
ABSTRACT
The regulation of cell cycle progression by steroid hormones and growth factors is important for maintaining normal cellular processes including development and cell proliferation. Deregulated progression through the G1/S and G2/M cell cycle transitions can lead to uncontrolled cell proliferation and cancer. The transcription factor E2F1, a key cell cycle regulator, targets genes encoding proteins that regulate cell cycle progression through the G1/S transition as well as proteins important in DNA repair and apoptosis. E2F1 expression and activity is inhibited by inorganic arsenic (iAs) that has a dual role as a cancer therapeutic and as a toxin that leads to diseases including cancer. An understanding of what underlies this dichotomy will contribute to understanding how to use iAs as a more effective therapeutic and also how to treat cancers that iAs promotes. Here, we show that quiescent breast adenocarcinoma MCF-7 cells treated with 17-β estradiol (E2) progress through the cell cycle, but few cells treated with E2 + iAs progress from G1 into S-phase due to a block in cell cycle progression. Our data support a model in which iAs inhibits the dissociation of E2F1 from the tumor suppressor, retinoblastoma protein (pRB) due to changes in pRB phosphorylation which leads to decreased E2F1 transcriptional activity. These findings present an explanation for how iAs can disrupt cell cycle progression through E2F1-pRB and has implications for how iAs acts as a cancer therapeutic as well as how it may promote tumorigenesis through decreased DNA repair.
Introduction
The transcription factor E2F1 and the tumor suppressor protein retinoblastoma (pRB) are 2 key regulators of cell cycle progression. Together they have roles in determining progression through checkpoints at G1/S and G2/M that dictate whether a cell can proceed with DNA replication and cell division. The phosphorylation of pRB by cyclin-dependent kinases (CDKs) at specific amino acid residues inhibits heterodimerization with E2F1, and allows E2F1 to be transcriptionally active, whereas dephosphorylation of pRB promotes heterodimerization with E2F1 and suppression of E2F1 activity.Citation1 Because E2F1 is a transcription factor for many of the key proteins that drive cells through the G1/S transition and S-phase, cell cycle progression will be blocked if E2F1 activity is inhibited by heterodimerization with pRB. Over-expression of E2F1 can result in uncontrolled cell proliferation that can lead to tumorigenesis and cancer.Citation2 A block in E2F1 expression can lead to developmental anomalies, disrupted DNA repair, and to tumor progression by inhibition of apoptosis.Citation3,4
Exposure of cells to inorganic sodium arsenite (NaAsO2 or iAs) or arsenic trioxide (ATO) affects multiple signaling pathways,Citation5 can induce apoptosis,Citation6 and can inhibit cell proliferation in the human adenocarcinoma breast cancer cell line MCF-7 by causing them to accumulate in the G1 phase. This suggests a block at the G1/S transitionCitation7,8 and data presented herein. Other cell types affected by iAs exposure include myeloid cells, melanoma cells, and prostate cancer cells.Citation9-11 Myeloid cells exposed to iAs are delayed in cell cycle progressionCitation12 in association with Cdc25a repression,Citation10 and melanoma cells arrest in G1 or G2/M.Citation13 In non-malignant bronchial epithelial cells, E2F1 expression is repressed by exposure to iAs.Citation14 Together, these data suggest that iAs may inhibit cell cycle progression by inhibition of E2F1-mediated transcription, but how the proteins and pathways that iAs deregulates mediate cell cycle inhibition has not been described. In line with an inhibition of cell cycle progression, treatment of hematopoietic cancers with arsenic has been successful although success has been marginal in treating solid tumors. ATO is an approved drug for the treatment of acute promyelocytic leukemia (APL) where it has been used successfully, both alone and in combination with all-trans retinoic acid (ATRA) or chemotherapy.Citation15,16 Paradoxically, long-term exposure to low levels of iAs in drinking water, is linked not only to developmental abnormalities but to a multitude of diseases ranging from bladder, lung and skin cancers to diabetes and is found in many parts of the world including the U.S.Citation17 Here we focus on how iAs inhibits cell cycling in MCF-7 cells that have been stimulated by 17-β estradiol (E2). Our results have implications for not only how iAs can act as a therapeutic but also furthers our understanding of how it may act as a cancer promoting agent.
E2F1 is part of a family of E2F proteins that can promote or repress breast cancer cell proliferationCitation18,19 with roles in DNA replication, DNA damage checkpoint control and in apoptosis.Citation3 E2Fs 1, 2, and 3 are activating members of the E2F family important in promoting both the G1/S and G2/M transitions,Citation20,21 and many of their activities are redundant.Citation22 They promote cell proliferationCitation23,24 by transcriptionally activating cyclin expression including Cyclin E1 and Cyclin E2 (CCNE1 & CCNE2),Citation25 Cyclin D1 (CCND1) and Cyclin A.Citation26 Cyclins E, A and D form active complexes with the cyclin-dependent kinases (CDKs), CDK4/6 and CDK2.Citation27,28 CDK-Cyclin complex formation is dependent on the dephosphorylation of the CDK by the dual-specificity phosphatase Cdc25A and E2F1 is also a transcription factor for Cdc25A.Citation29,30 Ultimately, active CDKs target pRB for phosphorylation that determines pRB activity.Citation31 Importantly, E2F1 binds to its own promoter and autoregulates it's own transcriptionCitation32 and E2F2 and E2F3 can also activate E2F1 transcription.Citation32,33 It is interesting to note that E2F1 has tumor suppressive activities as well as oncogenic potentialCitation34-36 similar to the dual outcomes associated with exposure to iAs.
The retinoblastoma tumor suppressor protein is a major regulator of E2F1 transcription and cell cycle progression.Citation1,37,38 It is a member of a family of proteins known as the “pocket proteins” that include pRB, p130 and p107.Citation39,40 It acts mainly through binding to E2F1 directly, and when bound, suppresses the growth promoting activity of E2F1 by inhibiting its transcriptional activity. The inactivation of the pRB pathway is almost universal to all cancers.Citation41 While there can be changes in pRB expression, the most frequent cause of pRB dysfunction is due to changes in post-translational modifications such as acetylation and phosphorylation,Citation38 the most well studied being phosphorylation. Changes in phosphorylation at specific sites in pRB determines pRB-E2F1 interaction,Citation42,43 and subsequent E2F1 activity. When pRB is hyperphosphorylated, heterodimerization with E2F1 is inhibited which allows E2F1 to activate transcription at it's targets. When hypophosphorylated, pRB forms a heterodimer with E2F1 and E2F1 activity is repressed which inhibits cell cycle progression.Citation44 Both CyclinD/Cdk4/6 and CyclinE/A/Cdk2 kinases target specific sites on pRB,Citation38,45,46 and induce structural changes that affect the binding affinity of pRB for E2Fs1–3.Citation42-44 Of the 16 potential amino acids that can be phosphorylated in pRB, 2 that are differentially phosphorylated are T373 and S608.Citation45,67 Phosphorylation of pRB-S608 structurally orders the pocket domain loop and competitively inhibits pRB binding to the E2F1 transactivation domain and phosphorylation at pRB-T373 causes an allosteric change in pRB that promotes interaction of the N-terminus and the pocket domain that disrupts heterodimerization with E2F1.Citation42,43 Here we show that phosphorylation at both T373 and S608 change in response to iAs exposure.
ERα is a transcriptional activator when bound by its ligand E2, and it activates specific cell cycle-associated genes including E2F1.Citation19,30,47,48 It has been reported that treatment with iAs can inhibit ERα expression in MCF-7 cells, other breast cancer cells and other cell types.Citation7,49-51 Here we present experiments to investigate the expression and activities of key proteins in the E2F1-pRb pathway that drive MCF-7 cells through the G1/S cell cycle transition in response to E2 activation with and without iAs. Our data support a model in which the phosphorylation of pRB at specific sites is inhibited by exposure to iAs that, in turn, inhibits the dissociation of the E2F1-pRB complex from the E2F1 promoter. This results in a block in E2F1 transcriptional activity and inhibition of the expression of E2F1 target genes.
Results
G1/S transition is inhibited by iAs
iAs inhibits cell cycle progression in MCF-7 breast cancer cells that lead to the accumulation of cells in G1 after exposure, which suggests a block at or before the G1/S transition.Citation7 We initially verified this in our MCF-7 cells by flow cytometry after cells were made quiescent by growth for 72 h in phenol red-free DMEM containing dextran/charcoal-stripped serum, which removes growth and estrogenic factors from the medium. This was followed by a 24 h treatment with 5 nM E2 ± 2.5 µM or 5 µM iAs (). As the amount of iAs with which cells were treated was increased, fewer cells entered S-phase (). Arsenic effects on cell cycle progression were then determined over a 48 h time course ( and ). Quiescent cells were treated with 5 nM E2 ± 5 µM iAs. Cells treated with E2 alone () began to move into S-phase by 14 h with the peak at 18 to 24 h. In contrast, few cells treated with E2 + iAs () moved into S-phase before 18–24 h and never reached the number of cells found in S-phase with E2 alone. If the rate of cell cycle progression was slowed by iAs, an expectation would be that the percentage of cells in S-phase would eventually increase to levels seen with E2 alone (compare the percent cells at 24 h with E2 alone versus E2 + iAs at later time points). By 42–48 h, most of the cells treated with E2 had moved through G2/M and returned to G1, whereas some iAs-treated cells entered S-phase by 18–24 hours and continued into G2/M, but were blocked again, as indicated by the higher percentage of cells in G2/M that did not cycle back into G1 (compare 48 h in and ). These data indicate that the primary block in MCF-7 cells in response to treatment with iAs + E2 is at or before the G1/S transition, with an additional block at G2/M.
Figure 1. Treatment of MCF-7 cells with iAs blocks cells in G1/S and G2/M transitions. (A) Quiescent cells were left untreated, or treated with 5 nM E2, E2 + 2.5 µM iAs, or E2 + 5 µM iAs for 24 hours, stained with propidium iodide, and analyzed by flow cytometry. Values indicated by “G1, S, and G2/M” shown on the graphs are percent cells found in each phase. Representative experiment shown, repeated 4 times. (B) Cells treated with 5 nM E2 and analyzed as in (A) for indicated times. (C) Cells treated with 5 nM E2 +5 µM iAs and analyzed as in (A) for indicated times. Both B & C show means and SEM (n = 3 individual experiments) for 0–38 h. No error bars are shown for 40–48 h because these points represent one experiment. (D-G) Flow analysis of MCF-7 cells treated for 24 h to determine the distribution of apoptotic vs. necrotic cells with No Treatment (D), 5 nM E2 (E), 5 nM E2 + 5 µM iAs (F) and 5 µM iAs alone (G). Quadrant labels indicated in (D) are the same in (E-G).
