
ABSTRACT
Viruses can interact with host cell molecules responsible for the recognition and repair of DNA lesions, resulting in dysfunctional DNA damage response (DDR). Cells with inefficient DDR are more vulnerable to therapeutic approaches that target DDR, thereby raising DNA damage to a threshold that triggers apoptosis. Here, we demonstrate that 2 Jurkat-derived cell lines with incorporated silent HIV-1 provirus show increases in DDR signaling that responds to formation of double strand DNA breaks (DSBs). We found that phosphorylation of histone H2AX on Ser139 (gamma-H2AX), a biomarker of DSBs, and phosphorylation of ATM at Ser1981, Chk2 at Thr68, and p53 at Ser15, part of signaling pathways associated with DSBs, are elevated in these cells. These results indicate a DDR defect even though the virus is latent. DDR-inducing agents, specifically high doses of nucleoside RT inhibitors (NRTIs), caused greater increases in gamma-H2AX levels in latently infected cells. Additionally, latently infected cells are more susceptible to long-term exposure to G-quadruplex stabilizing agents, and this effect is enhanced when the agent is combined with an inhibitor targeting DNA-PK, which is crucial for DSB repair and telomere maintenance. Moreover, exposing these cells to the cancer drug etoposide resulted in formation of DSBs at a higher rate than in un-infected cells. Similar effects of etoposide were also observed in population of primary memory T cells infected with latent HIV-1. Sensitivity to these agents highlights a unique vulnerability of latently infected cells, a new feature that could potentially be used in developing therapies to eliminate HIV-1 reservoirs.
Introduction
DNA damage response is an important part of cellular networks, responsible for maintaining genome stability. The system monitors DNA integrity, detects DNA lesions, activates cell cycle checkpoints and regulates DNA repair pathways.Citation1 Among the different types of DNA damage, DSBs are considered to be the most serious, as a single DSB is sufficient to induce cell death.Citation2 Improperly repaired DSBs can lead to chromosomal rearrangements resulting in translocations, or deletions, both contributing to cell death or oncogenic transformation.Citation3 DSBs can be caused by direct interaction with a damaging agent, reactive oxygen species (ROS), metabolic processes, impaired DNA damage repair processes, and telomere erosion.Citation4-8
Relationships between viruses and DDR in the host are well described and show that viruses modulate cell metabolism and interact with cellular molecules to activate or deactivate different pathways in DNA damage repair.Citation9-12 Virus-induced dysfunction in DNA repair may result from up- or downregulation of key factors involved in DDR. There is also growing evidence that HIV-1 influences DDR during active infection, with most studies focusing on the accessory protein Vpr targeting different factors essential for host genome integrity and cell cycle arrest.Citation13-22 However, nothing is known about DDR in HIV-1 reservoirs, where the virus remains latent.
Here, we used 2 Jurkat-derived cell lines with incorporated HIV-1 provirus to determine whether latently infected cells have specific DDR characteristics that distinguish them from un-infected cells. We asked whether these cells, which do not express the virus, exhibit a greater degree of DSB formation and activated cellular signaling associated with detection of DSBs, when exposed to agents targeting DNA or DDR. Long-term exposure of latent cells to an agent targeting telomeres increased cell death, an effect enhanced by a DNA repair inhibitor. High doses of NRTIs caused greater increases in the DSB biomarker in latent cells, indicating defective repair of induced damage. Moreover, exposing latent cells to the DSB-inducing cancer drug etoposide produced more DSBs than in un-infected cells. Similar effect of etoposide were also observed in population of primary memory T cells infected with latent HIV-1, a latency model, which closely resembles the environment in vivo. We discuss how changes in signaling in response to DSB formation, derived from deficient DNA repair and telomere erosion, suggest that HIV-1 latency influences the ability of the host to efficiently respond to DNA damage.
Results
Latently infected cells are more susceptible to agents targeting G-quadruplexes and an inhibitor of DSB repair
Previous reports demonstrated that G-quadruplexes (G4) are formed in HIV-1 sequences and that G4-stabilizing agents, BRACO19 and TMPyP4 have activity against G-rich HIV-1 sequences, inhibiting viral replication.Citation23-28 The cellular effects of G4 binding agents have been intensively investigated in anticancer research.Citation29,30 Studies showed that G4-stabilizing agents can induce DSBs, and sensitize cells to inhibitors of DNA repair, triggering apoptosis.Citation31,32 This characteristic prompted us to investigate whether cells infected with latent HIV-1 could be more susceptible to G4-stabilizing agents and inhibitors of DNA repair, than un-infected cells. We first tested antiviral activity of BRACO19 and TMPyP4 on replication competent infectious HIV-1 virus propagating in PM1 cell culture. Consistent with a previous report, BRACO19 showed strong antiviral activity at as low as 6 µM concentration, much lower than previously reported,Citation26 inhibiting HIV-1 replication to an undetectable level (p24 ELISA) in a cell culture maintained for 1 w (). TMPyP4 at 25 µM was also able to inhibit HIV-1 replication and reduced the level of p24 about 100-fold. Viability of PM1 cells was not affected at indicated concentrations of tested G4-stabilizing agents (data not shown). The strong antiviral activity of BRACO19 was also observed in long-term treatment (). Here, the level of virus replication dropped rapidly when infected PM1 cells were exposed to the agent on day 5 post-infection and virus was undetectable by p24 ELISA for up to 3 w post-infection. These data confirm that 2 different G4-stabilizing agents affect HIV-1 replication, suggesting that the agents directly target HIV-1 G-rich sequences capable of forming G-quadruplexes.
Figure 1. Latently infected cells are susceptible to G4 binding agents and an inhibitor of DSB repair. (A) Antiviral activity of TMPyP4 and BRACO19. Left panel, PM1 cells were infected with replication-competent HIV-1 NL4–3 and exposed to 6 µM BRACO19 (B) or 25 µM TMPyP4 (T). Samples were collected on days 5, 6, 7 and 8 post-infection and HIV-1 gag p24 was assayed in cell-free culture supernatants by ELISA. N – not treated infected PM1 cells. Right panel, BRACO19 displays strong antiviral activity. The level of virus replication dropped rapidly when infected PM1 cells were exposed to the agent on day 5 post-infection and virus was undetectable by p24 ELISA for up to 3 w post-infection. (B) Representative contour plots of flow cytometric analyses showing that Jurkat-derived HIV-1 latently infected cells CA5 and EF7 show increased susceptibility to G4-binding agents and a DNA repair inhibitor. The cultures were maintained in the presence of 6 µM BRACO19 (BR), 15 µM TMPyP4 (TM), and in combination with 1 µM NU7441 (NU), an inhibitor of DNA-PK involved in DSB repair and telomere maintenance. Apoptosis was analyzed at day 6 (left panel), and day 8 (right panel). Live (Square III), early apoptotic (IV), and late apoptotic/dead cells (II) were discriminated based on binding of Annexin V APC and the uptake of 7AAD. (C) The graph shows changes in a population of cells, which stained positively with Annexin V APC (mean of triplicate experiments). NT – not treated cells.
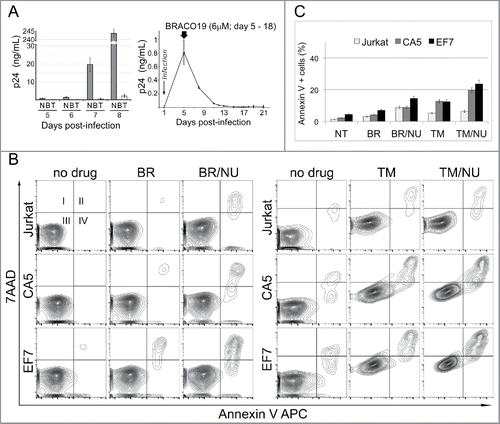
To test the effects of G4-stabilizing agents and a DNA repair inhibitor on HIV-1 latently infected cells, we used 2 Jurkat-derived T cell lines, CA5 and EF7 with established HIV-1 latency.Citation33,34 Both cell lines have an integrated single copy of a full-length HIV-1 genome, which is not expressed, but can be activated upon induction with TNFα producing infectious replication-competent virions. We first tested susceptibility of CA5 cells to G4-stabilizing agents at different concentrations and in combination with a DNA repair inhibitor by analysis of cell viability using a Vi-CELL Cell Viability Analyzer. Cells were seeded in the presence of BRACO19 (3 µM and 6 µM) or TMPyP4 (5 µM and 15 µM), and also in the presence or absence of the inhibitor 2-N-morpholino-8-dibenzothiophenyl-chromen-4-one (NU7441, 1.5 µM), targeting DNA-dependent protein kinase (DNA-PK).Citation31 DNA-PK is required for the non-homologous end-joining (NHEJ) pathway of DNA repair, which rejoins double-strand breaks. The number of live cells was determined 48h later. No changes in cell viability were observed at all tested concentrations of G4 binding agents alone or in combination with the DNA-PK inhibitor (data not shown).
