
ABSTRACT
Timely recruitment of DNA damage response proteins to sites of genomic structural lesions is very important for signaling mechanisms to activate appropriate cell cycle checkpoints but also repair the altered DNA sequence to suppress mutagenesis. The eukaryotic cell is characterized by a complex cadre of players and pathways to ensure genomic stability in the face of replication stress or outright genomic insult by endogenous metabolites or environmental agents. Among the key performers are molecular motor DNA unwinding enzymes known as helicases that sense genomic perturbations and separate structured DNA strands so that replacement of a damaged base or sugar-phosphate backbone lesion can occur efficiently. Mutations in the BLM gene encoding the DNA helicase BLM leads to a rare chromosomal instability disorder known as Bloom’s syndrome. In a recent paper by the Sengupta lab, BLM’s role in the correction of double-strand breaks (DSB), a particularly dangerous form of DNA damage, was investigated. Adding to the complexity, BLM appears to be a key ringmaster of DSB repair as it acts both positively and negatively to regulate correction pathways of high or low fidelity. The FANCJ DNA helicase, mutated in another chromosomal instability disorder known as Fanconi Anemia, is an important player that likely coordinates with BLM in the balancing act. Further studies to dissect the roles of DNA helicases like FANCJ and BLM in DSB repair are warranted.
Bloom’s syndrome and its molecular pathology
The complexity of the DNA damage response reflects a multiplicity of repair pathways invoked to handle different forms of DNA damage in a manner that is dependent on sophisticated regulatory mechanisms. This theme is upheld in a recently published paper by Tripathi et al. in Nature Communications on 9 March 2018. Sengupta and scientists from the National Institute of Immunology in New Delhi and Indian Institute of Science in Bangalore collaborated to study the recruitment and repair functions of the BLM helicase at double-strand breaks (DSBs), a potentially lethal form of DNA damage that poses a great source of genomic instability [Citation1]. Bi-allelic mutations in the BLM gene, which encodes a molecular motor DNA unwinding enzyme, are linked to a rare disorder known as Bloom’s syndrome (BS) [Citation2] (). BS is characterized by prenatal and postnatal growth disorder, sun sensitivity, erythematous skin rashes, accelerated aging, immune-deficiency and predisposition to a wide spectrum of cancers [Citation3]. Cells from BS patients display a very high rate of sister chromatid exchange (SCE), a form of chromosomal instability attributed to defects in homologous recombination (HR) repair. The underlying basis for the distinct BS clinical phenotypes including short stature, immune dysfunction, and a wide range of cancers remains poorly understood. At the cellular and molecular level, aberrant processing of genomic DNA structures during replication and recombination are believed to underlie the aberrant phenotypes of BS [Citation4], as well as other hereditary chromosomal instability disorders defective in key DNA repair helicases [Citation5].
Table 1. Properties of the BLM and FANCJ DNA helicases.
Competing pathways in double-strand break repair
DSBs in the DNA double helix arise due to various cellular processes including meiosis, V(D)J recombination, or class switch recombination. Exogenous sources such as ionizing radiation (IR) and chemicals that directly introduce strand breaks (e.g., bleomycin) or replicative stress (e.g., hydroxyurea (HU)) can also give rise to deleterious DSBs. HR is a high-fidelity process by which DSBs are restored using homologous sequence from the donor DNA strand of the sister chromatid duplex for DNA synthesis and repair. During the S and G2 phases of the cell cycle, the proximity of the sister chromatids favors HR as the primary form of recombinogenic repair. In contrast, canonical nonhomologous end-joining (NHEJ) occurs throughout the cell cycle (including G1) to repair DSBs (). However, NHEJ compared to HR is a more error-prone process of DNA repair that can lead to loss of nucleotides [Citation6]. Other than these two major pathways (HR and NHEJ) for DSB repair, cells can also employ the highly error-prone process of microhomology mediated end-joining (MMEJ) also known as alternative NHEJ (alt-NHEJ) [Citation7]. This pathway utilizes complementary DNA sequences of limited length (5–25 bp) at the partly resected strand ends to enact joining of the DNA ends.
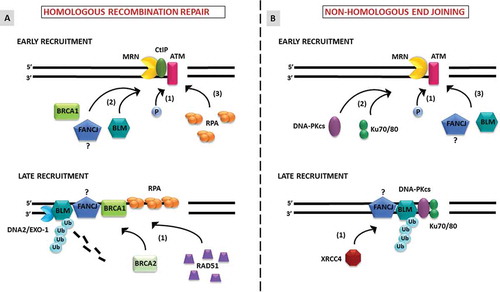
Figure 1. Models depicting the recruitment of BLM, FANCJ and other DNA repair proteins to double-strand breaks. A, Homologous recombination repair. Early recruitment: Early responders including the MRN complex recruit to the DSB and activate ATM during S or G2 phases of the cell cycle. 1) ATM is activated by autophosphorylation which in-turn can amplify MRN-mediated signaling. MRN with its protein partner CTIP begin resection at the DSB, thereby initiating DSB repair by HR. 2) BLM is recruited to the DSB along with BRCA1 and FANCJ. It is unclear whether FANCJ and BRCA1 form a complex in solution before recruitment to the DSB or interact at the DSB. The interaction between FANCJ-BRCA1 and BLM may potentiate DSB repair by the HR pathway. 3) The short DNA overhangs created by the MRE11 nuclease of the MRN complex are stabilized by RPA. Late recruitment: Long 5ʹ strand resection occurs through the BLM-DNA2 or EXO1 pathway. FANCJ may functionally interact with BLM and MRE11 leading to the formation of RPA nucleated stable 3ʹ ssDNA. 1) The long RPA filament is replaced by RAD51 and this replacement is facilitated by BRCA2. Both BLM and FANCJ can negatively regulate HR by inhibiting RAD51 interaction with ssDNA filaments. Further, BLM’s retention at the DNA is aided by ubiquitylation, which is important for interaction of NBS1 with the MRN complex. B, Non-homologous end-joining. Early recruitment: Early responders including ATM and the MRN complex recruit to the DSB and set up a cascade for NHEJ during any phase of the cell cycle. 1) Upon detection of DSBs, ATM becomes phosphorylated and in turn phosphorylates other proteins functioning later in NHEJ. 2) The recruitment of DNA-PKcs followed by KU 70/80 complex at DSBs displaces MRN complex and initiates NHEJ. 3) BLM and potentially FANCJ may be recruited at this stage. Late recruitment: Whether BLM and FANCJ interact to modulate NHEJ is an important area of investigation. 1) BLM regulates the recruitment of the repair protein XRCC4 which enhances the joining activity of LIG4.
The manner in which a DSB is repaired is a complex process in which the chromatin compaction, structure of the DNA overhangs, phase of the cell cycle, and DNA break repair/recognition proteins all play a role in order to favor a particular repair process [Citation8,Citation9]. However, it is important to remember that various DNA repair processes are constitutively active during most stages of cell cycle and certain DNA repair/recognition proteins actively participate in both HR and NHEJ. Furthermore, to maintain genomic integrity and cellular homeostasis, the choice of DNA repair pathway to repair DSBs is coupled with regulatory players/events that promote or inhibit early events in the two types of DNA repair processes [Citation10].
Antagonistic roles for BLM helicase in double-strand break repair
From a historical perspective, understanding the role of BLM in the maintenance of genomic stability has been complex. The elevated SCE characteristic of cells from BS patients suggested a role of the BLM DNA helicase in HR. Through its catalytic DNA unwinding function (), BLM is presumed to regulate HR repair of DSBs to either suppress illegitimate recombination or promote repair of homologous DNA sequences [Citation11]. Indeed, the purified recombinant BLM helicase was shown to branch-migrate four-stranded DNA structures known as Holliday Junctions (HJ), a key intermediate of HR repair [Citation12]. Further work showed that BLM was capable of branch-migrating D-loop structures, an early intermediate of DNA strand exchange by the major strand recombinase Rad51 [Citation13]. This activity in principle may either promote or dissuade HR repair. An important advance in the field was made by the Hickson lab when they demonstrated that a multi-protein BLM complex with a cellular topoisomerase was capable of catalyzing double HJ dissolution in vitro, a reaction that in principal would suppress SCE [Citation14].