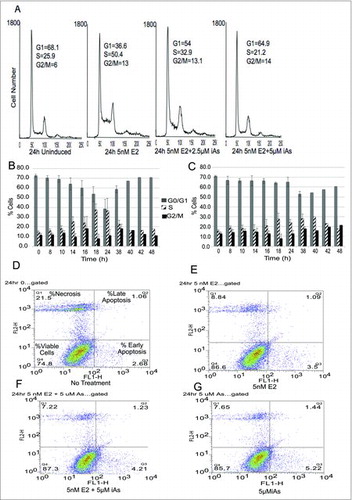
Treatment with iAs alone can induce apoptosis in various cell types,Citation52,53 and in cancer cellsCitation13,54 but effects are cell type and iAs-concentration dependent.Citation53,55 To determine if the decrease in iAs-treated cells entering S-phase was due to cell death, both necrosis and apoptosis were measured in an AnnexinV/propidium iodide assay. Quiescent cells were treated with 5 nM E2 ± 5 µM iAs, or 5 µM iAs alone for 24 h (). Some cell death (necrosis) was observed but there was little difference between treatments in the first 24 hours. Likewise little difference in either early or late apoptosis was observed. In cells treated with E2 + iAs for 48 h to 96 h more of the cells (about 8–10%) were apoptotic by 96 h (data not shown). Thus, in the first 24 h of treatment with 5 nM E2 ± 5 µM iAs, neither apoptosis nor cell death can account for the treatment-related differences in cell cycle distribution. shows that the average fraction of live cells at 8, 16 and 24 hours of treatment was about 80% with an average of about 20% cell death in all treatments and a small percentage due to apoptosis. A staurosporine control was done to show that apoptosis can be induced in these cells but it occurred later (96 h) than expected (data not shown).
Table 1. Percentage of live versus necrotic or apoptotic cells.
Arsenic inhibits Cyclin E2 (CCNE2) Expression
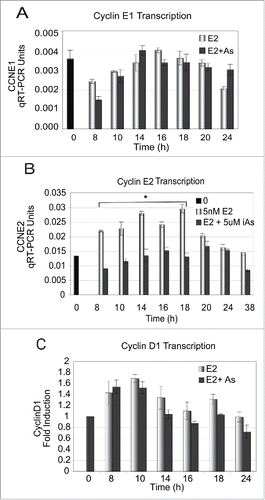
The expression of both CyclinE1 and CyclinE2 are increased by estrogen in MCF-7 cellsCitation56 and both cyclins associate with Cdk2 kinase to activate kinase activity in late G1, an activity that is important in moving the cells through the G1/S transition. Park et al.Citation57 showed that in renal carcinoma cells the expression of Cdk2, Cdk6 kinases and cyclins D1, E and A were decreased in response to arsenic trioxide (ATO). ATO has similar effects in our cells as the sodium arsenite used in our study. In addition, in Beas-2B lung cancer cells, CyclinD1 (CCND1) expression was increased and CCNE2 decreased in response to ATO.Citation14
To determine whether iAs affected cyclin expression that could be involved in inhibiting cell cycle progression, quiescent MCF-7 cells were treated with 5 nM E2 ± 5 µM iAs for 24 to 38 h. Cyclin mRNA samples were collected every 2 to 4 hours, starting at 8 h, and levels of cyclin mRNAs was determined by qRT-PCR (). CCNE1 mRNA expression was slightly decreased in response to E2 + iAs at 8 h but there was no significant difference from treatment with E2 alone after 8 h (). In contrast, CCNE2 mRNA was significantly decreased in cells treated with E2 + iAs vs. E2 alone from 8h to 18 h (). Not only was there a statistically significant effect of the iAs treatment compared with E2 alone but the dependence on time of the effect of the treatment was also statistically significant. Note that by 20 h of treatment there is no difference in CCNE2 expression. In we show that CCND1 levels peak earlier than CCNE1 or CCNE2 and there is no significant difference in expression between treatments. Thus, although CCND1 has been shown to be involved in the activation of CCNE2 in response to estrogen,Citation56 it is unlikely to have a role in iAs inhibition of CCNE2 expression or cell cycling in MCF-7 cells. We conclude that CCNE2 is the primary cyclin target of iAs vs. CCNE1 or CCND1, and that the effect of iAs treatment on CCNE2 is time dependent, occurring during G1 and up to the G1/S transition.
Figure 2. Gene expression of CCNE1 and CyclinD1 does not change but CCNE2 decreases in response to iAs across the cell cycle. (A-C) Quiescent cells were treated with 5nM E2 or 5 nM E2 + 5 µM iAs for indicated times and expression of mRNAs was evaluated by qRT-PCR with the appropriate primer set. All data shown is from the same representative experiment done in triplicate using primer sets to CCNE1, CCNE2 or CyclinD1 and the experiment was repeated 4 times with all primers shown. (n = 3; Error bars = SEM; *p-Value < 0.001 for CCNE2 at 8–18 h).
CDK2 kinase activity and Cdc25a expression are decreased in response to iAs
Because the cyclins bind to and activate the kinase activity of the CDK kinases that help drive the G1/S transition and CCNE2 expression was decreased in cells treated with iAs, we determined whether the activity of CDK2 kinase was also decreased. We chose CDK2 because CCNE2, the primary cyclin targeted by iAs that we identified, heterodimerizes with CDK2 to activate its kinase activity.Citation58 CDK4/6 is also an active kinase during the early G1 phase of the cell cycle but its primary partner is CyclinD and CCND1 expression was not decreased by treatment with iAs. shows that CDK2 kinase activity in cells treated for 14h with 5 nM E2 + 5 µM iAs was decreased compared with cells treated with 5 nM E2 alone where kinase activity increased.
Figure 3. Arsenic inhibits Cdk2 kinase activity and the expression of the phosphatase Cdc25a. (A) Cdk2 kinase was immunoprecipitated from MCF-7 cells after no treatment or a 14 h treatment with 5 nM E2 or 5 nM E2 + 5 µM iAs. The immunoprecipitated protein was incubated with [γ32P] ATP and 1 µg histone H1 in a kinase assay. Proteins were then separated by gel electrophoresis, transferred to a filter and visualized by PhosphorImager analysis (phosphor histone H1). The filter was then subjected to western blot analysis with an antibody to Cdk2. (B) Quiescent cells were treated with 5 nM E2 ± 5 µM iAs, mRNA was isolated and qRT-PCR was done with primers to Cdc25a. Representative experiment repeated 4 times as above (n = 3; Error bars = SEM; *p-Value < 0.001 at 8–18 h). (C) Cells were treated as in (B) and harvested at 8 h or 14 h of treatment. Western blot analysis was done with an antibody to CDK2 phosphoT14.
![Figure 3. Arsenic inhibits Cdk2 kinase activity and the expression of the phosphatase Cdc25a. (A) Cdk2 kinase was immunoprecipitated from MCF-7 cells after no treatment or a 14 h treatment with 5 nM E2 or 5 nM E2 + 5 µM iAs. The immunoprecipitated protein was incubated with [γ32P] ATP and 1 µg histone H1 in a kinase assay. Proteins were then separated by gel electrophoresis, transferred to a filter and visualized by PhosphorImager analysis (phosphor histone H1). The filter was then subjected to western blot analysis with an antibody to Cdk2. (B) Quiescent cells were treated with 5 nM E2 ± 5 µM iAs, mRNA was isolated and qRT-PCR was done with primers to Cdc25a. Representative experiment repeated 4 times as above (n = 3; Error bars = SEM; *p-Value < 0.001 at 8–18 h). (C) Cells were treated as in (B) and harvested at 8 h or 14 h of treatment. Western blot analysis was done with an antibody to CDK2 phosphoT14.](/cms/asset/22c7240a-ebe1-477a-8020-aa215e932394/kccy_a_1338221_f0003_b.gif)
Cdc25a is a member of a family of dual-specificity phosphatases whose expression is activated by E230 and it has a role in regulating cell cycle progression.Citation59 Specifically, Cdc25a dephosphorylates CDK2 at threonine 14 (T14) and tyrosine 15 (Y15) and this promotes CDK2-CCNE2 complex formation and kinase activity.Citation60 We reasoned that the decrease in CCNE2 could account for the decreased kinase activity but that a decrease in Cdc25a phosphatase activity could also contribute to the decrease in CDK2 kinase activity if CDK2 remained phosphorylated which would inhibit CCNE2-CDK2 heterodimerization. We determined whether Cdc25a expression was inhibited by iAs and found that between 8h and 18h of treatment with 5 nM E2 + 5 µM iAs Cdc25a mRNA expression was decreased in cells relative to those exposed to E2 alone (). We then examined whether the phosphorylation of CDK2 at T14 was decreased. shows that at 14 h the untreated cells (indicated by 0) remained phosphorylated as did the CDK2-phosphoT14 from iAs treated cells. Cells treated with E2 were dephosphorylated at residue T14. Together these data indicate that CDK2 kinase activity is inhibited by iAs, most likely due to a decrease in the expression of its binding partner CCNE2 and to less phosphatase activity of Cdc25a. This would account for the continued phosphorylation of CDK2 that inhibits CCNE2-CDK2 heterodimerization necessary for CDK2 activity.
Inhibition of E2F expression by iAs
E2F1 is transcribed by ligand-bound ERα and is itself a transcriptional activator of CCNE2, other G1-phase cyclins, Cdc25a phosphatase,Citation29 and of itself.Citation19,25,33,47 Because we found the expression of CCNE2, Cdc25a and CDK2 kinase activity downregulated in the presence of iAs ( and ) and that there is a major block in cell cycle progression leading to the G1/S transition () we next investigated whether E2F1 expression was decreased in cells treated with iAs. Quiescent cells were treated with 5 nM E2 ± 5 µM iAs and expression was examined over the following 38h. In response to E2 alone, E2F1 mRNA was maximal by 14–18 h (). This finding correlates well with the timing of the transition into S-phase (). After treatment with E2 + iAs the expression of E2F1 mRNA was significantly decreased by 4 h to less than basal levels (zero time point), indicating a probable inhibition of E2F1 transcription by iAs. E2F1 protein expression was also inhibited by 4–8 h when compared with treatment with E2 alone ().
