2006, 79:131-140; PMID:17940623
- Kasinski AL, Kelnar K, Stahlhut C, Orellana E, Zhao J, Shimer E, Dysart S, Chen X, Bader AG, Slack FJ. A combinatorial microRNA therapeutics approach to suppressing non-small cell lung cancer. Oncogene 2014; PMID:25174400
- Trang P, Wiggins JF, Daige CL, Cho C, Omotola M, Brown D, Weidhaas JB, Bader AG, Slack FJ. Systemic delivery of tumor suppressor microRNA mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol Ther 2011, 19:1116-1122; PMID:21427705; http://dx.doi.org/10.1038/mt.2011.48
- Wiggins JF, Ruffino L, Kelnar K, Omotola M, Patrawala L, Brown D, Bader AG. Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res 2010, 70:5923-5930; PMID:20570894; http://dx.doi.org/10.1158/0008-5472.CAN-10-0655
- Trang P, Medina PP, Wiggins JF, Ruffino L, Kelnar K, Omotola M, Homer R, Brown D, Bader AG, Weidhaas JB, Slack FJ. Regression of murine lung tumors by the let-7 microRNA. Oncogene 2010, 29:1580-1587; PMID:19966857; http://dx.doi.org/10.1038/onc.2009.445
- Bader AG. miR-34 - a microRNA replacement therapy is headed to the clinic. Front Genet 2012, 3:120; PMID:22783274; http://dx.doi.org/10.3389/fgene.2012.00120
- A Multicenter Phase I Study of MRX34, MicroRNA miR-RX34 Liposomal Injection [http://clinicaltrials.gov/show/NCT01829971], accessed 11/25/2014
- Zhao J, Kelnar K, Bader AG. In-depth analysis shows synergy between erlotinib and miR-34a. PloS One 2014, 9:e89105; PMID:24551227; http://dx.doi.org/10.1371/journal.pone.0089105
- Zhou JY, Chen X, Zhao J, Bao Z, Chen X, Zhang P, Liu ZF, Zhou JY. MicroRNA-34a overcomes HGF-mediated gefitinib resistance in EGFR mutant lung cancer cells partly by targeting MET. Cancer Lett 2014, 351:265-271; PMID:24983493; http://dx.doi.org/10.1016/j.canlet.2014.06.010
- Singh M, Lima A, Molina R, Hamilton P, Clermont AC, Devasthali V, Thompson JD, Cheng JH, Bou Reslan H, Ho CC, et al. Assessing therapeutic responses in Kras mutant cancers using genetically engineered mouse models. Nat Biotechnol 2010, 28:585-593; PMID:20495549; http://dx.doi.org/10.1038/nbt.1640
- Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 2006, 58:621-681; PMID:16968952
- Glisson BS, Kurie JM, Perez-Soler R, Fox NJ, Murphy WK, Fossella FV, Lee JS, Ross MB, Nyberg DA, Pisters KM, et al. Cisplatin, etoposide, and paclitaxel in the treatment of patients with extensive small-cell lung carcinoma. J Clin Oncol 1999, 17:2309-2315; PMID:10561292
- Kelnar K, Peltier HJ, Leatherbury N, Stoudemire J, Bader AG. Quantification of therapeutic miRNA mimics in whole blood from nonhuman primates. Anal Chem 2014, 86:1534-1542; PMID:24397447