The molecular explanation for BLM’s role in suppressing SCE was very satisfying; however, further studies indicated that BLM’s role in HR repair was more complex. BLM [Citation15,Citation16] and its yeast homolog, Sgs1 [Citation16–Citation18], operate with structure-specific nucleases DNA2 or EXO-1 to extensively resect DNA ends up to a 1000 bp or more in vitro, a critical biochemical activity required for the generation of 3ʹ single-stranded DNA at a chromosome break to initiate HR repair. Emerging evidence suggests that BLM has dual roles in DNA end-resection, modulating both HR and alt-NHEJ [Citation15,Citation18–Citation20] (). CTIP and MRE11 complex are important for the initiation of resection, promoting HR over NHEJ. The promotion of resection is controlled by BLM (or its yeast homolog Sgs1) and DNA2 or EXO-1 [Citation15,Citation17,Citation18], leading to HR-dependent DSB repair. However, BLM in conjunction with 53BP1 and the downstream RIF1 also inhibits CTIP-MRE11 dependent extensive resection of DNA ends thereby inhibiting alt-NHEJ [Citation19,Citation20]. In the absence of RIF1 and 53BP1, BLM mediates extensive resection of DNA ends to promote alt-NHEJ. Furthermore, HR was reduced in CTIP-depleted cells, as observed by reduced RPA foci and RAD51 filament formation, but the overall DSB repair kinetics are unchanged due to increased NHEJ [Citation21]. Other studies done with TRF1- and/or TRF2-deficient mouse embryonic fibroblasts with dysfunctional telomeres showed that both 53BP1 [Citation22] and RIF1 [Citation23] block extensive 5ʹ-resection. Both 53BP1 and RIF1 inhibit resection mediated by BLM and EXO-1 [Citation22,Citation23]. Depletion of BLM and EXO-1 also led to reduced HR as evidenced by the lack of RAD51 foci formation; this HR defect was alleviated by co-depletion of 53BP1 [Citation24]. The anti-recombinogenic role of 53BP1 has been substantiated by various other studies [Citation25–Citation27], but the role of BLM in regulating HR is more complex.
In the recently published work, Tripathi et al. employed an in vivo restriction endonuclease-targeting strategy coupled with cellular immunofluorescence microscopy to determine that BLM localizes to DSBs in a biphasic manner dependent on early responders to DSBs, namely the MRE11 nuclease and its interacting structural protein partner NBS1, as well as the DNA damage signaling kinase ATM [Citation1]. ATM kinase activity and MRE11 exonuclease activity were shown to be required for BLM’s early recruitment to the DSB, whereas MRE11 and post-translational modification of BLM by ubiquitylation, which is important for interaction with NBS1, are required for BLM’s second phase of recruitment and retention at the DSB. The authors next examined BLM’s role in recruitment of downstream factors to the DSB. Pharmacological inhibition of BLM helicase activity in human cells significantly decreased chromatin DSB recruitment of the major HR strand recombinase RAD51 in the DNA synthesis (S) phase and the NHEJ factor XRCC4 in the pre-replication G1 phase. To understand the dynamic role of BLM in DSB repair, BLM-depleted cells synchronized in S-phase or G1-phase were treated with a RAD51 or Ligase IV inhibitor that affected DNA binding by either protein. Interestingly, depletion of BLM rescued HR and NHEJ in S-phase and G1-/S-phases, respectively [Citation1]. Thus, the work provided evidence that BLM can also antagonize recombinogenic pathways of DSB repair. These findings present a complex scenario for BLM’s pro- and anti-recombinogenic modes of action in response to DSBs that are necessary to maintain optimal DNA repair and disease/cancer suppression. Presumably, BLM’s inhibition of repair pathways may help to suppress illegitimate or inappropriate recombinational repair, but the balancing act performed by BLM remains puzzling in both mechanism and timely purpose. Further studies are clearly required.
Involvement of BLM’s protein partner FANCJ helicase in pathway choice
Aside from BLM’s complex balancing antics, other important performers are likely to take part in the act. One strong candidate is FANCJ, another DNA helicase that is defective in a genetic chromosomal instability disorder known as Fanconi Anemia (FA) as well as breast and ovarian cancer (for review, see [Citation28,Citation29]) (). FANCJ was originally discovered as a protein that physically interacts with the tumor suppressor BRCA1, and this interaction is important for cellular resistance to ionizing radiation (IR) [Citation30]. FANCJ protein plays a crucial role in overcoming replication stress [Citation29,Citation31] especially DNA interstrand crosslinks (ICL) [Citation32–Citation34]. Treatment of FANCJ-deficient cells with mitomycin C, an ICL-inducing agent, caused increased genetic instability as determined by cytogenetic analysis of chromosome spreads. Patient-derived cells with mutant FANCJ accumulate in the G2/M phase, probably due to aberrant S-phase DNA repair. Sensitivity to a DNA cross-linking agent was also observed in chicken cell lines, in which treating FANCJ-deficient DT40 cells with cisplatin led to accumulation of cells in the S/G2 phase [Citation35]. Interestingly, BLM was shown to reside in a complex with proteins of the FA ICL repair pathway that operate upstream of FANCJ [Citation36,Citation37]. Subsequently, we determined that FANCJ physically and functionally interacts with BLM, and that the two helicases collaborate in the cellular response to replication stress [Citation38]. Therefore, BLM may modulate events upstream and downstream of FANCD2 mono-ubiquitination, a key activation step in the FA pathway.
Various studies have implicated FANCJ as an important player in repairing DSBs presumably by regulating DNA repair processes including HR, NHEJ and MMEJ [Citation32,Citation39–Citation41] (). Studies in human cancer or FA-J patient-derived cell lines show that the helicase domain of FANCJ is critical in the pathway of DSB repair and that inhibiting FANCJ helicase activity reduced HR-mediated repair by half [Citation41]. FANCJ helicase activity was also found to be important to confer cellular resistance to DNA damage induced by HU [Citation38] that can cause replication fork collapse and eventual DSBs in DNA [Citation42]. The physical interaction between BLM and FANCJ helps cells tolerate HU exposure [Citation38]. Thus, the FANCJ-BLM interaction is important for overcoming replication stress that potentially leads to DSBs. Although BLM and FANCJ were shown to functionally interact in vitro to unwind damaged dsDNA in a synergistic manner [Citation38], the physiological significance of the BLM-FANCJ interaction remains to be fully elucidated in vivo.
FANCJ has been shown to physically and functionally interact with MRE11 [Citation40]. As mentioned previously, MRE11 is a protein that is involved in the early detection and repair of DSBs via HR. We showed by live-cell imaging that FANCJ recruits rapidly to laser-induced DSBs shortly after MRE11 and its recruitment is dependent on the nuclease activity of MRE11 [Citation40], a finding that is analogous to what is reported for BLM in the latest study by Tripathi et al. [Citation1] (). Furthermore, we found that FANCJ directly interacts with MRE11 and negatively regulates its nuclease activity [Citation40], leaving in question if BLM might be involved in an early phase of DNA end-processing in addition to its known role in long-strand resection with the structure-specific nucleases DNA2 or EXO-1. Long strand resection is critical for the nucleation of RPA on the single-stranded DNA, which is subsequently replaced by RAD51 in a BRCA2-dependent manner. RAD51 filament formation and strand exchange with recipient homologous duplex is an absolute requirement for HR [Citation15]. Notably, BLM helicase and CTIP, also implicated in DNA end-processing [Citation43], are dependent on FANCJ for their efficient and timely recruitment to laser-induced DSBs [Citation40].