Figure 4. Expression of E2F1 mRNA and protein and E2F2 and E2F3 mRNA changes during the cell cycle following treatment with 5 nM E2 or 5 nM E2 + 5 µM iAs. (A) Quiescent cells were treated for indicated times and expression of E2F1 mRNA was measured by qRT-PCR. (n = 3; Error bars = SEM; *p-Value < 0.001 at 4–38 h). (B) Western blot analysis of E2F1 protein expression at times after treatment with 5 nM E2 ± 5 µM iAs. GAPDH is a loading control. (C) As in (A) but qRT-PCR was with primers to E2F2. Shown is a representative experiment repeated 3 times. (n = 3; Error bars = SEM; *p-Value < 0.001 at 10–38 h) (D) As in (A) but with primers to E2F3a. Shown is a representative experiment repeated 3 times. (n = 3; Error bars = SEM; *p-Value < 0.001 at 10–16 h). In (C) and (D) Black bar = Time zero, Light Gray = 5 nM E2, Dark Gray = 5 nM E2 + 5 µM iAs.
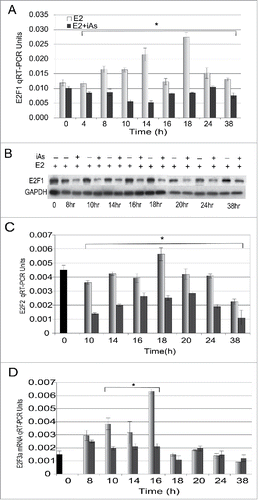
E2F2 and E2F3a are E2F1 transcriptional targets that are also involved in progression through G1 and the G1/S transition,Citation32,33 and all 3 E2Fs are transcriptional activators. These factors share some transcriptional targets but also have unique individual activities.Citation23,31 Because cell cycle progression and E2F1 expression were decreased in response to iAs, and E2F2 and E2F3a can compensate for E2F1, we predicted that the expression of one or both might be repressed in addition to E2F1. We found that E2F2 mRNA expression was repressed similarly to E2F1 (). Two isoforms of E2F3, E2F3a and E2F3b, are transcribed from independent promoters in the E2F3 gene.Citation61 PCR primers to a region that is conserved in both E2F3a and E2F3b showed no difference in expression between treatments (data not shown) but primers specific to E2F3a indicate that iAs inhibits its expression but only in the first 16 hours of treatment (). This suggests that the decrease in E2F2 and E2F3a may be due to the decrease in E2F1 because E2F3a is a transcriptional target of E2F1 but E2F3b is constitutively expressed and not an E2F1 target.Citation61 Thus the transcriptional repression of the 3 activating E2Fs, E2F1, F2F2 and E2F3a in response to iAs likely contributes to the iAs-associated block in the G1/S and possibly to the G2/M transition.
Is ERα associated with the decrease in E2F1 expression by iAs?
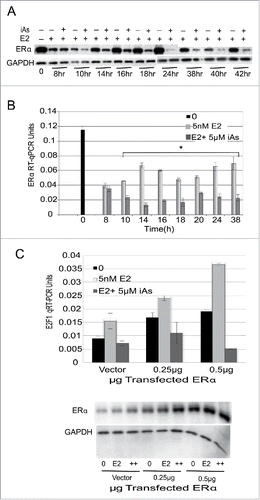
E2F1 transcription is activated by estrogen via ERα.Citation19,30,47,48 Because we saw an early decrease in E2F1 mRNA expression in iAs treated cells () we determined whether the decrease in E2F1 mRNA expression could be due to a decrease in ERα expression itself. The expression of ERα mRNA was determined in response to iAs over time by treating quiescent cells with 5 nM E2 ± 5 µM iAs and the expression of ERα protein and mRNA was determined (). In response to iAs the expression of ERα protein was decreased by 10 h () that is after the observed decrease in E2F1 protein expression at 8 h (compare to ). Likewise ERα mRNA expression did not decrease until 10 h (). Because the difference in the timing between changes in E2F1 expression (4–8 h) and ERα expression (10 h) was only 2 to 6 hours we investigated whether ERα overexpression would restore the expression of E2F1 mRNA in transient transfections. We found that the overexpression of ERα resulted in a significant increase in the expression of E2F1 when cells were treated with E2 alone but not when treated with E2 + iAs (). Basal levels of E2F1 also increased with added ERα as was expected. These data suggest that although ERα expression decreases with iAs exposure and may affect the expression of E2F1 after 10 h, ERα expression is not likely responsible for the initial decrease in E2F1 expression in response to iAs.
Figure 5. Arsenic inhibits the expression of the ERα protein and mRNA across the cell cycle starting at 10 h but overexpression of ERα does not restore E2F1 expression. (A) Western blot analysis of ERα protein expression at times after treatment of quiescent cells with 5 nM E2 ± 5 µM iAs. GAPDH is a loading control. (B) The expression of ERα mRNA from the same experiment as in (A) measured by qRT-PCR. (n = 3; Error bars = SEM; *p-Value < 0.001 at 10–38 h). Experiment was repeated 3 times. (C) Transient transfection with 0.25 µg or 0.5 µg ERα in cells treated with 5nM E2 ± 5 µM iAs for 14 h and analyzed for expression of E2F1 mRNA by qRT-PCR. (n = 3; Error bars = SEM; *p-Value < 0.001 at 10–38 h). Below is shown the Western blot analysis of the expression of ERα protein in the cells incubated with an antibody to ERα with GAPDH as a loading control.
pRB phosphorylation and the iAs-mediated decrease in E2F1 expression
Because the initial decrease in E2F1 expression by iAs was not likely due to changes in ERα expression (), we focused on the possible involvement of pRB in the iAs-mediated inhibition of E2F1 expression. Heterodimerization of E2F1 with pRB blocks E2F1 transcriptional activity. Our data showing that CDK2 kinase activity was reduced with iAs treatment () suggested the possible involvement of pRB because pRB is a substrate for phosphorylation by CDK2/CCNE2 kinase and the underphosphorylation of pRB promotes pRB-E2F1 heterodimerization. If pRB phosphorylation by CDK2 kinase was inhibited then we would expect pRB to be less phosphorylated at specific amino acids and would remain in a heterodimeric complex with E2F1.
Our initial experiment was a ChIP assay to determine if pRB and E2F1 were associated with the E2F1 promoter when cells were treated with E2 ± iAs. If both E2F1 and pRB were found at the E2F1 promoter it would suggest the possibility of heterodimerization between the 2 proteins that could lead to inhibition of E2F1 transcription and expression. Quiescent cells were treated for 16 hours before they were cross-linked and harvested for ChIP analysis. We found that in untreated cells and in cells treated with E2 alone there was significantly less of both E2F1 and pRB at the E2F1 promoter than in cells treated with E2 + iAs (), where we found significantly more of both at the promoter. Because transcription is a dynamic process in which a transcription factor binds to a promoter and then traverses the DNA in the process of transcriptional elongation, we interpret this result as an indication that with iAs treatment, E2F1 binds to its own promoter but transcription may not progress because E2F1 has formed a heterodimeric complex with pRB and remains “locked’ in position, unable to activate its own transcription. In contrast, with E2 alone some E2F1 and pRB are found at the promoter but because there is active transcription and elongation there is significantly less E2F1 and pRB then when cells have been treated with iAs.
Figure 6. E2F1 and pRB heterodimerize at the E2F1 promoter in association with a decrease in pRB phosphorylation. (A) ChIP analysis of pRB and E2F1 bound to the E2F1 promoter after 16h treatment of cells with 5nME2 ± 5 µM iAs expressed as % InPut E2F1. Representative experiment, repeated twice, Error Bars = SEM from qRT-PCR triplicate analysis. (B) Western blot analysis of pRB and phosphor-T373 pRB across the cell cycle in cells treated with 5nM E2 ± 5 µM iAs. GAPDH is a loading control. Dots to the left indicate mobility changes in pRB. (C) Quantification of western blot shown in (B) with phosphor-T373 in each lane normalized to the amount of pRB expressed in the corresponding lane (ratio T373/pRB). Blot was hybridized with antibody to pRB, stripped and re-hybridized with antibody to T373. GAPDH is a loading control. (D) Quantification of western blot (not shown) incubated with antibody to phosphor-S608-pRB normalized to the amount of pRB expressed in the corresponding lane as in (B)(ratio S608/pRB). Blot was hybridized to antibody to pRB, stripped and re-hybridized with antibody to S608-pRB. GAPDH is a loading control. Both (C) and (D) experiments were repeated 3 times.
