
The lymphotropic gammaherpesvirus Kaposi's sarcoma-associated herpesvirus (KSHV) is the infectious cause of the endothelial cell tumor Kaposi's sarcoma (KS) and the B cell lymphomas primary effusion lymphoma (PEL) and multicentric Castleman's disease (MCD). Like all gammaherpesviruses, KSHV establishes long-term latent infection of lymphoid cells wherein the virus remains largely inconspicuous, transcribing a limited set of genes dedicated to maintenance of the latent viral episome and evasion of immune surveillance. Because latency typically predominates in tumor tissue, the products of the latent gene expression program have been subjected to intense scrutiny since the discovery of KSHV 20 years ago.Citation1
Over millennia of co-evolution between herpesviruses and ancestral primates a number of host genes have been ‘captured’ and have become fixed in viral genomes, where they accrue additional mutations that allow them to resist natural host regulatory mechanisms. These genes are often dispensable for viral replication in cell culture, but play important pathogenic roles in vivo. KSHV v-cyclin is a fascinating example of a pirated host gene. It is a homolog of human cyclin D2 that has gained additional properties through evolution; when complexed to host cyclin-dependent kinase CDK6, v-cyclin licenses phosphorylation of a remarkably broad range of substrate proteins, while simultaneously resisting control by host CDK inhibitor proteins.Citation2 In this way, v-cyclin profoundly dysregulates the cell cycle, pushing the “gas pedal” of cell proliferation while disabling the brakes.
Surprisingly, the dramatic effects of v-cyclin on the cell cycle did not immediately translate to a strong oncogenic phenotype in vivo; expression of v-cyclin in the lymphoid compartment of mixed CBA/C57BL/6 mice led to the development of low penetrance, late onset lymphomas.Citation3 Undeterred, Pekkonen and colleagues adapted this model by crossbreeding into the ICR (CD1) background.Citation4 In stark contrast to the previous model, these mice displayed a robust phenotype characterized by early onset T-cell lymphomas, despite relatively low v-cyclin expression levels in the thymus and an absence of additional mutations in common tumor suppressors. These findings provide strong evidence that v-cyclin can act as a true oncogene in the appropriate cellular context.
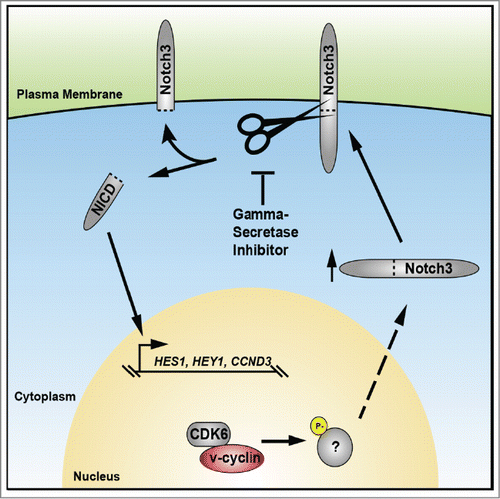
In the course of investigating the molecular mechanism of T-lymphoma development in this model, the authors discovered a new function for v-cyclin in controlling the Notch signal transduction pathway. The Notch pathway has a well-established role in controlling normal T-cell development and perturbations in Notch signaling have been previously linked to lymphoma initiation. In v-cyclin-induced thymic lymphomas, Pekkonen, et. al. observed an increase in the expression and activity of Notch3, with a corresponding increase in transcription of downstream targets Hes1, Hey1 and cyclin D3 (CCND3). Importantly, both in vivo and in vitro, v-cyclin-mediated Notch pathway activation was shown to be dependent on v-cyclin-CDK6 kinase activity (). Clearly, more work will be required to fully elucidate the mechanism of v-cyclin-mediated Notch pathway activation.
Figure 1. Model for KSHV v-cyclin-induced dysregulation of Notch signaling during lymphomagenesis. Expression of v-cyclin within T-cells reprograms the functions of its cognate kinase CDK6, an important regulator of lymphocyte development. By hijacking CDK6, v-cyclin alters intracellular signaling and up-regulates expression of Notch 3. Proteolytic cleavage of Notch at the cell surface by gamma-secretase causes accumulation of the Notch intracellular domain (NICD), which translocates to the nucleus and facilitates transcription of HES1, HEY1 and CCND3. The emerging role for Notch pathway dysregulation during KSHV infection suggests that therapeutics targeting this pathway, such as gamma-secretase inhibitors, may be used to combat KSHV-associated lymphomas.
There have been case reports linking KSHV infection to rare T-cell lymphomas, but the evidence at this time is too fragmentary to firmly establish an etiologic link. Nevertheless, the current study provides insight relevant to all KSHV-associated malignancies. It is clear that KSHV has invested heavily in the control of the Notch pathway; v-cyclin now joins other KSHV genes LANA, vFLIP, RTA, vGPCR and vIL6 in controlling the expression of Notch pathway receptors and ligands (recently reviewed in.Citation5 KSHV-induced malignancies display strong Notch pathway activation, and the growth of PEL cell linesCitation6 and xenografted PEL lesions in miceCitation7 are sensitive to inhibition of Notch signaling by gamma-secretase inhibitors (GSIs) that block the proteolytic processing of the full-length Notch receptor to the active Notch intracellular domain (NICD). There are currently few therapeutic options for advanced PEL. Pekkonen et. al. provide solid rationale for investigation of next-generation Notch pathway inhibitors in the treatment of KSHV-associated malignancies.
References
- Giffin L, Damania B. Adv Virus Res 2014; 88:111-59; PMID:24373311; http://dx.doi.org/10.1016/B978-0-12-800098-4.00002-7
- Verschuren EW, et al. J Gen Virol 2004; 85:1347-61; PMID:15166416; http://dx.doi.org/10.1099/vir.0.79812-0
- Verschuren EW, et al. Cancer Cell 2002; 2:229-41; PMID:12242155; http://dx.doi.org/10.1016/S1535-6108(02)00123-X
- Pekkonen P, et al. Cell Cycle 2014; 13:3670-84; PMID:25483078; http://dx.doi.org/10.4161/15384101.2014.964118
- Cheng F, et al. Future Microbiol 2012; 7:1191-205; PMID:23030424; http://dx.doi.org/10.2217/fmb.12.95
- Lan K, et al. J Virol 2006; 80:6411-9; PMID:16775329; http://dx.doi.org/10.1128/JVI.00239-06
- Lan K, et al. Cancer Biol Ther 2009; 8:2136-43; PMID:19783901; http://dx.doi.org/10.4161/cbt.8.22.9743