As mentioned earlier, BLM can aid HR repair of DSBs by resecting DNA ends and producing 3ʹ single-stranded DNA [Citation15,Citation44] (). This is accomplished via the recruitment of BLM/DNA2 or EXO1 by MRE11-Rad50–NBS1/CTIP complex at DSBs. The single-stranded DNA protrusion can then be stabilized by RPA, a crucial step in HR-mediated DSB repair. BLM has been shown to promote RPA foci formation at IR-induced DSBs [Citation20]. FANCJ potentially plays a part in this interaction, as RPA and FANCJ co-localize at DSBs after IR treatment and under conditions of replication stress [Citation45]. Furthermore, acetylation defective mutant-FANCJ cells treated with camptothecin have reduced RPA foci formation, suggesting an important role of the post-translational modification in DNA repair [Citation46]. Another key intermediate in HR repair is the RAD51 filament which forms downstream of RPA coating the resected single-stranded DNA end [Citation44,Citation47]. Interestingly, BLM can act as a negative regulator of HR by inhibiting RAD51 interaction with ssDNA filaments [Citation48,Citation49]. FANCJ can also inhibit RAD51 interaction with ssDNA filaments, thereby inhibiting RAD51 strand exchange capacity in vitro [Citation50]. Thus, BLM and FANCJ may balance their pro- and anti-recombinogenic activities in DSB repair by similar mechanisms.
The tumor suppressor BRCA1 that is implicated in DSB repair was shown to not only directly interact with FANCJ [Citation30], but also be associated with BLM and other DNA repair factors (MSH2, MSH6, MLH1, RPA, ATM, and MRE11-RAD50-NBS1) [Citation51], suggesting that BRCA1 is an important partner of both DNA helicases in recombinational repair. BRCA1 promotes HR repair by localizing to DSBs, where it may aid DNA end-resection [Citation52,Citation53]. Further, BRCA1 is involved in the stabilization of single-stranded DNA as evidenced by reduced RAD51 interaction at DSBs in BRCA1-depleted cells [Citation53,Citation54]. In a paper by the Bunting lab, it was shown that some residual activity of BRCA1 might be essential for HR-dependent repair [Citation49]. They utilized cells expressing a mutant version of BRCA1 with its exon 11 deleted [Citation55]. These hypomorphic BRCA1 cells (including, primary B cells and MEFs) had increased genomic instability and reduced cell proliferation. Depletion of BLM in cells expressing the mutant BRCA1, but not in cells completely devoid of BRCA1 expression, rescued the genomic instability by aiding RAD51 stability and in-turn HR repair [Citation49]. Additionally, disruption of HR via knockdown of other pro-recombinogenic proteins such as BRCA2 or XRCC2 also led to destabilized RAD51 filaments; BLM depletion could rescue RAD51 stability and HR repair of DSBs in these cases as well [Citation49]. Thus, BLM’s anti-recombinogenic mode of action is relevant when FANCJ’s interacting partner BRCA1 is absent. However, it is unclear if FANCJ collaborates with BLM to suppress HR in BRCA1-deficient cells or if BLM behaves as an anti-recombinase when FANCJ is deficient.
FANCJ is likely to influence the aforementioned BRCA1-mediated HR, as FANCJ is important in the formation and maintenance of BRCA1 foci at DSBs [Citation56]. Further, BRCA1 is important for the recruitment of FANCJ to UV laser induced DSBs [Citation57]. This complex interaction between FANCJ and BRCA1 is mediated through the BRCT domain of BRCA1 [Citation30,Citation58]. Disruption of the FANCJ-BRCA1 interaction in cells expressing a FANCJ mutant unable to bind the BRCA1 BRCT domain led to reduced HR [Citation59]. This attenuation in HR may be caused by reduced RAD51 filament formation. However, when the FANCJ-BRCA1 interaction is defective, FANCJ’s interaction with the DNA mismatch repair protein MLH1α is enriched. This in turn shifts the DNA damage repair to a low-fidelity Pol η-dependent translesion synthesis (TLS), thereby reducing HR [Citation59]. This finding illustrates the complexity of FANCJ’s role in DSB repair, mediated in part by its interactions with not only BRCA1 but other protein partners as well. Moreover, like BLM, FANCJ is likely to have both pro- and anti-recombinogenic functions.
FANCJ can influence DSB repair via non-HR processes including NHEJ and MMEJ [Citation40,Citation41]. Studies with cells deficient for FANCJ and/or MRE11 that examined DNA repair (HR and NHEJ) suggested that depletion of FANCJ led to a modest increase in NHEJ as detected by damage response to IR-induced DSBs [Citation40]. However, this modest increase in NHEJ due to FANCJ depletion was augmented by co-depleting MRE11. Perhaps deficiency of MRE11 leads to attenuated recruitment and retention of BLM at DSBs. The absence of BLM at the DNA ends would alleviate the inhibitory effect of BLM on NHEJ, as suggested by Tripathi et al. [Citation1]. Further studies are warranted to examine the complex role of BLM-FANCJ interaction in modulation of DSB repair via NHEJ.
Collectively, these results suggest that the DNA helicases FANCJ and BLM together coordinate with DNA damage signaling proteins, structure-specific nucleases, polymerases, RPA, and RAD51 to enable a delicate balance of HR and NHEJ to repair DSBs and ultimately preserve genomic stability ().
Perspective
The dynamic role of BLM in DNA damage repair and overall maintenance of genomic integrity is a multifactorial complex process. The most recent results from Tripathi et al. [Citation1] emphasize the pro- and anti-recombinogenic roles of BLM in DSB repair, consistent with the findings of other labs. The helicase function of BLM and perhaps its strand annealing/strand exchange activity [Citation60,Citation61], as well as its interactions with proteins implicated in recombinational repair (), provides opportunities for BLM to regulate cellular pathways to achieve an intricate balance of DSB repair activities for genome preservation. Altogether, it seems probable that the BLM and FANCJ helicases play early and late interactive roles in response to replication stress or DNA damage resulting in DSBs. For example, both BLM and FANCJ can use their motor ATPase to resolve three-stranded DNA intermediates [Citation13,Citation62] or strip RAD51 bound to DNA [Citation48,Citation50], activities that may serve to prevent recombination or help mature recombinant DNA molecules so that they can be resolved. In addition to HR, experimental evidence has suggested that BLM and FANCJ also modulate NHEJ pathways, which may contribute to DSB repair by an error-prone mechanism when either helicase is defective [Citation20,Citation41]. Genome-wide screens similar to those recently used to identify synthetic viable interactions between BLM and the FA pathway [Citation63] may be useful to investigate FANCJ’s interactive roles. BLM protein stability in human cells was shown to be strongly dependent on FANCJ status [Citation38], further emphasizing the complex relationship between the two DNA repair helicases.