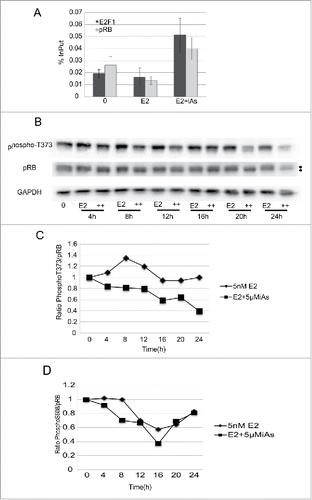
If the increase in pRB at the E2F1 promoter were due to heterodimerization, we would expect a decrease in phosphorylation of pRB in cells treated with E2 + iAs and an increase in phosphorylation with E2 treatment alone. Because we did not see changes in the expression of Cyclin D1 () and we did see a decrease in CDK2 kinase activity (), we focused on identifying phosphorylation differences on pRB amino acid residues that are specifically targeted for phosphorylation by Cyclin E/CDK2 kinase activity. Phosphorylation of pRB is extensive, and early (G1) phosphorylation events by CyclinD/Cdk4/6 likely cause structural changes in pRB that expose sites that can later (late G1) be targeted by Cyclin E/CDK2.Citation43,91 Thus specific phosphorylation changes are likely to be transient with phosphorylation patterns changing over time. Taking this into consideration, the experiments were done in time courses where quiescent cells were treated with E2 alone or E2 + iAs as in previous experiments, cells were harvested, and western blot analysis was done with antibodies to phosphorylated pRB amino acids. It has been shown that the amino acid threonine 373 (T373) when phosphorylated stabilizes a structural change between the pocket domain of pRB and its N-terminus that can dissociate preformed pRB-E2F1 complexes by allosteric inhibition.Citation43 We found that as early as 4–8 h after treatment with E2 alone, there was an increase in the phosphorylated form of T373 which was maintained throughout the time course, compared with T373 from cells treated with E2 + iAs where phosphorylation on pRB was significantly lower ( and ). Furthermore, a slight difference in gel mobility of pRB between the 2 treatments, was observed in lanes incubated with antibody to pRB (). A slower migrating form of pRB seen with E2 alone suggests that the E2 treated form of pRB is more phosphorylated than pRB treated with E2 + iAs, consistent with the observed increase in phosphorylated T373 with E2 alone. Together these data identify an amino acid, T373, in pRB that is less phosphorylated when cells are treated with E2 + iAs but is more phosphorylated with E2 alone and this phosphorylation can inhibit pRB from interacting with the transactivation domain of E2F1.Citation43 We also attempted to determine whether there was a change in S608 phosphorylation with time. There was a decrease in S608 phosphorylation at 8h with E2 + iAs compared with E2 alone () but no significant differences at later times as we saw with T373. These data suggest that at least 2 different phosphorylation events on pRB are inhibited by iAs, one, is a decrease in S608 phosphorylation and the other, a decrease in phosphorylation at T373. Dephosphorylation of both sites can contribute to stabilizing pRB heterodimerization with the E2F1 transactivation domain that would be consistent with our observation that both E2F1 and pRB are present together at the E2F1 promoter after iAs treatment (). E2F1-pRB heterodimerization leads to a decrease in the transactivation of E2F1 and its targets, including E2F1 itself. Because T373 phosphorylation is associated with an allosteric change in pRB that can displace E2F1 from pRBCitation43 this suggests that E2F1 transcriptional activity is repressed by iAs through changes in pRB phosphorylation that leads to the observed block in cell cycle progression. By identifying a change in the phosphorylation of T373 and S608 in the pRB protein, we have identified a new mechanism by which iAs can disrupt cell cycle progression.
Discussion
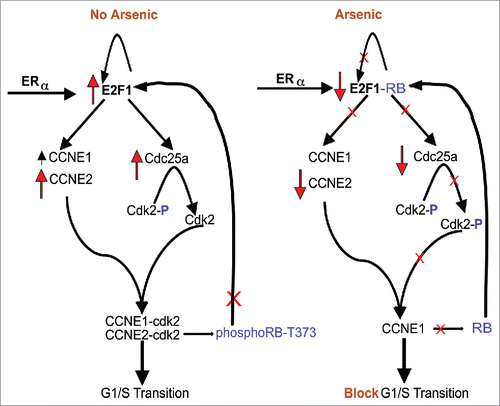
We present a model by which iAs inhibits cell cycle progression () based on the data presented and on what is known about the mechanisms that promote cell cycle progression through the G1/S transition. For clarity, our model is focused on the regulators our experiments directly address. We show that iAs inhibits the phosphorylation of pRB at specific sites by inhibiting the kinase activity of CyclinE2/CDK2, contributed to by the decrease in transcription of CCNE2 and additionally the decrease in phosphatase activity of CDC25a that prevents the dephosphorylation of CDK2. Through these experiments we have advanced the understanding of how iAs inhibits cell cycling mediated by estrogen and ERα that may apply to other mitogens that iAs inhibits. Our experiments did not address what the direct target iAs is that leads to the inhibition of cell cycling. There have been many studies done on how iAs affects signaling pathways such as the MAPK and p38 pathways that could affect pRB phosphorylationCitation38 but most of these studies also fail to identify a direct target for iAs. It is interesting to note that iAs can interact directly with Zinc-finger domains through cysteine and histidine residuesCitation90 and this can affect the enzymatic activity of proteins such as PARP-1 (poly(ADP-ribosyl)ation as well as protein-protein interactions and protein-DNA interactions that include transcription factors. This effect on Zinc-finger domains could very well have an effect in this system.
Figure 7. Model of how iAs affects pRB and E2F1 expression during G1 and the G1/S transition phases of the cell cycle that results in a block in cell cycling at the G1/S transition based on data shown herein. (See Discussion for more explanation.)
E2F1 is an estrogen responsive geneCitation47 that when knocked down renders MCF-7 cells unresponsive to the proliferative effect of estrogen. Because ERα is a transcriptional activator for E2F1 we focused our efforts on determining if iAs leads to a decrease in ERα expression, and if ERα overexpression could restore E2F1 expression. It has been reported that treatment with iAs can inhibit ERα expression in MCF-7, other breast cancer cells and in other cell types.Citation49,50,51 We found that iAs did inhibit ERα expression but not until after the decrease in E2F1 expression (compare and ). We cannot rule out that the significant decrease in ERα protein after iAs treatment does not contribute to the decrease in E2F1 expression at 10 h or later, but it does not explain how early E2F1 expression was inhibited. It is interesting to note that phosphorylation of ERα by CyclinA/CDK2 on Ser118 is associated with an increase in the transcriptional activity of ERα, whereas, inhibition of CDK2 and CDK7 kinases inhibits ERα transcriptional activity.Citation62-64 We have shown here that the CDK2 phosphatase, Cdc25a, is inhibited before the decrease in ERα expression (compare and ), and CDK2 kinase activity is also inhibited. We have also seen a decrease in CyclinA2 expression in response to iAs (unpublished data). It is thus possible that repression of the transcriptional activity of ERα vs. a decrease in ERα expression contributes to the early decrease in E2F1 expression in response to iAs but this requires further investigation.
It is likely that the significant decreases we see in CCNE2 and in Cdc25a occur in response to the decrease in the E2F1 transcription factor and it's activity because both have E2F1 binding sites and are transcriptionally regulated by E2F1.Citation65 CCNE1 and CCNE2 are regulated independently from different promotersCitation56 and CCNE2 may be more sensitive to transcriptional activation by E2F1 than is CCNE1.Citation66 This is consistent with our results that show that CCNE2 is inhibited in a similar time frame to E2F1, but CCNE1 is less susceptible to changes in E2F1 expression ( and ). This would account for the greater sensitivity of CCNE2 expression to iAs.
Because E2F1 can autoregulate it's own transcription,Citation32,33 we examined whether E2F1 interacts with it's own promoter in the presence of iAs and E2 using ChIP analysis, and found that there was more E2F1 at the promoter when cells were exposed to iAs + E2 than when exposed to E2 alone (). Further investigation showed that both pRB and E2F1 were at the promoter after 14 to 16h of treatment with E2 + iAs and in similar amounts (). These data led us to the hypothesis that E2F1 and pRB heterodimerize at the E2F1 promoter when iAs is present and block transcription by E2F1. We found more evidence for E2F1-pRB heterodimer formation when we used 2 different antibodies to pRB in the ChIP assay. One antibody was from Becton-Dickinson (BD) and one from Cell Signaling Technology (CST). We could detect pRB at the E2F1 promoter after iAs treatment with the CST-antibody but not with the BD-antibody. This suggests a potential iAs-associated conformational change in pRB that blocks recognition by the BD-antibody but not the CST-antibody. The recognition epitope for the BD-antibody is amino acids 332–344 which is part of a linker region just N-terminal to the highly structured pRB pocket domain.Citation42-44 It is a region in pRB that when bound to E2F1 undergoes an allosteric change that would likely block antibody recognition of the BD epitope. The CST-antibody recognizes an epitope within the C-terminal amino acids, 701–928, a largely unstructured region of pRB. This epitope would likely be available for recognition when E2F1 and pRB are heterodimerized. It should also be noted that both antibodies worked equally well in western blot analysis vs. in the ChIP analysis. Together these data make an argument for changes in pRB conformation, that likely result from an iAs-mediated decrease in pRB phosphorylation and the heterodimerization of E2F1-pRB.
It is interesting that in a study by Narasimha etal.Citation91 in which isoelectric focusing was used to separate monophosphorylated forms of pRB, pRB is monophosphorylated in early G1 at both S608 and T373 and monophosphorylated S608 pRB binds to E2F1 slightly less well than monophosphorylated T373. Furthermore the monophosphorylation is mediated by CyclinD/CDK4/6 in early G1. Our data show () that the residue T373 on pRB is phosphorylated at time zero but to a lesser extent than at later times. By 4 h in iAs treated cells T373 is just slightly less phosphorylated than E2 treated cells. Narasimha etal. show that rather than a progressive phosphorylation by CCNE2/CDK2 in G1 there is a rapid hyperphosphorylation of pRB in late G1 that ranges from 10 h-16 h, depending on the experimental conditions. In our experiments the difference in phosphorylation between treatments is greater by 8 h than at 4 h and by 8 h phosphorylation in E2-treated cells is maximal. This timing fits well with our data showing that CyclinD1 expression is maximal early, before cells move into S-phase at 14 h and CCNE2 expression is highest at 8 h to 18 h when pRB becomes hyperphosphorylated by CCNE2/CDK2 in cells treated with E2 alone. We also see that at 14 h CDK2 kinase activity is higher than at time zero () and that CDC25a expression is highest at 8 h-14 h () when it is important for CDK2 to be dephosphorylated to allow heterodimerization with CyclinE2. Thus the timing of expression of cyclins and of CDK activity in our experiments would be consistent with early monophosphorylation and late hyperphosphorylation. However, it could also be argued that our data shows a progressive increase in pRB phosphorylation with E2 if the difference at 4h is significant. Because western blots can be used for qualitative and not quantitative measure, we can only determine whether pRB is phosphorylated or not from our data but not the degree to which it is phosphorylated.