Next, we wanted to know whether long-term exposure to G4-stabilizing agents and the DNA repair inhibitor would affect the viability of latent cells. The cultures were maintained in the presence of these drugs for 1–2 w and were monitored for viability and apoptosis by flow cytometry at days 6 and 8. The long-term exposure to 6 µM BRACO19 resulted in a sharp decline in viability at a similar rate for all cells after 13–16 d (data not shown). The combination of BRACO19 with NU7441 (1 µM) affected EF7 more (about 14.5% apoptotic/dead cells) than CA5 and Jurkat (about 8%), which showed increased susceptibility by day 6 (). However, both latent cell lines showed increased sensitivity to TMPyP4 (15 µM). The susceptibility of the cells to TMPyP4 was analyzed at day 8 (). The apoptosis assay indicated that latently infected cells were entering apoptosis and death at a faster rate (12% of Annexin V positive cells), than un-infected cells (5%) (). Importantly, this effect was further accelerated by the presence of 1 µM NU7441. Here, about 6% of un-infected Jurkat cells were apoptotic/dead, compared with 19–23% of the latently infected cells.
In conclusion, cells infected with latent HIV-1 are more susceptible to agents targeting G-quadruplex structures when used alone and this effect is exacerbated by the combination with the DNA repair inhibitor. Results also indicate that the 2 G4-stabilizing agents have different antiviral activities in cells, and that tested latent cell lines show different levels of susceptibility to compounds targeting DNA and DDR. These differences may result from different affinities of these 2 drugs for G-quadruplexes formed by RNA and DNA sequences. Finally, increased susceptibility to agents targeting DNA and DDR suggests that the DNA damage response in infected cells is affected by HIV-1 latency.
DDR in latently infected cells responds less efficiently to NRTI-induced DNA damage
To determine whether efficiency of DDR is affected by HIV-1 latency, we analyzed cellular response to a high concentration of nucleoside analogues (NAs). NAs are used in treatment of cancer and viral infections. Antiviral NAs are designed to inhibit replication of the viral genome, but at high concentrations they induce DDR in cells.Citation35–37 This response is related to mobilization of the cellular DNA repair system to remove NAs, which become incorporated into host genomic DNA. NAs also affect telomerase activity and induce production of ROS, leading to further DNA damage. Cells with compromised DDR should display an increased level of DNA damage resulting from inefficient DNA repair of lesions produced by NAs. We analyzed DDR by exposing cells to high doses of the anti-HIV class of NAs called nucleoside reverse transcriptase inhibitors (NRTIs), choosing AZT (azidothymidine, zivudine) and d4T (stavudine), which are known to incorporate into genomic DNA, affect telomerase and to induce oxidative stress.Citation36,38-46
The effects of high doses of NRTIs on DNA damage response in latent cells were analyzed by assessing the level of gamma-H2AX (γH2AX), which is a biomarker of DSBs. The γH2AX protein is a histone H2A variant, H2AX, phosphorylated on serine 139 in response to DSBs. Jurkat and Jurkat-derived latently infected cell lines were initially treated with AZT at 5, 25, 125 µM for 1–2h, and the levels of γH2AX were measured by flow cytometry (data not shown). Greater increases in the level of γH2AX were observed for both CA5 and EF7 cells at the highest tested concentration of AZT, evident by a right shift of fluorescence intensity of the fluorophore BV421 conjugated with anti-γH2AX antibody. After 24h of treatment, up to 53–60% of latent cells showed the highest fluorescence intensity, whereas only 26% of Jurkat cells responded (). Similar increases were observed when these same cells were exposed to high doses (100 µM) of d4T (). Surprisingly, we observed that both Jurkat-derived latently infected HIV-1 cell lines show increases in γH2AX levels before NRTI exposure, when compared with un-infected cells. Here, roughly 18–22% of CA5 and 9–11% of EF7 populations showed highest fluorescence intensity of γH2AX-BV421, compared with only about 2–5% of the un-infected Jurkat cell population.
Figure 2. DNA damage response in latently infected cells to high doses of NRTIs. (A) Representative counter plots of 3 experiments showing that the level of γH2AX, a DSB biomarker, is increased in untreated CA5 and EF7 cells, and 24h after exposure to high concentrations of AZT (125 µM; left) and d4T (100 µM; right). The cells were stained with anti-H2AX (pS139) mAb conjugated to BV421 to detect γH2AX and analyzed by flow cytometry. The gate represents the plot area with the highest fluorescence intensity determined by BV421 labeling. The numbers indicate the percentage of the γH2AX positive population with the highest fluorescence intensity. Below, histograms show differences in fluorescence intensities between Jurkat and the 2 latent HIV-1 cell lines before and after treatment with high doses of NRTIs. (B) Population of γH2AX positive events detected for Jurkat and latent cells, and in response to high doses of AZT and d4T. (C) NRTIs-induced increases in γH2AX level are related to DNA damage. Distribution of γH2AX in cell cycle phases was analyzed by staining intensity for γH2AX vs. DNA content (7-AAD). Plots show that high doses of AZT and d4T caused a shift toward high fluorescence intensity of BV421 (in red) detecting γH2AX in each cell cycle phase (G1, S and G2), which is consistent with cellular response to DNA damage. Significant accumulation of signal observed for S-phase suggests that the cell cycle arrest derives from replication inhibition.
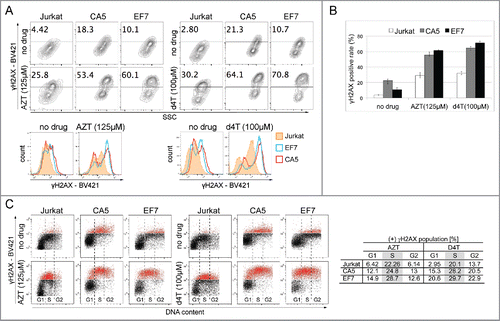
To confirm that the NRTI-induced effects are related to DDR, the levels of γH2AX were analyzed in each phase of the cell cycle by checking γH2AX distributions versus DNA content.Citation47 Results show that in response to the AZT and d4T treatments, the levels of the DSB biomarker were elevated in every phase (G1, S, G2), which is consistent with an alteration of the DDR. In addition, accumulation of the γH2AX signal in S-phase indicates that high doses of NRTIs caused cell cycle arrest in the context of DNA damage. This is consistent with previous reports indicating that AZT causes replication inhibition and accumulation of cells in S-phase.Citation38
In conclusion, greater increases in the level of γH2AX in response to high doses of NAs indicate that latent cells do not process NA-induced damage in DNA at the same rate as un-infected cells. This suggests that DDR is likely less efficient in latently infected cells. In fact, untreated latently infected cells initially appear to have small increases in the level of γH2AX, indicating that defective DDR is a fundamental property of latently infected cells.