In addition to DSB repair, both BLM and FANCJ are implicated in the replication stress response, as evidenced by the hypersensitivity of the corresponding mutant cell lines to the replication inhibitor HU [Citation38,Citation64]. In fact, BLM and FANCJ strongly co-localize with each other after HU exposure, and experimental evidence suggests that the physical interaction of BLM with FANCJ is important for HU resistance [Citation38]. It remains to be seen if the collaboration of BLM and FANCJ is relevant to the suppression of DSB formation, execution of DSB repair, or both. How might BLM and FANCJ work together to prevent DSBs forming in the first place? FANCJ [Citation65,Citation66] and BLM [Citation67,Citation68] have the ability to resolve G-quadruplex non-B DNA structures (G4) which are thought to perturb DNA replication and affect gene expression [Citation69,Citation70]. Exposure of FANCJ-deficient or BLM-deficient cells to a G-quadruplex stabilizing ligand impairs DNA replication and results in the nuclear accumulation of immunoreactive G4 DNA structures [Citation65,Citation71,Citation72]. A recent study attributed the pattern of increased SCEs observed in BS to recombination at transcribed genes near the site of G4 sequences [Citation73]. It is unclear if aberrant G4 DNA metabolism contributes to the elevated SCE observed in FANCJ-deficient patient fibroblasts [Citation38]. Large genomic deletions close to G4-forming genomic regions have been reported in cells from FA complementation group J (FA-J) patients [Citation66]. Furthermore, in chicken cells FANCJ collaborates with BLM (as well as WRN and the TLS polymerase REV1) to replicate past G4 DNA structures, thereby preserving epigenetic stability [Citation74]. BLM and FANCJ are proving to be increasingly relevant in G4 metabolism, and in turn the maintenance of genomic stability and cellular homeostasis. For a discussion of the cross-talk between FANCJ and BLM in replication past G4, see [Citation75]. From a more general standpoint, the poor growth of FANCJ-/- BLM-/- cells and their accumulation in sub-G1 suggests that BLM and FANCJ at least partially compensate for each other even under conditions of endogenous replication stress [Citation76].
Perhaps somewhat underappreciated are the developmental or tissue-specific regulatory mechanisms which influence BLM and FANCJ expression and protein stability. The distribution of BLM varies vastly between normal and neoplastic tissue [Citation77]. BLM is highly expressed in the lymphoid tissues such as spleen, tonsils, and the cortical region of the thymus gland. In normal tissue, BLM was most greatly expressed in regions with high cellular proliferation, such as digestive tract and skin. Cancer cells of both lymphoid and epithelial origin, including various lung, breast and lymph node cancers showed elevated expression of BLM [Citation77]. Thus, there appears to be a high correlation between BLM expression and rate of cell proliferation. To our knowledge, the expression pattern of FANCJ in human tissues has not been reported. Such an analysis and comparison to BLM may be informative. Genetic backgrounds of tumors have become highly relevant in personalized medicine to better understand the basis for the resistance of cancer cells to chemotherapeutic DNA damaging treatments and radiation, and potential value of selective small molecule DNA repair inhibitors [Citation78,Citation79].
A study done exclusively with breast cancer cells found that BLM expression not only correlated with high-proliferative breast cancer subtypes but was also indicative of a poor survival rate [Citation80]. This was further substantiated by the observation that low proliferative breast cancer subtypes display lower BLM expression. Remarkably, in the aggressive sub-types of breast cancer cells the localization of BLM changed from the nucleus to the cytoplasm [Citation80], emphasizing the need to factor in the subcellular environmental cues regulating BLM function in different cell types. In addition to its role in breast cancer, BLM mutations [Citation81,Citation82] and overexpression [Citation83] have been associated with ovarian cancer. Interestingly, several studies have shown that functional mutations in FANCJ lead to increased risk of breast cancer [Citation30,Citation84–Citation86] and ovarian cancer [Citation87–Citation89]. Given the destabilizing effect of FANCJ deficiency on BLM proteinin FA-J patient fibroblasts or RNAi-depleted HeLa cells [Citation38], it will be important to assess BLM protein levels in breast cancer and ovarian cancer cell lines. Whether other DNA helicases compensate for the lack of FANCJ and/or BLM in these tumors is an important area of investigation.
BS patients are highly susceptible to the formation of lymphomas [Citation90]. Also, a study done in Fancj-null mice showed increased occurrence of lymphomas [Citation91]. Mouse embryonic fibroblasts derived from these Fancj-null mice exhibited elevated level of microsatellite instability (MSI). Further, the extent of MSI was increased with aphidicolin or HU-induced replication stress in FANCJ-deficient cells [Citation91,Citation92]. Interestingly, an earlier study done with immortalized B-cell lymphoma from BS patient showed increased microsatellite instability [Citation93]. These findings raise the possibility that FANCJ and BLM coordinately control microsatellite instability, particularly in lymphomas.
Functional changes in both BLM and FANCJ were observed in colorectal cancers as well. FANCJ was overexpressed in colorectal tumor cells and this increased expression was linked with greater resistance to treatment with 5-fluorouracil [Citation94], a chemotherapeutic drug that blocks thymidine synthesis leading to replication stress [Citation95]. This resistance to 5-fluorouracil treatment was observed in cells expressing MLH1 but not in cells deficient of MLH1. Additionally, there was an increased correlation between inactivating mutations of BLM and elevated risk of colorectal cancer [Citation96]. It is of interest to determine if FANCJ is overexpressed in MLH1-proficient colorectal cancer cells characterized by BLM mutations. If so, an increased FANCJ-MLH1 interaction may be associated with defective HR [Citation59] and this HR defect could be compounded by the inactivation of BLM. Further work is necessary to understand the complex interaction of BLM and FANCJ in DNA repair that is critical to genome homeostasis and suppression of mutagenesis found in certain cancers.
Various missense or truncating mutations in the coding sequence of the BLM gene are linked to BS, a disease characterized by pronounced disposition to cancers of all types [Citation90]. Notably, certain missense mutations linked to BS are located within or immediately downstream of the conserved helicase core domain [Citation97]. Such missense mutations in BLM cause partial or total loss of helicase function by attenuating DNA binding or its ATPase activity [Citation98]. Loss of BLM catalytic activity was shown to negatively affect its ability to resolve HR structural DNA intermediates [Citation98]. Consistent with these findings, the ATP-dependent branch-migration activity of BLM is required for resolution of HJs, a key intermediate in HR repair [Citation12,Citation14,Citation99,Citation100]. As is the case for BLM helicase inactivating mutations, defects in one or several pathways of HR-dependent DNA repair can cause genomic instability and contribute to the pathophysiology of and susceptibility to cancer [Citation101]. Ashkenazi Jews are a population of people with high occurrence of BS [Citation102] and suffer from a loss of function mutation in BLM referred to as blmAsh [Citation103]. These blmAsh patients have deletion of 6 nt and insertion of 7 nt in BLM causing frameshift and truncated mutations [Citation2,Citation103] that in-turn can increase their risk of developing certain cancers [Citation104,Citation105].
FA patients usually have congenital defects, develop bone marrow failure and are predisposed to cancer, particularly acute myeloid leukemia and head and neck cancers [Citation106]. In contrast to FA patients, individuals with BS display all types of cancers, especially leukemia, lymphoma and gastrointestinal tract tumors [Citation107]. Mutations in multiple FA genes are associated with the development of FA [Citation108]. Mutation in FANCJ accounts for 2 percent of the total FA patients [Citation109]. In a recent paper from our lab, we characterized two helicase core domain missense mutations linked to FA corresponding to amino acid substitutions H396D or R707C in FANCJ, and determined that both mutations compromised ATP hydrolysis, DNA binding, and helicase activity of the FANCJ mutant recombinant proteins [Citation76]. Moreover, neither mutant allele expressed in fancj-/- cells conferred resistance to agents that induce ICLs, a critical lesion thought to underlie the molecular pathology of FA [Citation76]. The aforementioned mutations in FANCJ led to accumulation of single-stranded DNA and RAD51 foci in cells treated with the crosslinking agent cisplatin [Citation76] and may eventually affect HR repair of DNA damage [Citation29]. In this work, we also determined that only partial catalytic activity of FANCJ-R707C was sufficient to overcome replication stress caused by the DNA polymerase inhibitor aphidicolin or DNA damage caused by the strand breaker bleomycin [Citation76]. The fact that FANCJ-R707C is a disease-causing mutation linked to FA suggests that DNA damage specific to ICL-inducing agents underlie the molecular pathology of FA.