The phosphorylation of pRB S608/S612 occurs earlier than T373 phosphorylationCitation67 but we only detected a slight decrease in phosphorylation at S608 at 4–8 hours of treatment with E2 + iAs and no increase in phosphorylation with E2 treatment. It thus remains possible that in addition to a decrease in phosphorylation due to less active CDK2 in iAs-treated cells, there is an active dephosphorylation of pRB at S608 triggered by iAs. Two protein phosphatases, protein phosphatase 1 (PP1) and protein phosphatase 2A (PP2A) dephosphorylate pRB at specific sites,Citation68,69 and S608 has been identified as a target for PP1.Citation68,69 Whether iAs affects the phosphatase activity of PP1 and whether this can account for the decrease in S608 phosphorylation by iAs remains to be determined. It is also interesting to note that PP2A can be activated at times of oxidative stress to dephosphorylate pRBCitation69 and iAs has effects on ROS accumulation that leads to accumulation of H2O2.Citation71
Apoptosis was not observed in cells treated with E2 alone or E2+ iAs in the first 24 h of treatment (). Apoptosis may be slower to occur in the MCF-7 cells because they lack caspase 3Citation70 and iAs can induce apoptosis through activation of caspase 3.Citation71 E2 alone can also inhibit apoptosis in MCF-7 cells by inducing the expression of BCL-2 mRNACitation72 but conversely, iAs alone can stimulate apoptosis by repression of BCL-2 and activation of Bax in bone marrow mesenchymal stem cellsCitation52 although in MCF-7 cells we did not observe increased apoptosis with iAs alone (). Lastly, both E2F1 and E2F3 can induce an apoptotic response in a p53-dependent or independent manner when overexpressed, and apoptosis by E2F3 is dependent on E2F1.Citation24,73,74 Because we see decreased expression of E2F1 by iAs, this may contribute to the repression of apoptosis in MCF-7 cells in our experiments but further investigation into how apoptosis is regulated by E2 ± iAs is warranted.
The data presented here is the first to support a role for E2F1-pRB as an underlying factor in the inhibition of cell cycling by iAs and suggests a mechanism by which iAs can act as a cancer therapeutic. It is an approved therapeutic used in the treatment of all-trans retinoic acid (ATRA)-resistant acute promyelocytic leukemia (APL)Citation75-77 and is used as a monotherapeutic or in combination therapy.Citation15 It is significant that iAs represses all 3 activating E2Fs as well as CCNE2 and Cdk2 as a consequence of changes in E2F1-pRB interactions and suggests increased potential for therapeutic value. In lung, prostate, and bladder tumors, E2F3 is overexpressed and in other cancers E2Fs 1–3a can be overexpressed.Citation78 Cyclin overexpression in many cancers, including breast cancer, prostate cancer and ovarian cancer, correlates with poor patient outcome.Citation79-81 CCNE2 and CCNE1 have been tested as prognostic indicators in breast tumors and CCNE2 expression correlated with poor prognostic outcome in ERα-positive breast tumors whereas CCNE1 was prognostic in both ERα -positive and negative tumors.Citation80 Because CCNE1 expression was not repressed by iAs but CCNE2 was, this suggests a potential for therapeutic value in some cancers but not others. It is of note that ATO (arsenic trioxide-As2O3) has been shown to down-regulate the expression of E2F1 in several lung adenocarcinoma cell lines and in a mouse xenograft model that over-expresses E2F1.Citation82 Additionally, sodium arsenite treatment results in repression of E2F1 and E2F2 expression in bronchial epithelial cellsCitation14 as well as in MCF-7 cells as shown here. Myeloid cells exposed to ATO are delayed in cell cycle progressionCitation12 in association with Cdc25a repression,Citation10 and melanoma cells arrest in G1 or G2/M in response to ATO.Citation13 We, and others, have shown that ATO has a similar inhibitory effect on E2F1 expression as the sodium arsenite used here, but the effect is greater at a lower concentration of ATO. There has been an interest in the potential therapeutic value of iAs not only in hematologic cancers but also in solid tumorsCitation83 where therapeutic results have been less promising,Citation84 in part because of lack of effective delivery in solid tumors.
In addition to being a therapeutic, iAs has long been considered a toxin that leads to disease or death, and chronic exposure to low levels of iAs is a major human health hazard throughout the world.Citation17 The duality of iAs exposure may be due, in part, to iAs-mediated deregulation of E2F1-pRB and this may be important in understanding how iAs has serious health effects including cancer but also has therapeutic effects. While arsenic-mediated inhibition of E2F1 activity and expression provides a rationale for the therapeutic effect of iAs, E2F1 also has an important function in DNA repairCitation4 which likely contributes to E2F1s role in tumorigenesis. Some of the transcriptional targets of E2F1 are the DNA damage response proteins such as damage-specific DNA binding protein (DDB2), xeroderma pigmentosa complementation group C (XPC), the DNA recombinase, Rad51, and excision repair cross complement 1 (ERCC1).Citation36 The expression of these proteins would likely be repressed by iAs repression of E2F1 transcriptional activity. Interestingly, it has been shown that many of these DNA damage response proteins are downregulated by chronic low level exposure to iAs.Citation85,86 This provides a plausible rationale for how iAs might cause disease by an inhibition of DNA repair through the repression of E2F1 transcriptional activity and it's targets. Additionally, E2F1 itself accumulates at DNA double-strand breaks, and DNA damaged by exposure to UV light, and stimulates double strand break repair and nucleotide excision repair (NER).Citation88,89 It is interesting that iAs can bind to PARP-1 directly to inhibit it's activity.Citation90 PARP-1 contributes to the dissociation of protein at damaged DNA to allow access of repair enzymesCitation86 and this could affect the availability of E2F1 at sites of DNA damage. Thus the potential decrease in DNA repair as well as in apoptosis through the inhibition of E2F1 activity and expression by pRB, suggests a plausible route to iAs-associated tumorigenesis. In conclusion, the data we have presented, in combination with what is already known of E2F1 activities and its regulation by pRB, supports an interesting and viable hypothesis that the duality of iAs function as a therapeutic and its association with the development of specific cancers, is likely contributed to by iAs effects on the E2F1-pRB axis.
Materials and methods
Cells and reagents
MCF-7 cells were purchased from ATCC (ATCC, #HTB-22) and maintained in DMEM (InVitrogen, #11965–126) supplemented with 10% Cosmic Calf serum (CCS) (HyClone, #SH30087.03) plus pyruvate (Gibco, #11360–070) and 1% Penicillin-Strepomycin (InVitrogen, #15149–122). Prior to treatment cells were grown for 3 d in phenol red-free DMEM (Gibco, #21063–029) + 5% charcoal-stripped BGS (Hyclone #SH30541.03 before treatment with E2 (Calbiochem, #3301) ± sodium arsenite (NaAsO2) (ScienceLab.com, #SLS2332) or arsenic trioxide (Aldrich, #202673).
Antibodies
ChIP and western blot analysis-E2F1(Millipore/Upstate #05–379), ERα (Western Blot) (Thermo Scientific/Labvision #MS-1071 & Santa Cruz #sc-543), Cdk2 (Santa Cruz #sc-163), CDK2-T14 (Abcam #ab68265), RB 4H1 for ChIP analysis (Cell Signaling Technology, #9309), RB for protein gels (Becton-Dickinson, #554136), T373 (Abcam, #ab52975), S608 (Abcam, #ab172975), GAPDH (American Research Products, #05–50118), non-immune IgG (Sigma #I5381).
Primers
All primers are from Integrated DNA Technology (IDT). Unless indicated, the primers are to coding region.
CCNE1 (F-5′TACCCAAACTCAACGTGCAA; R-5′CATGATTTTCCAGACTTCCTCTC),
CCNE2 (F-5′CCCCAAGAAGCCCAGATAAT; R-5′CTGATGTTTCTTGGTGACCTC), CCND1 (F-5′TCCTCTCCAAAATGCCAGAG; R-5′GGCGGATTGGAAATGAACT), CDC25A (F-5′GCACTCGGTCAGTGTTGAAG; R-5′CATGGGCCTTCTCTGGATTA), E2F1 (F-5′CCATCCAGGAAAAGGTGTGA; R-5′GCTCAGCAGCTCCAGGAA),
E2F1 promoter (F-5′TTCGCGGCAAAAAGGATTT; R- 5′GCCGCTGCCTGCAAAGT),
E2F2 (F-5′GCTGCTCACCAAGAAGTTCATTT; R-5′GGTCCAGGACCCCATCCT),
E2F3 (F-5′CCTCAGAAAAAACGCGGTATG;R-5′CAGGAGCTGAATGAACTTCTTGGT), E2F3A (F-5′CCTCACCACGAACACTTCCAC; R-5′CCGCTTTGGAGGGAGGAG),
ERα (F-5′GTTCGGCTCCAACGGCCTGG; R-5′CCGTGGGGCTGCAGGAAAGG), GAPDH (F-5′TCCACCCATGGCAAATTCC; R-5′TCGCCCCACTTGATTTTGG)
Gel electrophoresis and western blot analysis
Proteins were lysed in the presence of protease inhibitor cocktail (EMD Millipore #539134) and phosphatase inhibitor HALT (Fisher, #P178420) were separated on 7.5–10% polyacrylamide gels (BioRad #4561026S) or NuPAGE Novex BisTris 4–20% gradient gels (Thermo #NP0323) in recommended buffers, transferred to Immobilon-P 0.45 µm PVDF membrane (Millipore, #IPVH00010), blocked with Superblock (Pierce, #37545) in TBS-0.05% Tween and visualized by HRP-based chemiluminescence, SuperSignal FemtoWest (Thermo-Pierce, #34095) or Immun-Star WesternC (BioRad, #170–5070). Imaging was on film (Kodak, BioMax AR #165 1454) or a BioRad ChemiDocMP Imaging System and ImageLab 5.1 software (BioRad).
Chromatin immunoprecipitation
Cells were harvested and cross-linked with 1.5mM Ethylene Glycol-bis Succinimidylsuccinate (EGS) (Thermo, #PI521565) at 25°C for 25 min followed by 1.0% formaldehyde (Thermo #28960) at 25°C for 10 min. Nuclei were isolated by centrifugation through a glycerol pad plus tritonX-100, dounce homogenized, and DNA digested with micrococcal nuclease (0.0375U/μg nucleic acid) (Roche, #107921). ChIP was with antibodies to E2F1 and pRB and incubation was overnight at 4°C. Immune complexes were isolated with Protein A/G coupled magnetic beads (New England Biolabs, #S1430S). Four washes, a high salt, a low salt with 0.5% deoxycholate and 2x with Tris pH 7.0 were done and cross-links were reversed 4–12 h at 65°C, DNA was purified with MinElute columns (Qiagen, #28204). ChIP controls used non-immune IgG. ChIP DNA was analyzed by qRT-PCR on a MJ Research Chromo4 RealTime PCR Detector DNA Engine (Bio-Rad, Hercules, CA) with SSoFast EvaGreen SuperMix (BioRad #172–5201) for promoter DNAs or IQ SYBR Green Supermix (BioRad, #170–8880) for cDNA. E2F1 promoter qRT-PCR values were quantified by the Comparative C(t) method,Citation87 normalized to GAPDH and expressed as percent InPut.