Latently infected cells are more susceptible to an agent that promotes formation of DSBs
An increased level of the DSB marker in response to high doses of NAs suggests that mechanisms of DNA damage repair and/or telomere maintenance are antagonized by HIV-1 latency. DNA repair deficiency is true and well described for different types of cancer cells, and, therefore, is a major consideration in anti-tumor drug development efforts. To determine whether HIV-1 latency affects cellular sensitivity to a cancer drug that induces DSBs in cells, we exposed latently infected cells to etoposide, an inhibitor of DNA topoisomerase II. The cells were treated with 50 µM etoposide for 1 to 2h, then harvested and analyzed for the level of etoposide-induced γH2AX (). Flow cytometry analyses of the γH2AX levels show that after about 1h of treatment in both latently infected cell lines most of detected events (77–87%) were recorded with the high fluorescence intensity of fluorophore BV421 corresponding to positive detection of γH2AX. However, for Jurkat cells, about 45% of all events were still not detected in the range of high fluorescence intensity. After 2h of exposure, most of the events were recorded in the range of high fluorescence intensity for all cells. However, the distribution of fluorescence intensities observed on histograms showed that right shifts toward high fluorescence intensities of fluorophore BV421 were greater for latent cells than for un-infected cells suggesting that the number of DNA lesions per cell had been greater in the latent cells.
Figure 3. Effect of etoposide on cells infected with latent HIV-1. Jurkat cells and cells latently infected with HIV-1, CA5 and EF7 were exposed to 50 µM etoposide (ET) for about 1h (left) and 2h (right). Collected cells were washed and prepared for analysis of the γH2AX levels by flow cytometry. The gate represents the plot area with the highest fluorescence intensity determined by BV421 labeling. The numbers indicate the percentage of the γH2AX positive population with the highest fluorescence intensity. Below, histograms show differences between Jurkat and the 2 latent HIV-1 cell lines in distribution of fluorescence intensities of fluorophore BV421 used for detection of γH2AX.
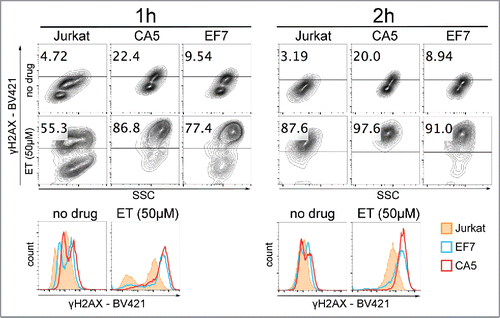
These results indicate that exposing latently infected cells to etoposide caused formation of DSBs at higher rate than in un-infected cells. In conclusion, increased levels of the DSB biomarker observed in untreated cells and after etoposide treatment suggest that cells with established HIV-1 latency have deficiencies in DNA repair mechanisms, and are more susceptible to targeting of DDR.
Signaling of DNA damage is activated in cells infected with latent HIV-1
The elevated levels of γH2AX in cell lines infected with latent HIV-1 suggest that these cells are experiencing more DNA damage than the uninfected parental ones, which is triggering a cellular response to conduct repairs. To determine whether signaling of DNA damage is chronically activated in latently infected cells, we analyzed the proteins Ataxia telangiectasia mutated (ATM), serine/threonine kinase Chk2, and tumor suppressor p53, all involved in specific signaling in response to DSBs. ATM is a serine/threonine kinase that regulates cell cycle checkpoints and DNA repair.Citation48 It is a primary DSB sensor recruited to DNA lesions to phosphorylate the histone H2AX on Ser139. In response to DSBs, ATM is activated by autophosphorylation on Ser1981.Citation49 The protein further recruits other factors including the effector protein kinase Chk2 and the tumor suppressor p53. ATM phosphorylates Chk2 at Threonine 68, and this Thr68-phosphorylated form of Chk2 localizes at sites of DNA breaks.Citation50,51 p53 becomes activated in response to different stressors, but in the case of DNA damage, phosphorylation of Serine 15 on p53 is required to permit local acetylation of histones and relaxation of chromatin.Citation52-55
To determine whether this signaling pathway associated with DNA damage is activated in latently infected cells, we analyzed the levels of phosphorylated forms of ATM at Ser1981, Chk2 at Thr68 and p53 at Ser15 (). These levels were assessed by flow cytometry after staining with monoclonal antibodies that specifically detect only phosphorylated forms of these factors. Our data show that phosphorylated forms of the analyzed factors are elevated in cells infected with latent HIV-1. This is evident from a right shift of fluorescence intensity of detected fluorophore-conjugate antibodies in the 2 latently infected cell types, compared with intensities detected in un-infected cells.
Figure 4. Activation of the DNA damage response pathway in latently infected cells. Representative histograms illustrating increases in levels of phosphorylated forms of ATM(Ser1981), Chk2(Thr68) and p53(Ser15) in 2 latently infected cells, CA5 and EF7, as compared with un-infected parental Jurkat cells. To detect phosphor-p53(Ser15), samples were stained with mAb conjugated with Alexa Fluor® 647. To detect phosphor-ATM(Ser1981) and phosphor-Chk2(Thr68), samples were stained with specific mAb, followed by staining with secondary antibody conjugated with Alexa Fluor® 647. A right shift of fluorescence intensities detected for the 2 latently infected cells corresponds to increases in the level of analyzed factors involved in detection of DSBs. On right, levels of phosphorylated forms of ATM, Chk2 and p53 in cells were compared using median of fluorescence intensities (MFI). The graph shows relative MFIs calculated from 3 experiments using MFI values of unstained samples as controls in the p53(Ser15) analysis; and samples stained only with secondary antibodies conjugated with Alexa Fluor® 647 in analysis of ATM(Ser1981) and Chk2(Thr68). Significance (p-value) was determined by 2-tailed paired-samples t test analysis.
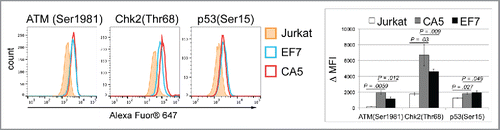
In summary, increases in the level of the DSB biomarker γH2AX, as well as phosphorylated forms of ATM, Chk2 and p53, indicate that signaling linked to DNA damage is induced in cells infected with latent HIV-1.
Primary memory T cells infected with latent HIV-1 are more susceptible to etoposide
We also used a HIV-1 latency model, which closely resembles the environment in vivo to determine whether latent HIV-1 infection influences DNA damage response. Here, we used a method developed by Bosque and Planelles Citation56,57 to generate latently HIV-1 infected cells derived from human primary CD4+ T lymphocytes (). The efficiency of established latency in this model varies from about 15% to 50%, depending on the blood donor.Citation56 Peripheral blood mononuclear cells (PBMC) isolated from healthy blood donors were used for negative selection of primary naïve CD4+ T cells. The cells were primed toward differentiation into nonpolarized (NP) cells, which resemble the central memory T cells (TCM) phenotype characterized by simultaneous expression of CD27 and CCR7 ().Citation58,59 Infection with HIV-1 was performed when cells were still in an activated state (Day 0, ), and latency was established by progression of activated cells to a quiescent, memory-like state. For infections, we used VSV-G pseudotyped single cycle HIV-1 virus with defective env (DHIV), and HIV-GFP, a NL4–3 virus with EGFP incorporated into env. Progression toward a quiescent state was monitored by flow cytometry analysis of the p24 or GFP level in cells, which reflects the fraction of cells still in an active state ( and Supplement Fig. 1A). By day 10, cultured cells exposed to HIV-1 were in a quiescent state with only about 1% of cells expressing viral genes.
Figure 5. Effect of etoposide on the population of primary memory T cells infected with latent HIV-1. (A) Procedure used for the generation of cultured TCM cells latently infected with HIV-1. Naïve CD4 T cells selected from PBMC were activated with αCD3/αCD28 beads in the presence of αIL-4, αIL-12 and TGF-β1 for 3 d (Day-7 to -4), and then cultured in the presence of IL-2 to differentiate into nonpolarized (NP) cells. On day 0, cells were infected with VSV-G-pseudotyped HIV-1 that expresses GFP reporter (HIV-GFP), or without it (DHIV). The cultures were maintained for another 2 w in the presence of IL-2. (B) Cultured cells show simultaneous expression of CD27 and CCR7 on day 0 and 7, which also characterizes TCM cells. (C) Cells infected with DHIV (shown for donor 2) have characteristics of central memory T cells. The cells return to a quiescent state by day 10 post-infection. (D) Cultured TCM cells were prepared from 7 donors and at day 12 or 14 post-infection, HIV infected and un-infected cells were assessed for the level of γH2AX after 1h treatment with 25 µM etoposide (shown for 3 donors). The numbers in the gates indicate the percentage of cells recorded with the high fluorescence intensity corresponding to positive detection of γH2AX. Values corresponding to 7 donors are shown in panel (E), where each symbol represents a different donor and horizontal lines indicate media values. Significance was determined by 2-tailed paired-samples t test analysis (p-value).