While BS and FA are distinct genetic disorders, they both share increased cancer occurrences [Citation29,Citation110] and growth retardation/failure [Citation111,Citation112]. Indeed, multiple studies have established important protein interactions and pathway intersection between BS and FA [Citation36,Citation37,Citation113–Citation116]. FA-J cells exposed to ICL-inducing agents display increased chromosomal abnormalities including radial structures [Citation32,Citation35], as observed in BS [Citation117]. Certainly, shared or partially overlapping functions of BLM and FANCJ helicases at the molecular and cellular levels are apparent. Moreover, the catalytic functions and protein interactions of BLM and FANCJ are important for their roles in regulating recombinational repair, suggesting that the balancing act to preserve the genome involves multiple talents and performers, akin to a traveling circus. Further analyses are clearly required, and their importance is emphasized by the still poorly understood molecular deficiencies underlying hereditary helicase disorders and cancers characterized by chromosomal instability.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Tripathi V, Agarwal H, Priya S, et al. MRN complex-dependent recruitment of ubiquitylated BLM helicase to DSBs negatively regulates DNA repair pathways. Nat Commun. 2018 Mar 9;9(1):1016. PubMed PMID: 29523790; PubMed Central PMCID: PMCPMC5844875.
- Ellis NA, Groden J, Ye TZ, et al. The Bloom’s syndrome gene product is homologous to RecQ helicases. Cell. 1995 Nov 17;83(4):655–666. PubMed PMID: 7585968
- German J. Bloom syndrome: a mendelian prototype of somatic mutational disease. Medicine. 1993 Nov;72(6):393–406. PubMed PMID: 8231788.
- Bohm S, Bernstein KA. The role of post-translational modifications in fine-tuning BLM helicase function during DNA repair. DNA Repair. 2014 Oct;22:123–132. PubMed PMID: 25150915; PubMed Central PMCID: PMCPMC4175148.
- Brosh RM. DNA helicases involved in DNA repair and their roles in cancer. Nat Rev Cancer. 2013 07 11;13(8):542–558. . PubMed PMID: PMC4538698.
- Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. . PubMed PMID: 20192759; PubMed Central PMCID: PMCPMC3079308.
- McVey M, Lee SE. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet. 2008 Nov;24(11):529–538. . PubMed PMID: 18809224; PubMed Central PMCID: PMCPMC5303623.
- Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol. 2008 Apr;9(4):297–308. . PubMed PMID: 18285803.
- Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012 Aug 24;47(4):497–510. PubMed PMID: 22920291.
- Ceccaldi R, Rondinelli B, D’Andrea AD. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016 Jan;26(1):52–64. PubMed PMID: 26437586; PubMed Central PMCID: PMCPMC4862604.
- Cheok CF, Bachrati CZ, Chan KL, et al. Roles of the Bloom’s syndrome helicase in the maintenance of genome stability. Biochem Soc Trans. 2005 Dec;33(6):1456–1459. PubMed PMID: 16246145.
- Karow JK, Constantinou A, Li JL, et al. The Bloom’s syndrome gene product promotes branch migration of holliday junctions. Proc Natl Acad Sci U S A. 2000 Jun 6;97(12):6504–6508. PubMed PMID: 10823897; PubMed Central PMCID: PMCPMC18638.
- Bachrati CZ, Borts RH, Hickson ID. Mobile D-loops are a preferred substrate for the Bloom’s syndrome helicase. Nucleic Acids Res. 2006;34(8):2269–2279. PubMed PMID: 16670433; PubMed Central PMCID: PMCPMC1456333.
- Wu L, Hickson ID. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature. 2003 Dec 18;426(6968):870–874. PubMed PMID: 14685245.
- Nimonkar AV, Genschel J, Kinoshita E, et al. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011 Feb 15;25(4):350–362. PubMed PMID: 21325134; PubMed Central PMCID: PMCPMC3042158.
- Gravel S, Chapman JR, Magill C, et al. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008 Oct 15;22(20):2767–2772. PubMed PMID: 18923075; PubMed Central PMCID: PMCPMC2569880.
- Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008 Oct 9;455(7214):770–774. PubMed PMID: 18806779; PubMed Central PMCID: PMCPMC3818707.
- Zhu Z, Chung WH, Shim EY, et al. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008 Sep 19;134(6):981–994. PubMed PMID: 18805091; PubMed Central PMCID: PMCPMC2662516.
- Kumar R, Cheok CF. RIF1: a novel regulatory factor for DNA replication and DNA damage response signaling. DNA Repair. 2014 Mar;15:54–59. PubMed PMID: 24462468.
- Grabarz A, Guirouilh-Barbat J, Barascu A, et al. A role for BLM in double-strand break repair pathway choice: prevention of CtIP/Mre11-mediated alternative nonhomologous end-joining. Cell Rep. 2013 Oct 17;5(1):21–28. PubMed PMID: 24095737.
- Shibata A, Conrad S, Birraux J, et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011 Mar 16;30(6):1079–1092. PubMed PMID: 21317870; PubMed Central PMCID: PMCPMC3061033.
- Sfeir A, de Lange T. Removal of shelterin reveals the telomere end-protection problem. Science. 2012 May 4;336(6081):593–597. PubMed PMID: 22556254; PubMed Central PMCID: PMCPMC3477646.
- Zimmermann M, Lottersberger F, Buonomo SB, et al. 53BP1 regulates DSB repair using Rif1 to control 5ʹ end resection. Science. 2013 Feb 8;339(6120):700–704. PubMed PMID: 23306437; PubMed Central PMCID: PMCPMC3664841.
- Misenko SM, Patel DS, Her J, et al. DNA repair and cell cycle checkpoint defects in a mouse model of ‘BRCAness’ are partially rescued by 53BP1 deletion. Cell Cycle. 2018 May 15:1–11. DOI:10.1080/15384101.2018.1456295. PubMed PMID: 29620483.
- Tripathi V, Nagarjuna T, Sengupta S. BLM helicase-dependent and -independent roles of 53BP1 during replication stress-mediated homologous recombination. J Cell Biol. 2007 Jul 2;178(1):9–14. . PubMed PMID: 17591918; PubMed Central PMCID: PMCPMC2064412.
- Tripathi V, Kaur S, Sengupta S. Phosphorylation-dependent interactions of BLM and 53BP1 are required for their anti-recombinogenic roles during homologous recombination. Carcinogenesis. 2008 Jan;29(1):52–61. . PubMed PMID: 17984114; PubMed Central PMCID: PMCPMC2365705.
- Bunting SF, Callen E, Wong N, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010 Apr 16;141(2):243–254. PubMed PMID: 20362325; PubMed Central PMCID: PMCPMC2857570.
- Cantor SB, Nayak S. FANCJ at the FORK. Mutat Res. 2016 Jun;788:7–11. PubMed PMID: 26926912; PubMed Central PMCID: PMCPMC4887407.
- Brosh RM Jr., Cantor SB. Molecular and cellular functions of the FANCJ DNA helicase defective in cancer and in Fanconi anemia. Front Genet. 2014;5:372. PubMed PMID: 25374583; PubMed Central PMCID: PMCPMC4204437.
- Cantor SB, Bell DW, Ganesan S, et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell. 2001 Apr 6;105(1):149–160. PubMed PMID: 11301010
- Wu Y, Brosh RM Jr. FANCJ helicase operates in the Fanconi Anemia DNA repair pathway and the response to replicational stress. Curr Mol Med. 2009 May;9(4):470–482. PubMed PMID: 19519404; PubMed Central PMCID: PMCPMC2763586.
- Litman R, Peng M, Jin Z, et al. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell. 2005 Sep;8(3):255–265. PubMed PMID: 16153896.