Quantitative RT-PCR (qRT-PCR)
Quantification by qRT-PCR was done on mRNAs (E2Fs1–3, CCNE1, CCND1, and CDC25A). RNA was isolated from cell lysates with the RNeasy kit (Qiagen, #74106), on column DNAseI (Roche, #04716728001) and quantified on a NanoDrop Spectrophotometer (ThermoScientific). cDNA for qRT-PCR was synthesized with M-MuLV reverse transcriptase (NEB,#M0253S), random primers (Roche, #11034731001) and dNTPs (Roche, #1181432601). PCR was with iQ SyberGreen SuperMix (BioRad, #170–8880S) and normalized to GAPDH mRNA by the Comparative C(t) method,Citation87 expressed as percent InPut.
Cell cycle analysis
Cells were synchronized in G0/G1 by growth in phenol red-free DMEM (Gibco, #21063–029) supplemented with 5% charcoal stripped BGS (Hyclone #SH30541.03) for 72 hours at 37°C in 5% CO2. After treatment with 5nM E2 or 5nME2 ± 5 µM iAs, cells were harvested and a portion reserved for RNA isolation using the RNeasy Kit (Qiagen, #74106) and the rest fixed with 95% EtOH and stained with propidium iodide (Sigma, #P4170) solution containing DNase-free RNaseA (Qiagen, #19101). Analysis was by FACScan Flow Cytometer (Becton-Dickinson) and FlowJo,LLC software (Ashland, OR).
Apoptosis
Cells plated at 1.75 × 105 cells/well in 6-well plates were synchronized as above and treated. Cells were harvested with phenol-red free trypsin, washed with binding buffer (BB) (10 mM HEPES pH 7.4, 140 mM NaCl, 2.5 mM CaCl2), resuspended in BB plus 0.5 ug/mL Annexin V-FITC (Clontech, ApoAlert AnnexinV-FITC, #630109), and incubated for 15 minutes. Cells were washed and resuspended in BB plus 2.4 ug/ml propidium iodide (PI) (Sigma, #P4170) and incubated15 min at room temperature. Analysis was on a FACScan flow cytometer (BD Biosciences) using bivariant plots of FL2 (PI) vs. FL1 (FITC) fluorescence. Non-apoptotic cells were negative and late apoptotic positive for Annexin V-FITC and PI. Early apoptotic cells exhibited Annexin V-FITC binding but no PI signal and only these were counted in this analysis. Cell necrosis was measured by Trypan Blue uptake (Dead cells) and live cells were counted with a hemocytometer.
Kinase assay
Cells were treated for 14 hr and lysed in 50mM Tris 7.5,140mM NaCl, 5 mM EDTA, 1%NP-40, 50 mM NaF, 0.1 mM Na3VO4, protease inhibitor cocktail (Calbiochem #539134). Cdk2 was immunoprecipitated for 1 h 4°C from 200 µg of total cell protein and isolated with Protein G-coupled magnetic beads. Beads were washed in kinase buffer and then incubated in 30 µl kinase buffer (50 mM Tris 7.4, 10mM MgCl2) with 25 µM ATP, 10 µCi [γ32P] ATP and 1 µg histone H1(Roche) for 30 minutes at 30°C and stopped by addition of 10 µl 4x SDS-PAGE sample buffer. Proteins were separated on a 15% polyacrylamide gel, transferred to Immobilon-P PVDF membrane and visualized by PhosphorImager analysis (Storm Typhoon Molecular Dynamics/GE) followed by incubation with antibody to Cdk2 and analysis by HRP-based chemiluminescence on an Alpha Innotek Fluorochem 8900 (San Leandro, CA).
Plasmid transfections
Plasmids were transfected with Lipofectamine Plus (Invitrogen, #18324–012). Cells recovered for 24 h and medium was replaced with 1% stripped serum (HyClone) in DMEM. Cells were treated with E2 ± iAs 72 h post transfection and were harvested by lysis in 50 mM Tris pH7.5, 1% NP-40, NaCl, EDTA followed by Bradford protein assays (Bio-Rad, #500–0006).
Statistical analyses
Data are expressed as means and Standard Deviation (SD) Standard Error of the Mean (SEM) as indicated. Statistical significance of the treatment of cells with E2 + iAs vs. E2 alone, as well as the significance of the dependence on time of treatment, was determined by a 2-way Analysis of Variance using the General Linear Model in the SPSS Statistical Analysis Program Version 21 (IBM).
Abbreviations
| ChIP | = | chromatin immunoprecipitation |
| qRT-PCR | = | quantitative real time polymerase chain reaction |
| G1/S | = | growth1/synthesis phase of cell cycle |
| G2/M | = | Growth2/mitotic phase of cell cycle |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
I would like to thank Shelby Stormann for her excellent technical expertise in all aspects of this project and Fiona Barr for excellent technical assistance in the early cell cycle experiments. I also thank Eric Pietrowicz for the flow analysis of apoptosis. My thanks to Dr. Geza Fejes-Toth for kindly providing the human ERα expression plasmid, to Dr. Ben Steele for assistance with the statistical analyses, and to Dr. Charles N. Cole and Dr. Todd Miller for helpful suggestions on the manuscript. Lastly, I would like to acknowledge and thank Dr. Allan Munck for his remarkable and unflagging encouragement, support and mentoring over the years.
Funding
This work was funded by NIH-NIEHS RO1ES013168.
References
- Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev 1998; 12:2245-62; PMID:9694791; https://doi.org/10.1101/gad.12.15.2245
- Kent LN, Bae S, Tsai SY, Tang X, Srivastava A, Koivisto C, Martin CK, Ridolfi E, Miller GC, Zorko SM, et al. Dosage-dependent copy number gains in E2f1 and E2f3 drive hepatocellular carcinoma. J Clin Invest 2017; 127(3):830-42; https://doi.org/10.1172/JCI87583
- Muller H, Bracken AP, Vernell R, Moroni MC, Christians F, Grassilli E, Prosperini E, Vigo E, Oliner JD, Helin K. E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev 2001; 15:267-85; PMID:11159908; https://doi.org/10.1101/gad.864201
- Poppy Roworth A, Ghari F, La Thangue NB. To live or let die - complexity within the E2F1 pathway. Mol Cell Oncol 2015; 2:e970480; PMID:27308406; https://doi.org/10.4161/23723548.2014.970480
- Bode AM, Dong Z. Signal transduction pathways in cancer development and as targets for cancer prevention. Prog Nucleic Acid Res Mol Biol 2005; 79:237-97; PMID:16096030
- Platanias LC. Biological responses to arsenic compounds. J Biol Chem 2009; 284:18583-7; PMID:19363033; https://doi.org/10.1074/jbc.R900003200
- Chow SK, Chan JY, Fung KP. Suppression of cell proliferation and regulation of estrogen receptor alpha signaling pathway by arsenic trioxide on human breast cancer MCF-7 cells. J Endocrinol 2004; 182:325-37; PMID:15283693; https://doi.org/10.1677/joe.0.1820325
- Baj G, Arnulfo A, Deaglio S, Mallone R, Vigone A, De Cesaris MG, Surico N, Malavasi F, Ferrero E. Arsenic trioxide and breast cancer: analysis of the apoptotic, differentiative and immunomodulatory effects. Breast Cancer Res Treat 2002; 73:61-73; PMID:12083632; https://doi.org/10.1023/A:1015272401822
- Maeda H, Hori S, Ohizumi H, Segawa T, Kakehi Y, Ogawa O, Kakizuka A. Effective treatment of advanced solid tumors by the combination of arsenic trioxide and L-buthionine-sulfoximine. Cell Death Differ 2004; 11:737-46; PMID:15002036; https://doi.org/10.1038/sj.cdd.4401389
- Lehmann GM, McCabe MJ, Jr. Arsenite slows S phase progression via inhibition of cdc25A dual specificity phosphatase gene transcription. Toxicol Sci 2007; 99:70-8; PMID:17545210; https://doi.org/10.1093/toxsci/kfm142
- McNeely SC, Belshoff AC, Taylor BF, Fan TW, McCabe MJ, Jr, Pinhas AR, States JC. Sensitivity to sodium arsenite in human melanoma cells depends upon susceptibility to arsenite-induced mitotic arrest. Toxicol Appl Pharmacol 2008; 229:252-61; PMID:18328521; https://doi.org/10.1016/j.taap.2008.01.020
- McCollum G, Keng PC, States JC, McCabe MJ, Jr. Arsenite delays progression through each cell cycle phase and induces apoptosis following G2/M arrest in U937 myeloid leukemia cells. J Pharmacol Exp Ther 2005; 313:877-87; PMID:15722406; https://doi.org/10.1124/jpet.104.080713
- Park WH, Seol JG, Kim ES, Hyun JM, Jung CW, Lee CC, Kim BK, Lee YY. Arsenic trioxide-mediated growth inhibition in MC/CAR myeloma cells via cell cycle arrest in association with induction of cyclin-dependent kinase inhibitor, p21, and apoptosis. Cancer Res 2000; 60:3065-71; PMID:10850458
- Chilakapati J, Wallace K, Ren H, Fricke M, Bailey K, Ward W, Creed J, Kitchin K. Genome-wide analysis of BEAS-2B cells exposed to trivalent arsenicals and dimethylthioarsinic acid. Toxicology 2009; 268:31-9; PMID:19945496; https://doi.org/10.1016/j.tox.2009.11.018
- Sanz MA, Grimwade D, Tallman MS, Lowenberg B, Fenaux P, Estey EH, Naoe T, Lengfelder E, Buchner T, Dohner H, et al. Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2009; 113:1875-91; PMID:18812465; https://doi.org/10.1182/blood-2008-04-150250