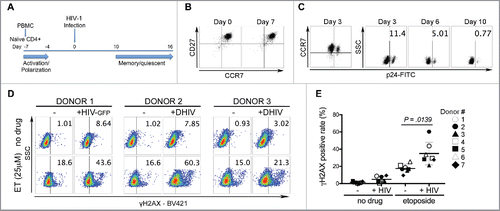
To determine whether HIV-1 latency affects DNA damage response in primary memory CD4+ T, we generated latently infected and un-infected cells from 7 blood donors and treated them with etoposide to assess cellular sensitivity to a DNA targeting drug. At day 12 or 14 post-infection, cells were exposed to 25 µM etoposide for 1 h, than washed and processed for analysis of the level of γH2AX. All infected cells showed greater increases in the γH2AX level in response to etoposide when compared with un-infected cells from the same donor ( and Supplementary Fig. 1C). Among 7 analyzed blood donors, latently infected cells generated from 2 donors showed greater increases of 40–60% in the levels of γH2AX, whereas for un-infected cells the increases were in a range of 16–18% in response to etoposide treatment ().
These results support the conclusion that HIV-1 latency negatively influences DNA damage response in infected cells.
Discussion
Our studies focused on determining whether cells with incorporated silent HIV-1 provirus have specific characteristics related to the DNA damage response that distinguish them from un-infected cells. We initially focused on G4 binding agents that stabilize G-quadruplexes, since they were demonstrated to have activity toward G-rich RNA and DNA sequences of HIV-1.Citation25-27 In anticancer research, G4 binding agents were shown to trigger formation of DSBs and to induce synthetic lethality when combined with DNA repair inhibitors.Citation31,32 We hypothesized that similar effects might be induced in HIV-1 latent reservoirs by targeting HIV-1 provirus incorporated into the host chromosome. We found that 2 different G-quadruplex-stabilizing agents, BRACO19 and TMPyP4 have strong antiviral activity against replication-competent HIV-1, suggesting that G4-stabilizing agents directly target G-rich sequences in HIV-1. To determine whether the presence of the provirus in latent cells can render the cells more susceptible to G4-stabilizing agents and DNA repair inhibitors, we used 2 Jurkat cell lines latently infected with HIV-1. We found that both cell lines show an increased rate of apoptosis/death compared with un-infected cells when cultures are maintained for 1 w in the presence of TMPyP4. The effect further increased when the agent was combined with a DNA repair inhibitor targeting DNA-PK crucial for DSB repair and telomere maintenance.Citation60,61 Intriguingly, BRACO19 had a smaller effect compared with TMPyP4 against latent cells despite its ability to completely block spreading infection in PM1 cell cultures. These different effects indicate that BRACO19 and TMPyP4 may have different affinities to G-quadruplexes formed in RNA and DNA. Increased susceptibility of latent cells to TMPyP4 suggests that this G4 agent targets proviral sequences and induces DSBs, sensitizing latent cells. Alternatively or additionally, a slow decline in cellular viability observed during long-term exposure to TMPyP4 may occur because its inhibitory effects are associated with targeting telomeres, not proviruses. The deleterious effects on telomeres would be observed after a delay, since their targeting causes telomeres to become shorter, and eventually induces apoptosis or growth arrest. This reasoning suggests that potential defects in telomere maintenance in latently infected cells sensitized them to TMPyP4 targeting of telomeres. Such a result would indicate that HIV-1 latency influences the cellular environment well beyond the site of integration.
Activation of DDR by exposing cells to high doses of NAs is evident from the level of phosphorylated Ser139 on histone H2AX, known as γH2AX. This effect in cells is related to DNA lesions induced directly by NAs incorporated into genomic DNA, or indirectly by targeting telomerase or ROS-induced oxidative damage.Citation35-46 The phosphorylation occurs soon after DNA damage and accumulates around the break site to attract proteins involved in repair and chromatin remodeling. We found that the level of γH2AX is much greater in both cell lines infected with latent HIV-1, than in un-infected cells, when they were exposed to high concentrations of the NRTI-type of NAs that we tested. This indicates that latently infected cells have lower ability to deal with deleterious effects caused by high concentrations of NRTIs. Moreover, exposing HIV-1 latent cells to etoposide resulted in formation of DSBs at a faster rate than in un-infected cells. We also observed similar susceptibility of latently infected cells to etoposide in generated latently infected memory T cells derived from human primary CD4+ T lymphocytes, indicating that normal cells with established HIV-1 latency have deficiencies in DNA repair mechanisms. In fact, our data show that the impact of HIV-1 latency on DNA damage or DDR is already evident in untreated cells, where not only γH2AX is elevated, but also phosphorylated forms of ATM at Ser1981, Chk2 at Thr68 and p53 at Ser15 are present, indicating DNA damage. This phosphorylation signaling is linked to formation of DSBs, but other types of DNA damage would also induce phosphorylation of these factors, for example oxidative DNA damage, formation of covalent DNA adducts, replication stress and SSBs.Citation62-66 This indicates that in latently infected cells, the DDR signaling pathway is activated in response to DNA damage suggesting that DNA repairs are not processed efficiently, or there is a factor raising the background level of DNA damage.
The relationship between viruses and DDR is well described.Citation9 Viruses interact with cellular mechanisms responsible for the recognition and repair of DNA lesions, and activate or deactivate individual DNA damage response pathways. They can induce genetic instability by interfering with factors responsible for recognition and repair of DNA damage. DNA damage is also caused by virus-induced oxidative stress.Citation10,11 Deregulation of DNA repair by viruses facilitates their replication and may contribute to development of cancer. Particularly, studies on the retrovirus human T cell leukemia virus (HTLV) show the magnitude of virus ability to deregulate DDR in its host.Citation9 The virus was found to affect each type of DNA damage repair mechanism. In addition, viruses cause telomere dysfunction, which may result in shortening of telomeres causing chromosome instability, or they may enhance telomere maintenance and bypass cellular senescence.Citation12 In the case of HIV-1, the virus-encoded protein Vpr was found to influence DNA damage response through several mechanisms. For example, Vpr targets uracil DNA glycosylase (Ung2) for degradation.Citation15,16 Ung2 initiates base excision repair (BER) to remove uracil from DNA. Interestingly, Vpr was found to induce formation of γH2AX, BRCA1 and 53BP1 nuclear foci, but without detection of DSBs. Together with Vpr-induced ATR activation and chromatin binding of replication protein A (RPA), this signaling is linked to activation of G2 cell cycle arrest due to replication stress.Citation13,14,21,22,67 It was also demonstrated that Vpr induces the premature activation of SLX4com, which coordinates the repair of replication-born DSBs.Citation17-19 This Vpr-induced activation of SLX4com was proposed to lead to abnormal cleavage of DNA replication intermediates.