- Levran O, Attwooll C, Henry RT, et al. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nat Genet. 2005 Sep;37(9):931–933. PubMed PMID: 16116424.
- Levitus M, Waisfisz Q, Godthelp BC, et al. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group. J. Nat Genetics. 2005 Sep;37(9):934–935. PubMed PMID: 16116423.
- Bridge WL, Vandenberg CJ, Franklin RJ, et al. The BRIP1 helicase functions independently of BRCA1 in the Fanconi anemia pathway for DNA crosslink repair. Nat Genet. 2005 Sep;37(9):953–957. PubMed PMID: 16116421.
- Deans AJ, West SC. FANCM connects the genome instability disorders Bloom’s Syndrome and Fanconi Anemia. Mol Cell. 2009 Dec 25;36(6):943–953. . PubMed PMID: 20064461.
- Meetei AR, Sechi S, Wallisch M, et al. A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol Cell Biol. 2003 May;23(10):3417–3426. PubMed PMID: 12724401; PubMed Central PMCID: PMCPMC164758.
- Suhasini AN, Rawtani NA, Wu Y, et al. Interaction between the helicases genetically linked to Fanconi anemia group J and Bloom’s syndrome. EMBO J. 2011 Feb 16;30(4):692–705. PubMed PMID: 21240188; PubMed Central PMCID: PMCPMC3041957.
- Wu W, Togashi Y, Johmura Y, et al. HP1 regulates the localization of FANCJ at sites of DNA double-strand breaks. Cancer Sci. 2016 Oct;107(10):1406–1415. PubMed PMID: 27399284; PubMed Central PMCID: PMCPMC5084677.
- Suhasini AN, Sommers JA, Muniandy PA, et al. Fanconi anemia group J helicase and MRE11 nuclease interact to facilitate the DNA damage response. Mol Cell Biol. 2013 Jun;33(11):2212–2227. PubMed PMID: 23530059; PubMed Central PMCID: PMCPMC3648079.
- Dohrn L, Salles D, Siehler SY, et al. BRCA1-mediated repression of mutagenic end-joining of DNA double-strand breaks requires complex formation with BACH1. Biochem J. 2012 Feb 1;441(3):919–926. PubMed PMID: 22032289.
- Petermann E, Orta ML, Issaeva N, et al. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell. 2010 Feb 26;37(4):492–502. PubMed PMID: PMC2958316.
- Syed A, Tainer JA. The MRE11-RAD50-NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu Rev Biochem. 2018 Apr 25;87:263–294. PubMed PMID: 29709199.
- Krejci L, Altmannova V, Spirek M, et al. Homologous recombination and its regulation. Nucleic Acids Res. 2012 July 1;40(13):5795–5818. PubMed PMID: PMC3401455.
- Gupta R, Sharma S, Sommers JA, et al. FANCJ (BACH1) helicase forms DNA damage inducible foci with replication protein A and interacts physically and functionally with the single-stranded DNA-binding protein. Blood. 2007 Oct 1;110(7):2390–2398. PubMed PMID: 17596542; PubMed Central PMCID: PMCPMC1988918.
- Xie J, Peng M, Guillemette S, et al. FANCJ/BACH1 acetylation at lysine 1249 regulates the DNA damage response. PLoS Genet. 2012 Jul;8(7):e1002786. PubMed PMID: 22792074; PubMed Central PMCID: PMCPMC3390368.
- Candelli A, Holthausen JT, Depken M, et al. Visualization and quantification of nascent RAD51 filament formation at single-monomer resolution. Proc Natl Acad Sci U S A. 2014 Oct 21;111(42):15090–15095. PubMed PMID: 25288749; PubMed Central PMCID: PMCPMC4210327.
- Bugreev DV, Yu X, Egelman EH, et al. Novel pro- and anti-recombination activities of the Bloom’s syndrome helicase. Genes Dev. 2007 Dec 01;21(23):3085–3094. PubMed PMID: 18003860; PubMed Central PMCID: PMCPMC2081975.
- Patel DS, Misenko SM, Her J, et al. BLM helicase regulates DNA repair by counteracting RAD51 loading at DNA double-strand break sites. J Cell Biol. 2017 Nov 6;216(11):3521–3534. PubMed PMID: 28912125; PubMed Central PMCID: PMCPMC5674892.
- Sommers JA, Rawtani N, Gupta R, et al. FANCJ uses its motor ATPase to destabilize protein-DNA complexes, unwind triplexes, and inhibit RAD51 strand exchange. J Biol Chem. 2009 Mar 20;284(12):7505–7517. PubMed PMID: 19150983; PubMed Central PMCID: PMCPMC2658046.
- Wang Y, Cortez D, Yazdi P, et al. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000 Apr 15;14(8):927–939. PubMed PMID: 10783165; PubMed Central PMCID: PMCPMC316544
- Chen L, Nievera CJ, Lee AY, et al. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J Biol Chem. 2008 Mar 21;283(12):7713–7720. PubMed PMID: 18171670.
- Schlegel BP, Jodelka FM, Nunez R. BRCA1 promotes induction of ssDNA by ionizing radiation. Cancer Res. 2006 May 15;66(10):5181–5189. PubMed PMID: 16707442.
- Bhattacharyya A, Ear US, Koller BH, et al. The breast cancer susceptibility gene BRCA1 Is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross-linking agent cisplatin. J Biol Chem. 2000 August 4;275(31):23899–23903. PubMed PMID: 10843985.
- Xu X, Qiao W, Linke SP, et al. Genetic interactions between tumor suppressors Brca1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat Genet. 2001 07 01 online;28:266–271. PubMed PMID: 11431698.
- Peng M, Litman R, Jin Z, et al. BACH1 is a DNA repair protein supporting BRCA1 damage response. Oncogene. 2006 Apr 6;25(15):2245–2253. PubMed PMID: 16462773.
- Greenberg RA, Sobhian B, Pathania S, et al. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006 Jan 1;20(1):34–46. PubMed PMID: 16391231; PubMed Central PMCID: PMCPMC1356099.
- Yu X, Chini CC, He M, et al. The BRCT domain is a phospho-protein binding domain. Science. 2003 Oct 24;302(5645):639–642. PubMed PMID: 14576433.
- Xie J, Litman R, Wang S, et al. Targeting the FANCJ-BRCA1 interaction promotes a switch from recombination to poleta-dependent bypass. Oncogene. 2010 Apr 29;29(17):2499–2508. PubMed PMID: 20173781; PubMed Central PMCID: PMCPMC2909592.
- Cheok CF, Wu L, Garcia PL, et al. The Bloom’s syndrome helicase promotes the annealing of complementary single-stranded DNA. Nucleic Acids Res. 2005;33(12):3932–3941. . PubMed PMID: 16024743; PubMed Central PMCID: PMCPMC1176015.
- Machwe A, Xiao L, Groden J, et al. RecQ family members combine strand pairing and unwinding activities to catalyze strand exchange. J Biol Chem. 2005 Jun 17;280(24):23397–23407. PubMed PMID: 15845538.
- Gupta R, Sharma S, Sommers JA, et al. Analysis of the DNA substrate specificity of the human BACH1 helicase associated with breast cancer. J Biol Chem. 2005 Jul 08;280(27):25450–25460. PubMed PMID: 15878853.
- Moder M, Velimezi G, Owusu M, et al. Parallel genome-wide screens identify synthetic viable interactions between the BLM helicase complex and Fanconi anemia. Nat Commun. 2017 Nov 1;8(1):1238. PubMed PMID: 29089570; PubMed Central PMCID: PMCPMC5663702.
- Davies SL, North PS, Dart A, et al. Phosphorylation of the Bloom’s syndrome helicase and its role in recovery from S-phase arrest. Mol Cell Biol. 2004 Feb;24(3):1279–1291. PubMed PMID: 14729972; PubMed Central PMCID: PMCPMC321429.