- Murgo AJ. Clinical trials of arsenic trioxide in hematologic solid tumors: overview of the NCI cooperative research and development studies. www.TheOncologist.com 2001; 6(suppl 2):22-8
- Naujokas MF, Anderson B, Ahsan H, Aposhian HV, Graziano JH, Thompson C, Suk WA. The broad scope of health effects from chronic arsenic exposure: update on a worldwide public health problem. Environ Health Perspect 2013; 121:295-302; PMID:23458756; https://doi.org/10.1289/ehp.1205875
- Wu L, de Bruin A, Wang H, Simmons T, Cleghorn W, Goldenberg LE, Sites E, Sandy A, Trimboli A, Fernandez SA, et al. Selective roles of E2Fs for ErbB2- and Myc-mediated mammary tumorigenesis. Oncogene 2015; 34:119-28; PMID:24276244; https://doi.org/10.1038/onc.2013.511
- Stender JD, Frasor J, Komm B, Chang KC, Kraus WL, Katzenellenbogen BS. Estrogen-regulated gene networks in human breast cancer cells: involvement of E2F1 in the regulation of cell proliferation. Mol Endocrinol 2007; 21:2112-23; PMID:17550982; https://doi.org/10.1210/me.2006-0474
- Wu L, Timmers C, Maiti B, Saavedra HI, Sang L, Chong GT, Nuckolls F, Giangrande P, Wright FA, Field SJ, et al. The E2F1-3 transcription factors are essential for cellular proliferation. Nature 2001; 414:457-62; PMID:11719808; https://doi.org/10.1038/35106593
- Zhu W, Giangrande PH, Nevins JR. Temporal control of cell cycle gene expression mediated by E2F transcription factors. Cell Cycle (Georgetown, Tex) 2005; 4:633-6; PMID:15876877; https://doi.org/10.4161/cc.4.5.1650
- Kong LJ, Chang JT, Bild AH, Nevins JR. Compensation and specificity of function within the E2F family. Oncogene 2007; 26:321-7; PMID:16909124; https://doi.org/10.1038/sj.onc.1209817
- Attwooll C, Lazzerini Denchi E, Helin K. The E2F family: specific functions and overlapping interests. Embo J 2004; 23:4709-16; PMID:15538380; https://doi.org/10.1038/sj.emboj.7600481
- Iaquinta PJ, Lees JA. Life and death decisions by the E2F transcription factors. Curr Opin Cell Biol 2007; 19:649-57; PMID:18032011; https://doi.org/10.1016/j.ceb.2007.10.006
- Ohtani K, DeGregori J, Nevins JR. Regulation of the cyclin E gene by transcription factor E2F1. Proc Natl Acad Sci U S A 1995; 92:12146-50; PMID:8618861; https://doi.org/10.1073/pnas.92.26.12146
- Yang R, Muller C, Huynh V, Fung YK, Yee AS, Koeffler HP. Functions of cyclin A1 in the cell cycle and its interactions with transcription factor E2F-1 and the Rb family of proteins. Mol Cell Biol 1999; 19:2400-7; PMID:10022926; https://doi.org/10.1128/MCB.19.3.2400
- Satyanarayana A, Kaldis P. Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009; 28:2925-39; PMID:19561645; https://doi.org/10.1038/onc.2009.170
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009; 9:153-66; PMID:19238148; https://doi.org/10.1038/nrc2602
- Vigo E, Muller H, Prosperini E, Hateboer G, Cartwright P, Moroni MC, Helin K. CDC25A phosphatase is a target of E2F and is required for efficient E2F-induced S phase. Mol Cell Biol 1999; 19:6379-95; PMID:10454584; https://doi.org/10.1128/MCB.19.9.6379
- Ru Lee W, Chen CC, Liu S, Safe S. 17beta-estradiol (E2) induces cdc25A gene expression in breast cancer cells by genomic and non-genomic pathways. J Cell Biochem 2006; 99:209-20; PMID:16598773; https://doi.org/10.1002/jcb.20902
- DeGregori J, Johnson DG. Distinct and overlapping roles for E2F family members in transcription, proliferation and apoptosis. Curr Mol Med 2006; 6:739-48; PMID:17100600
- Neuman E, Flemington EK, Sellers WR, Kaelin WG Jr. Transcription of the E2F-1 gene is rendered cell cycle dependent by E2F DNA-binding sites within its promoter. Mol Cell Biol 1994; 14:6607-15; PMID:7935380; https://doi.org/10.1128/MCB.14.10.6607
- Johnson DG, Ohtani K, Nevins JR. Autoregulatory control of E2F1 expression in response to positive and negative regulators of cell cycle progression. Genes Dev 1994; 8:1514-25; PMID:7958836; https://doi.org/10.1101/gad.8.13.1514
- Pierce AM, Schneider-Broussard R, Gimenez-Conti IB, Russell JL, Conti CJ, Johnson DG. E2F1 has both oncogenic and tumor-suppressive properties in a transgenic model. Mol Cell Biol 1999; 19:6408-14; PMID:10454586; https://doi.org/10.1128/MCB.19.9.6408
- Johnson DG. The paradox of E2F1: oncogene and tumor suppressor gene. Mol Carcinog 2000; 27:151-7; PMID:10708476; https://doi.org/10.1002/(SICI)1098-2744(200003)27:3%3c151::AID-MC1%3e3.0.CO;2-C
- Putzer BM, Engelmann D. E2F1 apoptosis counterattacked: evil strikes back. Trends Mol Med 2013; 19:89-98; PMID:23219173; https://doi.org/10.1016/j.molmed.2012.10.009
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell 1995; 81:323-30; PMID:7736585; https://doi.org/10.1016/0092-8674(95)90385-2
- MacDonald JI, Dick FA. Posttranslational modifications of the retinoblastoma tumor suppressor protein as determinants of function. Genes Cancer 2012; 3:619-33; https://doi.org/10.1177/1947601912473305
- Giacinti C, Giordano A. RB and cell cycle progression. Oncogene 2006; 25:5220-7; PMID:16936740; https://doi.org/10.1038/sj.onc.1209615
- Stengel KR, Thangavel C, Solomon DA, Angus SP, Zheng Y, Knudsen ES. Retinoblastoma/p107/p130 pocket proteins: protein dynamics and interactions with target gene promoters. J Biol Chem 2009; 284:19265-71; PMID:19279001; https://doi.org/10.1074/jbc.M808740200
- Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell 2002; 2:103-12; PMID:12204530; https://doi.org/10.1016/S1535-6108(02)00102-2
- Burke JR, Liban TJ, Restrepo T, Lee HW, Rubin SM. Multiple mechanisms for E2F binding inhibition by phosphorylation of the retinoblastoma protein C-terminal domain. J Mol Biol 2014; 426:245-55; PMID:24103329; https://doi.org/10.1016/j.jmb.2013.09.031
- Burke JR, Hura GL, Rubin SM. Structures of inactive retinoblastoma protein reveal multiple mechanisms for cell cycle control. Genes Dev 2012; 26:1156-66; PMID:22569856; https://doi.org/10.1101/gad.189837.112
- Burke JR, Deshong AJ, Pelton JG, Rubin SM. Phosphorylation-induced conformational changes in the retinoblastoma protein inhibit E2F transactivation domain binding. J Biol Chem 2010; 285:16286-93; PMID:20223825; https://doi.org/10.1074/jbc.M110.108167
- Zarkowska T, Mittnacht S. Differential phosphorylation of the retinoblastoma protein by G1/S cyclin-dependent kinases. J Biol Chem 1997; 272:12738-46; PMID:9139732; https://doi.org/10.1074/jbc.272.19.12738
- Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 1999; 98:859-69; PMID:10499802; https://doi.org/10.1016/S0092-8674(00)81519-6
- Wang W, Dong L, Saville B, Safe S. Transcriptional activation of E2F1 gene expression by 17beta-estradiol in MCF-7 cells is regulated by NF-Y-Sp1/estrogen receptor interactions. Mol Endocrinol 1999; 13:1373-87; PMID:10446910
- Doisneau-Sixou SF, Sergio CM, Carroll JS, Hui R, Musgrove EA, Sutherland RL. Estrogen and antiestrogen regulation of cell cycle progression in breast cancer cells. Endocr Relat Cancer 2003; 10:179-86; PMID:12790780; https://doi.org/10.1677/erc.0.0100179
- Davey JC, Bodwell JE, Gosse JA, Hamilton JW. Arsenic as an endocrine disruptor: effects of arsenic on estrogen receptor-mediated gene expression in vivo and in cell culture. Toxicol Sci 2007; 98:75-86; PMID:17283378; https://doi.org/10.1093/toxsci/kfm013
- Bae-Jump VL, Zhou C, Boggess JF, Gehrig PA. Arsenic trioxide (As2O3) inhibits expression of estrogen receptor-alpha through regulation of the mitogen-activated protein kinase (MAPK) pathway in endometrial cancer cells. Reprod Sci (Thousand Oaks, Calif) 2008; 15:1011-7; PMID:19088370; https://doi.org/10.1177/1933719108324134
- Chen GC, Guan LS, Hu WL, Wang ZY. Functional repression of estrogen receptor a by arsenic trioxide in human breast cancer cells. Anticancer Res 2002; 22:633-8; PMID:12014631
- Yadav S, Shi Y, Wang F, Wang H. Arsenite induces apoptosis in human mesenchymal stem cells by altering Bcl-2 family proteins and by activating intrinsic pathway. Toxicol Appl Pharmacol 2010; 244(3):263-72; PMID:20083129; https://doi.org/10.1016/j.taap.2010.01.001
- Bode AM, Dong Z. The paradox of arsenic: molecular mechanisms of cell transformation and chemotherapeutic effects. Crit Rev Oncol hematol 2002; 42:5-24; PMID:11923065; https://doi.org/10.1016/S1040-8428(01)00215-3
- Davison K, Mann KK, Waxman S, Miller WH Jr. JNK activation is a mediator of arsenic trioxide-induced apoptosis in acute promyelocytic leukemia cells. Blood 2004; 103:3496-502; PMID:14701702; https://doi.org/10.1182/blood-2003-05-1412