HIV-1 Tat protein was also found to down-regulate expression of DNA-PKs, which are important in repair of DSBs and in maintaining telomeres.Citation20,68 Finally, HIV-1 infection negatively regulates the telomerase activity in CD4+ T cells, CD8+ T cells and Jurkat cells, but increases telomerase activity in infected macrophages.Citation69-72
In the case of latently infected cells, the modulation of DDR by HIV-1 may involve viral proteins expressed at a very low level (e.g. reservoirs established in macrophages).Citation73 Potentially, non-coding RNAs produced from pro-virus sequences may modulate DDR. This concept is new and as yet unexplored, but studies showed that HIV-1 TAR microRNA, expressed during latent infection, down-regulates ERCC1, which is involved in nucleotide excision repair (NER), and is also important for DSB repair.Citation74,75
Finally, oxidative stress induced by viral infections may contribute to DNA damage. Affected redox regulation is a common feature of retrovirus and RNA virus infections, and has been shown to be associated with HIV-1 infection.Citation11 During active HIV-1 infection, cells generate a large amount of RNS and ROS, and this is accompanied by the deregulation of antioxidant enzymes increasing the level of DNA damage.Citation76-78 Consistent with this notion, HIV-1 proteins gp120, Tat and Vpr are known to induce ROS.Citation79-81
In conclusion, our results indicate that cells infected with latent HIV-1 show affected DNA damage response and have increased susceptibility to different agents inducing damage in DNA and to agents targeting the DDR. To our knowledge this is the first report indicating that cells with silent HIV-1 infections have some impairment preventing them from making efficient repairs. This defect likely results from ability of the latent HIV-1 virus to modulate the cellular environment and interact with mechanisms directly or indirectly linked to DNA damage repair, a relationship already described for some DNA viruses and retroviruses. Our results suggest that NRTIs should eliminate HIV-1 reservoirs in patients on HAART, since they induce DNA damage. This is not the case, probably because NRTIs used in therapy are found in blood at much lower concentrations (at least 25–50 times), than concentrations we used in cell cultures.Citation82
Our results reveal new features of latently infected cells, which could be used in targeting to enhance strategies currently designed to eliminate HIV-1 reservoirs. They also suggest that HIV-1 reservoirs could potentially be eliminated without virus reactivation. There are many further questions regarding these new features of latently infected cells. First, future experiments should investigate what DNA damage repair mechanisms are affected. It would also be important to determine whether virus proteins or non-coding RNAs are associated with these defects during latency. Finally, it would be important to test whether pre-existing conditions related to less efficient DDR in cells support establishment of HIV-1 latency.
Material and methods
Reagents
BRACO19 (Sigma, Cat No. SML0560) and TMPyP4 (EMD Millipore, Cat No. 613560) were resuspended in water, aliquoted and stored in − 20°C. AZT (Matrix Scientific, Cat No. 069595), d4T (Sigma-Aldrich, Cat No. Y0000408), NU7441 (Apexbio Technology LLC, Cat No. A831525) and Etoposide (EMD Millipore, Cat No. 341205) were resuspended in DMSO and stored in − 20°C.
Cell culture and plasmids
Jurkat-derived latently infected T cells CA5 and EF7 have been described earlier.Citation33,34,83 Infectious molecular clone pNL4–3 and PM1 neoplastic CD4+ T cell line expressing both the CCR5 and CXCR4 co-receptors were obtained from Carrie Dykes. HIV-1-GFP (pNL4–3-deltaE-EGFP) was obtained from the AIDS Research and Reference Reagent Program, Division of AIDS, National Institutes of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH). DHIV was obtained from Vicente Planelles.Citation56 PM1, Jurkat, CA5 and EF7 cells were maintained in RPMI 1640 supplemented with 2 mM L-glutamine, 100 U of penicillin/ml, 100 μg of streptomycin/ml, and 10% heat-inactivated fetal bovine serum obtained from HyClone. 293T cells were maintained in Dulbecco's modified Eagle medium supplemented as for RPMI 1640.
Cultured TCM cells and their latent infection
Naïve CD4+ T cells were isolated from peripheral blood mononuclear cells (PBMC) from healthy donors and cultured TCM cells were generated and infected as described previously.Citation56,57
Generation of virus stocks
293 T cells were transiently transfected with 40 μg of pNL4–3 plasmid DNA by lipofection (SuperFect®, Qiagen, Cat No. 301305); supernatants were harvested after 72 h, and stored at - 80°C. HIV-1 virus capsid protein (p24) quantitation was performed on virus stocks using an ELISA (PerkinElmer, Cat No. NEK05000). Single cycle VSV-G pseudotyped viruses were generated as described elsewhere.Citation56,84
Infection and virus replication in the presence of G4-binding agents
Volumes of virus stock corresponding to a total of 25ng of p24 were used to infect 1 × 106 PM1 cells in 1ml medium at 37°C for 1h. After washing with PBS, the infected cells were resuspended in 5ml of medium without or with G4-binding agent BRACO19 or TMPyP4 at final concentration 6 µM and 25 µM, respectively. The cultures were incubated at 37°C in 5% CO2. At 3, 4, 5, 6, 7 and 8 d post-infection, 2.5ml of culture (cells and supernatant) was removed for testing and the same volume replaced with fresh medium with or without drugs. The culture supernatant was harvested after centrifugation for p24 measurement. In long-term exposure to 6 µM BRACO19, the agent was added to infected culture at day 5 post-infection. A half volume of culture (cells and supernatant) was removed each day to maintain cells density between 0.5 and 1 × 106 of cells, and the same volume replaced with fresh medium with drug. The culture was maintained for 3 w.
Flow cytometric analysis
Cell apoptosis was examined by flow cytometry analysis of APC Annexin V and 7-aminoactinomycin (7-AAD) staining (BD Biosciences). Sample fluorescence of 10,000 cells was analyzed by using a BD™LSRII flow cytometer (BD Biosciences). Cell populations were analyzed using FlowJo software (Version 10.2). The intracellular levels of γH2AX, and phosphorylated forms of ATM(Ser1981), Chk2(Thr68) and p53(Ser15) were detected in cells fixed with BD Cytofix™ Fixation Buffer (Cat No. 554655) and permeabilized with BD Phosflow™ Perm Buffer III (Cat No. 558050), then washed and blocked with BD Pharmingen™ Stain Buffer (Cat No. 554656). To detect γH2AX and phosphor-p53(Ser15) samples were stained with specific monoclonal antibodies conjugated to BV421 (BD Horizon, Cat No. 564720) and Alexa Fluor®647 (Cell Signaling Technology, Cat No. 8695), respectively. For detection of phosphor-ATM(Ser1981) and phosphor-Chk2(Thr68), samples were stained with specific rabbit mAb (Cell Signaling Technology, Cat No. 13050 and 2197), followed by staining with anti-rabbit secondary antibody, IgG (H+L), F(ab’)2 fragment conjugated with Alexa Fluor®647 (Cell Signaling Technology, Cat No. 4414). Intracellular p24 Gag expression was detected with monoclonal antibody conjugated with FITC (KC57-FITC, Coulter Clone, Cat No. 6604665) and analyzed as described previously.Citation56 The phenotype of cultured primary T cells was analyzed by staining with mAs CD27-PE (BD PharMingen, Cat No. 557330) and CCR7- Alexa Fluor®647 (BD PharMingen, Cat No. 560816). Sample fluorescence of 10,000 cells was analyzed by using a BD™LSRII flow cytometer (BD Biosciences) or Accuri C6 (BD Biosciences). Statistical analysis was performed with GraphPad Prism (GraphPad Software, La Jolla, CA). The 2-tailed paired-samples t test analysis was used to calculate the p- value (α = .05).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental_materials.zip
Download Zip (1.5 MB)Acknowledgment
We are grateful to Dr Olaf Kutsch (University of Alabama at Birmingham) for latently infected cell lines CA5 and EF7 used in these studies. We thank Dr. Vicente Planelles for providing the DHIV plasmid.
Funding
This research was supported by a grant from the University of Rochester Center for AIDS Research (CFAR), an NIH-funded program (P30 AI078498). This work was also funded in part by NIH grant R01 GM097334 to G.S; and NIH grants R01 NS054578 and R01 NS066801 to S.B.M.