- Wu Y, Shin-Ya K, Brosh RM Jr. FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol Cell Biol. 2008 Jun;28(12):4116–4128. . PubMed PMID: 18426915; PubMed Central PMCID: PMCPMC2423121.
- London TB, Barber LJ, Mosedale G, et al. FANCJ is a structure-specific DNA helicase associated with the maintenance of genomic G/C tracts. J Biol Chem. 2008 Dec 26;283(52):36132–36139. PubMed PMID: 18978354; PubMed Central PMCID: PMCPMC2662291.
- Sun H, Karow JK, Hickson ID, et al. The Bloom’s syndrome helicase unwinds G4 DNA. J Biol Chem. 1998 Oct 16;273(42):27587–27592. PubMed PMID: 9765292
- Wu WQ, Hou XM, Li M, et al. BLM unfolds G-quadruplexes in different structural environments through different mechanisms. Nucleic Acids Res. 2015 May 19;43(9):4614–4626. PubMed PMID: 25897130; PubMed Central PMCID: PMCPMC4482088.
- Bochman ML, Paeschke K, Zakian VA. DNA secondary structures: stability and function of G-quadruplex structures. Nat Rev Genet. 2012 Nov;13(11):770–780. . PubMed PMID: 23032257; PubMed Central PMCID: PMCPMC3725559.
- Mendoza O, Bourdoncle A, J-B B, et al. G-quadruplexes and helicases. Nucleic Acids Res. 2016 Mar 18;44(5):1989–2006. PubMed PMID: PMC4797304.
- Drosopoulos WC, Kosiyatrakul ST, Schildkraut CL. BLM helicase facilitates telomere replication during leading strand synthesis of telomeres. J Cell Biol. 2015 Jul 20;210(2):191–208. . PubMed PMID: 26195664; PubMed Central PMCID: PMCPMC4508891.
- Henderson A, Wu Y, Huang YC, et al. Detection of G-quadruplex DNA in mammalian cells. Nucleic Acids Res. 2014 Jan;42(2):860–869. PubMed PMID: 24163102; PubMed Central PMCID: PMCPMC3902944.
- van Wietmarschen N, Merzouk S, Halsema N, et al. BLM helicase suppresses recombination at G-quadruplex motifs in transcribed genes. Nat Commun. 2018 Jan 18;9(1):271. PubMed PMID: 29348659; PubMed Central PMCID: PMCPMC5773480.
- Sarkies P, Murat P, Phillips LG, et al. FANCJ coordinates two pathways that maintain epigenetic stability at G-quadruplex DNA. Nucleic Acids Res. 2012 Feb;40(4):1485–1498. PubMed PMID: 22021381; PubMed Central PMCID: PMCPMC3287192.
- Bharti SK, Awate S, Banerjee T, et al. Getting ready for the dance: FANCJ irons out DNA wrinkles. Genes. 2016 Jul 1;7(7):31. PubMed PMID: 27376332; PubMed Central PMCID: PMCPMC4962001.
- Bharti SK, Sommers JA, Awate S, et al. A minimal threshold of FANCJ helicase activity is required for its response to replication stress or double-strand break repair. Nucleic Acids Res. 2018 Jul 6; 46:6238-6256. PubMed PMID: 29788478.
- Turley H, Wu L, Canamero M, et al. The distribution and expression of the Bloom’s syndrome gene product in normal and neoplastic human cells. Br J Cancer. 2001 Jul 20;85(2):261–265. PubMed PMID: 11461087; PubMed Central PMCID: PMCPMC2364038.
- Hengel SR, Spies MA, Spies M. Small-molecule inhibitors targeting DNA repair and DNA repair deficiency in research and cancer therapy. Cell Chemical Biology. 2017 Sep 21;24(9):1101–1119. PubMed PMID: 28938088; PubMed Central PMCID: PMCPMC5679738.
- Velic D, Couturier AM, Ferreira MT, et al. DNA damage signalling and repair inhibitors: the long-sought-after achilles’ heel of cancer. Biomolecules. 2015 Nov 20;5(4):3204–3259. PubMed PMID: 26610585; PubMed Central PMCID: PMCPMC4693276.
- Arora A, Abdel-Fatah TM, Agarwal D, et al. Transcriptomic and protein expression analysis reveals clinicopathological significance of bloom syndrome helicase (BLM) in breast cancer. Mol Cancer Ther. 2015 Apr;14(4):1057–1065. PubMed PMID: 25673821.
- Panneerselvam J, Wang H, Zhang J, et al. BLM promotes the activation of Fanconi Anemia signaling pathway. Oncotarget. 2016 May 31;7(22):32351–32361. PubMed PMID: PMC5078018.
- Kanchi KL, Johnson KJ, Lu C, et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nat Commun. 2014;5:3156. . PubMed PMID: 24448499; PubMed Central PMCID: PMCPMC4025965.
- Birkbak NJ, Li Y, Pathania S, et al. Overexpression of BLM promotes DNA damage and increased sensitivity to platinum salts in triple-negative breast and serous ovarian cancers. Ann Oncol. 2018;29(4):903–909. PubMed PMID: 2945233.
- De Nicolo A, Tancredi M, Lombardi G, et al. A novel breast cancer-associated BRIP1 (FANCJ/BACH1) germ-line mutation impairs protein stability and function. Clin Cancer Res. 2008 Jul 15;14(14):4672–4680. PubMed PMID: 18628483; PubMed Central PMCID: PMCPMC2561321.
- Seal S, Thompson D, Renwick A, et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet. 2006 Nov;38(11):1239–1241. PubMed PMID: 17033622.
- Cantor S, Drapkin R, Zhang F, et al. The BRCA1-associated protein BACH1 is a DNA helicase targeted by clinically relevant inactivating mutations. Proc Natl Acad Sci U S A. 2004 Feb 24;101(8):2357–2362. PubMed PMID: 14983014; PubMed Central PMCID: PMCPMC356955
- Ramus SJ, Song H, Dicks E, et al. Germline mutations in the BRIP1, BARD1, PALB2, and NBN genes in women with ovarian cancer. J Natl Cancer Inst. 2015 Nov;107(11). DOI:10.1093/jnci/djv214. PubMed PMID: 26315354; PubMed Central PMCID: PMCPMC4643629.
- Rafnar T, Gudbjartsson DF, Sulem P, et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat Genet. 2011 10 02 online;43:1104. PubMed PMID: 21964575.
- Weber-Lassalle N, Hauke J, Ramser J, et al. BRIP1 loss-of-function mutations confer high risk for familial ovarian cancer, but not familial breast cancer. Breast Cancer Res. 2018 Jan 24;20(1):7. PubMed PMID: 29368626; PubMed Central PMCID: PMCPMC5784717.
- German J. Bloom’s syndrome. XX. The first 100 cancers. Cancer Genet Cytogenet. 1997 01 01;93(1):100–106. PubMed PMID: 9062585.
- Matsuzaki K, Borel V, Adelman CA, et al. FANCJ suppresses microsatellite instability and lymphomagenesis independent of the Fanconi anemia pathway. Genes & Development.. 2015;29(24):2532–2546. PubMed PMID: 26637282.
- Barthelemy J, Hanenberg H, Leffak M. FANCJ is essential to maintain microsatellite structure genome-wide during replication stress. Nucleic Acids Res. 2016 Aug 19;44(14):6803–6816. . PubMed PMID: 27179029; PubMed Central PMCID: PMCPMC5001596.
- Kaneko H, Inoue R, Yamada Y, et al. Microsatellite instability in B-cell lymphoma originating from Bloom syndrome. Int J Cancer. 1996 Dec 20;69(6):480–483. PubMed PMID: 8980251.