- Bi X, Gu J, Guo Z, Tao S, Wang Y, Tang L, Wu J, Mi Q. Different pathways are involved in arsenic-trioxide-induced cell proliferation and growth inhibition in human keratinocytes. Skin Pharmacol Physiol 2009; 23:68-78; PMID:20016248; https://doi.org/10.1159/000265677
- Caldon CE, Sergio CM, Schutte J, Boersma MN, Sutherland RL, Carroll JS, Musgrove EA. Estrogen regulation of cyclin E2 requires cyclin D1 but not c-Myc. Mol Cell Biol 2009; 29:4623-39; PMID:19564413; https://doi.org/10.1128/MCB.00269-09
- Hyun Park W, Hee Cho Y, Won Jung C, Oh Park J, Kim K, Hyuck Im Y, Lee MH, Ki Kang W, Park K. Arsenic trioxide inhibits the growth of A498 renal cell carcinoma cells via cell cycle arrest or apoptosis. Biochem Biophys Res Commun 2003; 300:230-5; PMID:12480548; https://doi.org/10.1016/S0006-291X(02)02831-0
- Lauper N, Beck AR, Cariou S, Richman L, Hofmann K, Reith W, Slingerland JM, Amati B. Cyclin E2: a novel CDK2 partner in the late G1 and S phases of the mammalian cell cycle. Oncogene 1998; 17:2637-43; PMID:9840927; https://doi.org/10.1038/sj.onc.1202477
- Donzelli M, Draetta GF. Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep 2003; 4:671-7; PMID:12835754; https://doi.org/10.1038/sj.embor.embor887
- Blomberg I, Hoffmann I. Ectopic expression of Cdc25A accelerates the G(1)/S transition and leads to premature activation of cyclin E- and cyclin A-dependent kinases. Mol Cell Biol 1999; 19:6183-94; PMID:10454565; https://doi.org/10.1128/MCB.19.9.6183
- Adams MR, Sears R, Nuckolls F, Leone G, Nevins JR. Complex transcriptional regulatory mechanisms control expression of the E2F3 locus. Mol Cell Biol 2000; 20:3633-9; PMID:10779353; https://doi.org/10.1128/MCB.20.10.3633-3639.2000
- Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 1995; 270:1491-4; PMID:7491495; https://doi.org/10.1126/science.270.5241.1491
- Trowbridge JM, Rogatsky I, Garabedian MJ. Regulation of estrogen receptor transcriptional enhancement by the cyclin A/Cdk2 complex. Proc Natl Acad Sci U S A 1997; 94:10132-7; PMID:9294175; https://doi.org/10.1073/pnas.94.19.10132
- Wesierska-Gadek J, Gritsch D, Zulehner N, Komina O, Maurer M. Roscovitine, a selective CDK inhibitor, reduces the basal and estrogen-induced phosphorylation of ER-alpha in human ER-positive breast cancer cells. J Cell Biochem 2011; 112:761-72; PMID:21328450; https://doi.org/10.1002/jcb.23004
- Katich SC, Zerfass-Thome K, Hoffmann I. Regulation of the Cdc25A gene by the human papillomavirus Type 16 E7 oncogene. Oncogene 2001; 20:543-50; PMID:11313986; https://doi.org/10.1038/sj.onc.1204130
- Caldon CE, Musgrove EA. Distinct and redundant functions of cyclin E1 and cyclin E2 in development and cancer. Cell Division 2010; 5:2; PMID:20180967; https://doi.org/10.1186/1747-1028-5-2
- Zarkowska T, U S, Harlow E, Mittnacht S. Monoclonal antibodies specific for underphosphorylated retinoblastoma protein identify a cell cycle regulated phosphorylation site targeted by CDKs. Oncogene 1997; 14:249-54; PMID:9010227; https://doi.org/10.1038/sj.onc.1200824
- Rubin E, Mittnacht S, Villa-Moruzzi E, Ludlow JW. Site-specific and temporally-regulated retinoblastoma protein dephosphorylation by protein phosphatase type 1. Oncogene 2001; 20:3776-85; PMID:11439341; https://doi.org/10.1038/sj.onc.1204518
- Kolupaeva V, Janssens V. PP1 and PP2A phosphatases–cooperating partners in modulating retinoblastoma protein activation. FEBS J 2013; 280:627-43; PMID:22299668; https://doi.org/10.1111/j.1742-4658.2012.08511.x
- Mooney LM, Al-Sakkaf KA, Brown BL, Dobson PR. Apoptotic mechanisms in T47D and MCF-7 human breast cancer cells. Br J Cancer 2002; 87:909-17; PMID:12373608; https://doi.org/10.1038/sj.bjc.6600541
- Miller WH Jr, Schipper HM, Lee JS, Singer J, Waxman S. Mechanisms of action of arsenic trioxide. Cancer Res 2002; 62:3893-903; PMID:12124315
- Perillo B, Sasso A, Abbondanza C, Palumbo G. 17beta -estradiol inhibits apoptosis in MCF-7 cells, inducing bcl-2 expression via two estrogen-responsive elements present in the coding sequence. Mol Cell Biol 2000; 20:2901; https://doi.org/10.1128/MCB.20.8.2890-2901.2000
- Lazzerini Denchi E, Helin K. E2F1 is crucial for E2F-dependent apoptosis. EMBO Rep 2005; 6:661-8; PMID:15976820; https://doi.org/10.1038/sj.embor.7400452
- Hallstrom TC, Mori S, Nevins JR. An E2F1-dependent gene expression program that determines the balance between proliferation and cell death. Cancer Cell 2008; 13:11-22; PMID:18167336; https://doi.org/10.1016/j.ccr.2007.11.031
- Cohen MH, Hirschfeld S, Flamm Honig S, Ibrahim A, Johnson JR, O'Leary JJ, White RM, Williams GA, Pazdur R. Drug approval summaries: arsenic trioxide, tamoxifen citrate, anastrazole, paclitaxel, bexarotene. Oncologist 2001; 6:4-11; PMID:11161223; https://doi.org/10.1634/theoncologist.6-1-4
- Chou WC, Dang CV. Acute promyelocytic leukemia: recent advances in therapy and molecular basis of response to arsenic therapies. Curr Opin Hematol 2005; 12:1-6; PMID:15604884; https://doi.org/10.1097/01.moh.0000148552.93303.45
- Wang L, Zhang J, An Y, Wang Z, Liu J, Li Y, Zhang D. A study on the thermochemotherapy effect of nanosized As2O3/MZF thermosensitive magnetoliposomes on experimental hepatoma in vitro and in vivo. Nanotechnology 2011; 22:315102; PMID:21730756; https://doi.org/10.1088/0957-4484/22/31/315102
- Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer 2009; 9:785-97; PMID:19851314; https://doi.org/10.1038/nrc2696
- Hwang HC, Clurman BE. Cyclin E in normal and neoplastic cell cycles. Oncogene 2005; 24:2776-86; PMID:15838514; https://doi.org/10.1038/sj.onc.1208613
- Sieuwerts AM, Look MP, Meijer-van Gelder ME, Timmermans M, Trapman AM, Garcia RR, Arnold M, Goedheer AJ, de Weerd V, Portengen H, et al. Which cyclin E prevails as prognostic marker for breast cancer? Results from a retrospective study involving 635 lymph node-negative breast cancer patients. Clin Cancer Res 2006; 12:3319-28; PMID:16740753; https://doi.org/10.1158/1078-0432.CCR-06-0225
- Payton M, Scully S, Chung G, Coats S. Deregulation of cyclin E2 expression and associated kinase activity in primary breast tumors. Oncogene 2002; 21:8529-34; PMID:12466974; https://doi.org/10.1038/sj.onc.1206035
- Lam SK, Li YY, Zheng CY, Leung LL, Ho JC. E2F1 downregulation by arsenic trioxide in lung adenocarcinoma. Int J Oncol 2014; 45:2033-43; PMID:25174355
- Grimm SA, Marymont M, Chandler JP, Muro K, Newman SB, Levy RM, Jovanovic B, McCarthy K, Raizer JJ. Phase I study of arsenic trioxide and temozolomide in combination with radiation therapy in patients with malignant gliomas. J Neuro Oncol 2012; 110:237-43; PMID:22875709; https://doi.org/10.1007/s11060-012-0957-6
- Dilda PJ, Hogg PJ. Arsenical-based cancer drugs. Cancer Treatment Rev 2007; 33:542-64; PMID:17624680; https://doi.org/10.1016/j.ctrv.2007.05.001
- Andrew AS, Burgess JL, Meza MM, Demidenko E, Waugh MG, Hamilton JW, Karagas MR. Arsenic exposure is associated with decreased DNA repair in vitro and in individuals exposed to drinking water arsenic. Environ Health Perspect 2006; 114:1193-8; PMID:16882524; https://doi.org/10.1289/ehp.9008
- Muenyi CS, Ljungman M, States JC. Arsenic disruption of DNA damage responses-potential role in carcinogenesis and chemotherapy. Biomolecules 2015; 5:2184-93; PMID:26404387; https://doi.org/10.3390/biom5042184
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 2001; 29:e45; PMID:11328886; https://doi.org/10.1093/nar/29.9.e45
- Guo R, Chen J, Zhu F, Biswas AK, Berton TR, Mitchell DL, Johnson DG. E2F1 localizes to sites of UV-induced DNA damage to enhance nucleotide excision repair. J Biol Chem 2010; 285:19308-15; PMID:20413589; https://doi.org/10.1074/jbc.M110.121939
- Chen J, Zhu F, Weaks RL, Biswas AK, Guo R, Li Y, Johnson DG. E2F1 promotes the recruitment of DNA repair factors to sites of DNA double-strand breaks. Cell Cycle 2011; 10:1287-94; PMID:21512314; https://doi.org/10.4161/cc.10.8.15341
- Cooper KL, King BS, Sandoval MM, Liu KJ, Hudson LG. Reduction of arsenite-enhanced ultraviolet radiation-induced DNA damage by supplemental zinc. ToxicolAppl Pharmacol 2013; 269:81-8
- Narasimha AM, Kaulich M, Shapiro GS, Choi YJ, Sicinski P, Dowdy SF. CyclinD activates the Rb tumor suppressor by mono-phosphorylation. Elife 2014; 3:e02872