Related Research Data
References
- Giglia-Mari G, Zotter A, Vermeulen W. DNA damage response. Cold Spring Harb Perspect Biol 2011; 3:a000745; PMID:20980439; http://dx.doi.org/10.1101/cshperspect.a000745
- Papamichos-Chronakis M, Peterson CL. Chromatin and the genome integrity network. Nat Rev Genet 2013; 14:62-75; PMID:23247436; http://dx.doi.org/10.1038/nrg3345
- Varga T, Aplan PD. Chromosomal aberrations induced by double strand DNA breaks. DNA Repair (Amst) 2005; 4:1038-46; PMID:15935739; http://dx.doi.org/10.1016/j.dnarep.2005.05.004
- Mehta A, Haber JE. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb Perspect Biol 2014; 6:a016428; PMID:25104768; http://dx.doi.org/10.1101/cshperspect.a016428
- Woodbine L, Brunton H, Goodarzi AA, Shibata A, Jeggo PA. Endogenously induced DNA double strand breaks arise in heterochromatic DNA regions and require ataxia telangiectasia mutated and Artemis for their repair. Nucleic Acids Res 2011; 39:6986-97; PMID:21596788; http://dx.doi.org/10.1093/nar/gkr331
- Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol 2014; 16:2-9; PMID:24366029; http://dx.doi.org/10.1038/ncb2897
- Khadaroo B, Teixeira MT, Luciano P, Eckert-Boulet N, Germann SM, Simon MN, Gallina I, Abdallah P, Gilson E, Geli V, et al. The DNA damage response at eroded telomeres and tethering to the nuclear pore complex. Nat Cell Biol 2009; 11:980-7; PMID:19597487; http://dx.doi.org/10.1038/ncb1910
- Ma W, Westmoreland JW, Gordenin DA, Resnick MA. Alkylation base damage is converted into repairable double-strand breaks and complex intermediates in G2 cells lacking AP endonuclease. PLoS Genet 2011; 7:e1002059; PMID:21552545; http://dx.doi.org/10.1371/journal.pgen.1002059
- Hollingworth R, Grand RJ. Modulation of DNA damage and repair pathways by human tumour viruses. Viruses 2015; 7:2542-91; PMID:26008701; http://dx.doi.org/10.3390/v7052542
- Higgs MR, Chouteau P, Lerat H. ‘Liver let die’: oxidative DNA damage and hepatotropic viruses. J Gen Virol 2014; 95:991-1004; PMID:24496828; http://dx.doi.org/10.1099/vir.0.059485-0
- Reshi ML, Su YC, Hong JR. RNA viruses: ROS-mediated cell death. Int J Cell Biol 2014; 2014:467452; PMID:24899897; http://dx.doi.org/10.1155/2014/467452
- Deng Z, Wang Z, Lieberman PM. Telomeres and viruses: common themes of genome maintenance. Front Oncol 2012; 2:201; PMID:23293769; http://dx.doi.org/10.3389/fonc.2012.00201
- Zhu Y, Gelbard HA, Roshal M, Pursell S, Jamieson BD, Planelles V. Comparison of cell cycle arrest, transactivation, and apoptosis induced by the simian immunodeficiency virus SIVagm and human immunodeficiency virus type 1 vpr genes. J Virol 2001; 75:3791-801; PMID:11264368; http://dx.doi.org/10.1128/JVI.75.8.3791-3801.2001
- Roshal M, Kim B, Zhu Y, Nghiem P, Planelles V. Activation of the ATR-mediated DNA damage response by the HIV-1 viral protein R. J Biol Chem 2003; 278:25879-86; PMID:12738771; http://dx.doi.org/10.1074/jbc.M303948200
- Bouhamdan M, Benichou S, Rey F, Navarro JM, Agostini I, Spire B, Camonis J, Slupphaug G, Vigne R, Benarous R, et al. Human immunodeficiency virus type 1 Vpr protein binds to the uracil DNA glycosylase DNA repair enzyme. J Virol 1996; 70:697-704; PMID:8551605
- Schrofelbauer B, Yu Q, Zeitlin SG, Landau NR. Human immunodeficiency virus type 1 Vpr induces the degradation of the UNG and SMUG uracil-DNA glycosylases. J Virol 2005; 79:10978-87; PMID:16103149; http://dx.doi.org/10.1128/JVI.79.17.10978-10987.2005
- Lahouassa H, Blondot ML, Chauveau L, Chougui G, Morel M, Leduc M, Guillonneau F, Ramirez BC, Schwartz O, Margottin-Goguet F. HIV-1 Vpr degrades the HLTF DNA translocase in T cells and macrophages. Proc Natl Acad Sci U S A 2016; 113:5311-6; PMID:27114546; http://dx.doi.org/10.1073/pnas.1600485113
- Laguette N, Bregnard C, Hue P, Basbous J, Yatim A, Larroque M, Kirchhoff F, Constantinou A, Sobhian B, Benkirane M. Premature activation of the SLX4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing. Cell 2014; 156:134-45; PMID:24412650; http://dx.doi.org/10.1016/j.cell.2013.12.011
- Blondot ML, Dragin L, Lahouassa H, Margottin-Goguet F. How SLX4 cuts through the mystery of HIV-1 Vpr-mediated cell cycle arrest. Retrovirology 2014; 11:117; PMID:25496524; http://dx.doi.org/10.1186/s12977-014-0117-5
- Sun Y, Huang YC, Xu QZ, Wang HP, Bai B, Sui JL, Zhou PK. HIV-1 Tat depresses DNA-PK(CS) expression and DNA repair, and sensitizes cells to ionizing radiation. Int J Radiat Oncol Biol Phys 2006; 65:842-50; PMID:16751065; http://dx.doi.org/10.1016/j.ijrobp.2006.02.040
- Belzile JP, Abrahamyan LG, Gerard FC, Rougeau N, Cohen EA. Formation of mobile chromatin-associated nuclear foci containing HIV-1 Vpr and VPRBP is critical for the induction of G2 cell cycle arrest. PLoS Pathog 2010; 6:e1001080; PMID:20824083; http://dx.doi.org/10.1371/journal.ppat.1001080
- Zimmerman ES, Chen J, Andersen JL, Ardon O, Dehart JL, Blackett J, Choudhary SK, Camerini D, Nghiem P, Planelles V. Human immunodeficiency virus type 1 Vpr-mediated G2 arrest requires Rad17 and Hus1 and induces nuclear BRCA1 and gamma-H2AX focus formation. Mol Cell Biol 2004; 24:9286-94; PMID:15485898; http://dx.doi.org/10.1128/MCB.24.21.9286-9294.2004
- Piekna-Przybylska D, Sullivan MA, Sharma G, Bambara RA. U3 region in the HIV-1 genome adopts a G-quadruplex structure in its RNA and DNA sequence. Biochemistry 2014; 53:2581-93; PMID:24735378; http://dx.doi.org/10.1021/bi4016692
- Piekna-Przybylska D, Sharma G, Bambara RA. Mechanism of HIV-1 RNA dimerization in the central region of the genome and significance for viral evolution. J Biol Chem 2013; 288:24140-50; PMID:23839990; http://dx.doi.org/10.1074/jbc.M113.477265
- Perrone R, Nadai M, Poe JA, Frasson I, Palumbo M, Palu G, Smithgall TE, Richter SN. Formation of a unique cluster of G-quadruplex structures in the HIV-1 Nef coding region: implications for antiviral activity. PLoS One 2013; 8:e73121; PMID:24015290; http://dx.doi.org/10.1371/journal.pone.0073121
- Perrone R, Butovskaya E, Daelemans D, Palu G, Pannecouque C, Richter SN. Anti-HIV-1 activity of the G-quadruplex ligand BRACO-19. J Antimicrob Chemother 2014; 69:3248-58; PMID:25103489; http://dx.doi.org/10.1093/jac/dku280
- Perrone R, Nadai M, Frasson I, Poe JA, Butovskaya E, Smithgall TE, Palumbo M, Palu G, Richter SN. A dynamic G-quadruplex region regulates the HIV-1 long terminal repeat promoter. J Med Chem 2013; 56:6521-30; PMID:23865750; http://dx.doi.org/10.1021/jm400914r
- Sundquist WI, Heaphy S. Evidence for interstrand quadruplex formation in the dimerization of human immunodeficiency virus 1 genomic RNA. Proc Natl Acad Sci U S A 1993; 90:3393-7; PMID:8475087; http://dx.doi.org/10.1073/pnas.90.8.3393
- Yang D, Okamoto K. Structural insights into G-quadruplexes: towards new anticancer drugs. Future Med Chem 2010; 2:619-46; PMID:20563318; http://dx.doi.org/10.4155/fmc.09.172
- Duchler M. G-quadruplexes: targets and tools in anticancer drug design. J Drug Target 2012; 20:389-400; PMID:22424091; http://dx.doi.org/10.3109/1061186X.2012.669384
- McLuckie KI, Di Antonio M, Zecchini H, Xian J, Caldas C, Krippendorff BF, Tannahill D, Lowe C, Balasubramanian S. G-quadruplex DNA as a molecular target for induced synthetic lethality in cancer cells. J Am Chem Soc 2013; 135:9640-3; PMID:23782415; http://dx.doi.org/10.1021/ja404868t
- Rodriguez R, Miller KM, Forment JV, Bradshaw CR, Nikan M, Britton S, Oelschlaegel T, Xhemalce B, Balasubramanian S, Jackson SP. Small-molecule-induced DNA damage identifies alternative DNA structures in human genes. Nat Chem Biol 2012; 8:301-10; PMID:22306580; http://dx.doi.org/10.1038/nchembio.780
- Kutsch O, Benveniste EN, Shaw GM, Levy DN. Direct and quantitative single-cell analysis of human immunodeficiency virus type 1 reactivation from latency. J Virol 2002; 76:8776-86; PMID:12163598; http://dx.doi.org/10.1128/JVI.76.17.8776-8786.2002
- Duverger A, Jones J, May J, Bibollet-Ruche F, Wagner FA, Cron RQ, Kutsch O. Determinants of the establishment of human immunodeficiency virus type 1 latency. J Virol 2009; 83:3078-93; PMID:19144703; http://dx.doi.org/10.1128/JVI.02058-08
- Ewald B, Sampath D, Plunkett W. Nucleoside analogs: molecular mechanisms signaling cell death. Oncogene 2008; 27:6522-37; PMID:18955977; http://dx.doi.org/10.1038/onc.2008.316
- Wu Q, Beland FA, Chang CW, Fang JL. Role of DNA repair pathways in response to zidovudine-induced DNA damage in immortalized human liver THLE2 cells. Int J Biomed Sci 2013; 9:18-25; PMID:23675285
- Olivero OA. Mechanisms of genotoxicity of nucleoside reverse transcriptase inhibitors. Environ Mol Mutagen 2007; 48:215-23; PMID:16395695; http://dx.doi.org/10.1002/em.20195
- Olivero OA, Tejera AM, Fernandez JJ, Taylor BJ, Das S, Divi RL, Poirier MC. Zidovudine induces S-phase arrest and cell cycle gene expression changes in human cells. Mutagenesis 2005; 20:139-46; PMID:15784690; http://dx.doi.org/10.1093/mutage/gei019
- Gomez DE, Armando RG, Alonso DF. AZT as a telomerase inhibitor. Front Oncol 2012; 2:113; PMID:22973556
- Hukezalie KR, Thumati NR, Cote HC, Wong JM. In vitro and ex vivo inhibition of human telomerase by anti-HIV nucleoside reverse transcriptase inhibitors (NRTIs) but not by non-NRTIs. PLoS One 2012; 7:e47505; PMID:23166583; http://dx.doi.org/10.1371/journal.pone.0047505
- de la Asuncion JG, Del Olmo ML, Gomez-Cambronero LG, Sastre J, Pallardo FV, Vina J. AZT induces oxidative damage to cardiac mitochondria: protective effect of vitamins C and E. Life Sci 2004; 76:47-56; PMID:15501479; http://dx.doi.org/10.1016/j.lfs.2004.06.020
- Szabados E, Fischer GM, Toth K, Csete B, Nemeti B, Trombitas K, Habon T, Endrei D, Sumegi B. Role of reactive oxygen species and poly-ADP-ribose polymerase in the development of AZT-induced cardiomyopathy in rat. Free Radic Biol Med 1999; 26:309-17; PMID:9895221; http://dx.doi.org/10.1016/S0891-5849(98)00199-3
- Sutliff RL, Dikalov S, Weiss D, Parker J, Raidel S, Racine AK, Russ R, Haase CP, Taylor WR, Lewis W. Nucleoside reverse transcriptase inhibitors impair endothelium-dependent relaxation by increasing superoxide. Am J Physiol Heart Circ Physiol 2002; 283:H2363-70; PMID:12388299; http://dx.doi.org/10.1152/ajpheart.00151.2002
- Kline ER, Bassit L, Hernandez-Santiago BI, Detorio MA, Liang B, Kleinhenz DJ, Walp ER, Dikalov S, Jones DP, Schinazi RF, et al. Long-term exposure to AZT, but not d4T, increases endothelial cell oxidative stress and mitochondrial dysfunction. Cardiovasc Toxicol 2009; 9:1-12; PMID:19067249; http://dx.doi.org/10.1007/s12012-008-9029-8
- Amatore C, Arbault S, Jaouen G, Koh AC, Leong WK, Top S, Valleron MA, Woo CH. Pro-oxidant properties of AZT and other thymidine analogues in macrophages: implication of the azido moiety in oxidative stress. ChemMedChem 2010; 5:296-301; PMID:20063338; http://dx.doi.org/10.1002/cmdc.200900464
- Nagiah S, Phulukdaree A, Chuturgoon A. Mitochondrial and oxidative stress response in HepG2 cells following acute and prolonged exposure to antiretroviral drugs. J Cell Biochem 2015; 116:1939-46; PMID:25740676; http://dx.doi.org/10.1002/jcb.25149
- Darzynkiewicz Z, Traganos F, Zhao H, Halicka HD, Li J. Cytometry of DNA replication and RNA synthesis: Historical perspective and recent advances based on “click chemistry.” Cytometry A 2011; 79:328-37; PMID:21425239; http://dx.doi.org/10.1002/cyto.a.21048
- Beucher A, Birraux J, Tchouandong L, Barton O, Shibata A, Conrad S, Goodarzi AA, Krempler A, Jeggo PA, Lobrich M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J 2009; 28:3413-27; PMID:19779458; http://dx.doi.org/10.1038/emboj.2009.276
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003; 421:499-506; PMID:12556884; http://dx.doi.org/10.1038/nature01368
- Zannini L, Delia D, Buscemi G. CHK2 kinase in the DNA damage response and beyond. J Mol Cell Biol 2014; 6:442-57; PMID:25404613; http://dx.doi.org/10.1093/jmcb/mju045
- Ward IM, Wu X, Chen J. Threonine 68 of Chk2 is phosphorylated at sites of DNA strand breaks. J Biol Chem 2001; 276:47755-8; PMID:11668173; http://dx.doi.org/10.1074/jbc.M009785200
- Loughery J, Cox M, Smith LM, Meek DW. Critical role for p53-serine 15 phosphorylation in stimulating transactivation at p53-responsive promoters. Nucleic Acids Res 2014; 42:7666-80; PMID:24928858; http://dx.doi.org/10.1093/nar/gku501
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998; 281:1674-7; PMID:9733514; http://dx.doi.org/10.1126/science.281.5383.1674
- Rubbi CP, Milner J. p53 is a chromatin accessibility factor for nucleotide excision repair of DNA damage. EMBO J 2003; 22:975-86; PMID:12574133; http://dx.doi.org/10.1093/emboj/cdg082
- Williams AB, Schumacher B. p53 in the DNA-damage-repair process. Cold Spring Harb Perspect Med 2016; 6:pii: a026070
- Bosque A, Planelles V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 2009; 113:58-65; PMID:18849485; http://dx.doi.org/10.1182/blood-2008-07-168393
- Bosque A, Planelles V. Studies of HIV-1 latency in an ex vivo model that uses primary central memory T cells. Methods 2011; 53:54-61; PMID:20970502; http://dx.doi.org/10.1016/j.ymeth.2010.10.002
- Messi M, Giacchetto I, Nagata K, Lanzavecchia A, Natoli G, Sallusto F. Memory and flexibility of cytokine gene expression as separable properties of human T(H)1 and T(H)2 lymphocytes. Nat Immunol 2003; 4:78-86; PMID:12447360; http://dx.doi.org/10.1038/ni872
- Rivino L, Messi M, Jarrossay D, Lanzavecchia A, Sallusto F, Geginat J. Chemokine receptor expression identifies Pre-T helper (Th)1, Pre-Th2, and nonpolarized cells among human CD4+ central memory T cells. J Exp Med 2004; 200:725-35; PMID:15381728; http://dx.doi.org/10.1084/jem.20040774
- Gilley D, Tanaka H, Hande MP, Kurimasa A, Li GC, Oshimura M, Chen DJ. DNA-PKcs is critical for telomere capping. Proc Natl Acad Sci U S A 2001; 98:15084-8; PMID:11742099; http://dx.doi.org/10.1073/pnas.261574698
- Williams ES, Klingler R, Ponnaiya B, Hardt T, Schrock E, Lees-Miller SP, Meek K, Ullrich RL, Bailey SM. Telomere dysfunction and DNA-PKcs deficiency: characterization and consequence. Cancer Res 2009; 69:2100-7; PMID:19244120; http://dx.doi.org/10.1158/0008-5472.CAN-08-2854
- Yan C, Lu J, Zhang G, Gan T, Zeng Q, Shao Z, Duerksen-Hughes PJ, Yang J. Benzo[a]pyrene induces complex H2AX phosphorylation patterns by multiple kinases including ATM, ATR, and DNA-PK. Toxicol In Vitro 2011; 25:91-9; PMID:20888899; http://dx.doi.org/10.1016/j.tiv.2010.09.012
- Khoronenkova SV, Dianov GL. ATM prevents DSB formation by coordinating SSB repair and cell cycle progression. Proc Natl Acad Sci U S A 2015; 112:3997-4002; PMID:25775545; http://dx.doi.org/10.1073/pnas.1416031112
- Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science 2010; 330:517-21; PMID:20966255; http://dx.doi.org/10.1126/science.1192912
- Yan S, Sorrell M, Berman Z. Functional interplay between ATM/ATR-mediated DNA damage response and DNA repair pathways in oxidative stress. Cell Mol Life Sci 2014; 71:3951-67; PMID:24947324; http://dx.doi.org/10.1007/s00018-014-1666-4
- Olive PL. Endogenous DNA breaks: gammaH2AX and the role of telomeres. Aging (Albany NY) 2009; 1:154-6; PMID:20157507; http://dx.doi.org/10.18632/aging.100025
- Lai M, Zimmerman ES, Planelles V, Chen J. Activation of the ATR pathway by human immunodeficiency virus type 1 Vpr involves its direct binding to chromatin in vivo. J Virol 2005; 79:15443-51; PMID:16306615; http://dx.doi.org/10.1128/JVI.79.24.15443-15451.2005
- Zhang SM, Zhang H, Yang TY, Ying TY, Yang PX, Liu XD, Tang SJ, Zhou PK. Interaction between HIV-1 Tat and DNA-PKcs modulates HIV transcription and class switch recombination. Int J Biol Sci 2014; 10:1138-49; PMID:25332688; http://dx.doi.org/10.7150/ijbs.10366
- Reynoso R, Minces L, Salomon H, Quarleri J. HIV-1 infection downregulates nuclear telomerase activity on lymphoblastoic cells without affecting the enzymatic components at the transcriptional level. AIDS Res Hum Retroviruses 2006; 22:425-9; PMID:16706619; http://dx.doi.org/10.1089/aid.2006.22.425
- Franzese O, Adamo R, Pollicita M, Comandini A, Laudisi A, Perno CF, Aquaro S, Bonmassar E. Telomerase activity, hTERT expression, and phosphorylation are downregulated in CD4(+) T lymphocytes infected with human immunodeficiency virus type 1 (HIV-1). J Med Virol 2007; 79:639-46; PMID:17387751; http://dx.doi.org/10.1002/jmv.20855
- Ballon G, Ometto L, Righetti E, Cattelan AM, Masiero S, Zanchetta M, Chieco-Bianchi L, De Rossi A. Human immunodeficiency virus type 1 modulates telomerase activity in peripheral blood lymphocytes. J Infect Dis 2001; 183:417-24; PMID:11133373; http://dx.doi.org/10.1086/318072
- Reynoso R, Wieser M, Ojeda D, Bonisch M, Kuhnel H, Bolcic F, Quendler H, Grillari J, Grillari-Voglauer R, Quarleri J. HIV-1 induces telomerase activity in monocyte-derived macrophages, possibly safeguarding one of its reservoirs. J Virol 2012; 86:10327-37; PMID:22787205; http://dx.doi.org/10.1128/JVI.01495-12
- Kumar A, Abbas W, Herbein G. HIV-1 latency in monocytes/macrophages. Viruses 2014; 6:1837-60; PMID:24759213; http://dx.doi.org/10.3390/v6041837
- Klase Z, Winograd R, Davis J, Carpio L, Hildreth R, Heydarian M, Fu S, McCaffrey T, Meiri E, Ayash-Rashkovsky M, et al. HIV-1 TAR miRNA protects against apoptosis by altering cellular gene expression. Retrovirology 2009; 6:18; PMID:19220914; http://dx.doi.org/10.1186/1742-4690-6-18
- Ahmad A, Robinson AR, Duensing A, van Drunen E, Beverloo HB, Weisberg DB, Hasty P, Hoeijmakers JH, Niedernhofer LJ. ERCC1-XPF endonuclease facilitates DNA double-strand break repair. Mol Cell Biol 2008; 28:5082-92; PMID:18541667; http://dx.doi.org/10.1128/MCB.00293-08
- Gil L, Martinez G, Gonzalez I, Tarinas A, Alvarez A, Giuliani A, Molina R, Tapanes R, Perez J, Leon OS. Contribution to characterization of oxidative stress in HIV/AIDS patients. Pharmacol Res 2003; 47:217-24; PMID:12591017; http://dx.doi.org/10.1016/S1043-6618(02)00320-1
- Pang X, Panee J, Liu X, Berry MJ, Chang SL, Chang L. Regional variations of antioxidant capacity and oxidative stress responses in HIV-1 transgenic rats with and without methamphetamine administration. J Neuroimmune Pharmacol 2013; 8:691-704; PMID:23546885; http://dx.doi.org/10.1007/s11481-013-9454-8
- Samikkannu T, Rao KV, Kanthikeel SP, Atluri VS, Agudelo M, Roy U, Nair MP. Immunoneuropathogenesis of HIV-1 clades B and C: role of redox expression and thiol modification. Free Radic Biol Med 2014; 69:136-44; PMID:24480751; http://dx.doi.org/10.1016/j.freeradbiomed.2013.12.025
- Kim SH, Smith AJ, Tan J, Shytle RD, Giunta B. MSM ameliorates HIV-1 Tat induced neuronal oxidative stress via rebalance of the glutathione cycle. Am J Transl Res 2015; 7:328-38; PMID:25893035
- Ferrucci A, Nonnemacher MR, Wigdahl B. Extracellular HIV-1 viral protein R affects astrocytic glyceraldehyde 3-phosphate dehydrogenase activity and neuronal survival. J Neurovirol 2013; 19:239-53; PMID:23728617; http://dx.doi.org/10.1007/s13365-013-0170-1
- Shah A, Kumar S, Simon SD, Singh DP, Kumar A. HIV gp120- and methamphetamine-mediated oxidative stress induces astrocyte apoptosis via cytochrome P450 2E1. Cell Death Dis 2013; 4:e850; PMID:24113184; http://dx.doi.org/10.1038/cddis.2013.374
- van Praag RM, van Weert EC, van Heeswijk RP, Zhou XJ, Sommadossi JP, Jurriaans S, Lange JM, Hoetelmans RM, Prins JM. Stable concentrations of zidovudine, stavudine, lamivudine, abacavir, and nevirapine in serum and cerebrospinal fluid during 2 years of therapy. Antimicrob Agents Chemother 2002; 46:896-9; PMID:11850283; http://dx.doi.org/10.1128/AAC.46.3.896-899.2002
- Wolschendorf F, Bosque A, Shishido T, Duverger A, Jones J, Planelles V, Kutsch O. Kinase control prevents HIV-1 reactivation in spite of high levels of induced NF-kappaB activity. J Virol 2012; 86:4548-58; PMID:22345467; http://dx.doi.org/10.1128/JVI.06726-11
- Ramirez PW, DePaula-Silva AB, Szaniawski M, Barker E, Bosque A, Planelles V. HIV-1 Vpu utilizes both cullin-RING ligase (CRL) dependent and independent mechanisms to downmodulate host proteins. Retrovirology 2015; 12:65; PMID:26215564; http://dx.doi.org/10.1186/s12977-015-0192-2