- Nakanishi R, Kitao H, Fujinaka Y, et al. FANCJ expression predicts the response to 5-fluorouracil-based chemotherapy in MLH1-proficient colorectal cancer. Ann Surg Oncol. 2012 Oct;19(11):3627–3635. PubMed PMID: 22526901.
- Longley DB, Harkin DP, Johnston PG. 5-Fluorouracil: mechanisms of action and clinical strategies [Review Article]. Nat Rev Cancer. 2003 May 01;3:330–338. PubMed PMID: 12724731.
- Gruber SB, Ellis NA, Scott KK, et al. BLM heterozygosity and the risk of colorectal cancer. Science. 2002 Sep 20;297(5589):2013. PubMed PMID: 12242432.
- German J, Sanz MM, Ciocci S, et al. Syndrome-causing mutations of the BLM gene in persons in the Bloom’s Syndrome Registry. Hum Mutat. 2007 Aug;28(8):743–753. PubMed PMID: 17407155.
- Guo RB, Rigolet P, Ren H, et al. Structural and functional analyses of disease-causing missense mutations in Bloom syndrome protein. Nucleic Acids Res. 2007;35(18):6297–6310. PubMed PMID: 17878217; PubMed Central PMCID: PMCPMC2094094.
- Xu YN, Bazeille N, Ding XY, et al. Multimeric BLM is dissociated upon ATP hydrolysis and functions as monomers in resolving DNA structures. Nucleic Acids Res. 2012 Oct;40(19):9802–9814. PubMed PMID: 22885301; PubMed Central PMCID: PMCPMC3479192.
- Huber MD, Lee DC, Maizels N. G4 DNA unwinding by BLM and Sgs1p: substrate specificity and substrate-specific inhibition. Nucleic Acids Res. 2002 Sep 15;30(18):3954–3961. PubMed PMID: 12235379; PubMed Central PMCID: PMCPMC137114.
- Helleday T. Homologous recombination in cancer development, treatment and development of drug resistance. Carcinogenesis. 2010;31(6):955–960. PubMed PMID: 20351092.
- Shahrabani-Gargir L, Shomrat R, Yaron Y, et al. High frequency of a common Bloom syndrome Ashkenazi mutation among Jews of Polish origin. Genet Test. 1998;2(4):293–296. . PubMed PMID: 10464606.
- Li L, Eng C, Desnick RJ, et al. Carrier frequency of the Bloom syndrome blmAsh mutation in the Ashkenazi Jewish population. Mol Genet Metab. 1998 Aug;64(4):286–290. PubMed PMID: 9758720.
- de Voer RM, Hahn MM, Mensenkamp AR, et al. Deleterious Germline BLM Mutations and the Risk for Early-onset Colorectal Cancer. Sci Rep. 2015 Sep 11;5:14060. . PubMed PMID: 26358404; PubMed Central PMCID: PMCPMC4566092.
- Schayek H, Laitman Y, Katz LH, et al. Colorectal and endometrial cancer risk and age at diagnosis in BLMAsh mutation carriers. Isr Med Assoc Journal: IMAJ. 2017 Jun;19(6):365–367. PubMed PMID: 28647934.
- Kee Y, D’Andrea AD. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 2010 Aug 15;24(16):1680–1694. . PubMed PMID: 20713514; PubMed Central PMCID: PMCPMC2922498.
- National Organization For Rare Disorders. Rare disease information/bloom syndrome [ cited 2018 24 August]. Available from: https://rarediseases.org/rare-diseases/bloom-syndrome/
- Tischkowitz MD, Hodgson SV. Fanconi anaemia. J Med Genet. 2003 Jan;40(1):1–10. PubMed PMID: 12525534; PubMed Central PMCID: PMCPMC1735271.
- Wang AT, Smogorzewska A. SnapShot: fanconi anemia and associated proteins. Cell. 2015 Jan 15;160(1–2):354–354.e1. PubMed PMID: 25594185.
- Cunniff C, Bassetti JA, Ellis NA. Bloom’s syndrome: clinical spectrum, molecular pathogenesis, and cancer predisposition. Mol Syndromol. 2017;8(1):4–23. PubMed PMID: 28232778.
- Bloom D. The syndrome of congenital telangiectatic erythema and stunted growth. J Pediatr. 1966 Jan;68(1):103–113. PubMed PMID: 5901336.
- Dasi-Carpio A, Casado JA, Sanz G, et al. Clinical characteristics of patients with fanconi anemia in complementation Group J. Blood.2006;108(11):3769.
- Ling C, Huang J, Yan Z, et al. Bloom syndrome complex promotes FANCM recruitment to stalled replication forks and facilitates both repair and traverse of DNA interstrand crosslinks. Cell Discovery. 2016;2:16047. . PubMed PMID: 28058110; PubMed Central PMCID: PMCPMC5167996.
- Chaudhury I, Sareen A, Raghunandan M, et al. FANCD2 regulates BLM complex functions independently of FANCI to promote replication fork recovery. Nucleic Acids Res. 2013 Jul;41(13):6444–6459. PubMed PMID: 23658231; PubMed Central PMCID: PMCPMC3711430.
- Hoadley KA, Xue Y, Ling C, et al. Defining the molecular interface that connects the Fanconi anemia protein FANCM to the Bloom syndrome dissolvasome. Proc Natl Acad Sci U S A. 2012 Mar 20;109(12):4437–4442. PubMed PMID: 22392978; PubMed Central PMCID: PMCPMC3311393.
- Suhasini AN, Brosh RM Jr. Fanconi anemia and Bloom’s syndrome crosstalk through FANCJ-BLM helicase interaction. Trends Genet. 2012 Jan;28(1):7–13. . PubMed PMID: 22024395; PubMed Central PMCID: PMCPMC3249464.
- van Brabant AJ, Stan R, Ellis NA. DNA helicases, genomic instability, and human genetic disease. Annu Rev Genomics Hum Genet. 2000;1:409–459. . PubMed PMID: 11701636.
- Rudolf J, Makrantoni V, Ingledew WJ, et al. The DNA repair helicases XPD and FancJ have essential iron-sulfur domains. Mol Cell. 2006 Sep 15;23(6):801–808. PubMed PMID: 16973432.
- Wu Y, Sommers JA, Suhasini AN, et al. Fanconi anemia group J mutation abolishes its DNA repair function by uncoupling DNA translocation from helicase activity or disruption of protein-DNA complexes. Blood. 2010 Nov 11;116(19):3780–3791. PubMed PMID: 20639400; PubMed Central PMCID: PMCPMC2981534.
- Karow JK, Chakraverty RK, Hickson ID. The Bloom’s syndrome gene product is a 3ʹ-5ʹ DNA helicase. J Biol Chem. 1997 Dec 5;272(49):30611–30614. PubMed PMID: 9388193.
- Popuri V, Bachrati CZ, Muzzolini L, et al. The human RecQ Helicases, BLM and RECQ1, display distinct DNA substrate specificities. J Biol Chem. 2008 June 27;283(26):17766–17776. PubMed PMID: 18448429.
- van Brabant AJ, Ye T, Sanz M, et al. Binding and melting of D-loops by the Bloom syndrome helicase. Biochemistry. 2000 Nov 28;39(47):14617–14625. PubMed PMID: 11087418
- Ralf C, Hickson ID, Wu L. The Bloom’s syndrome helicase can promote the regression of a model replication fork. J Biol Chem. 2006 Aug 11;281(32):22839–22846. . PubMed PMID: 16766518.
- Machwe A, Xiao L, Groden J, et al. The Werner and Bloom syndrome proteins catalyze regression of a model replication fork. Biochemistry. 2006 Nov 28;45(47):13939–13946. PubMed PMID: 17115688.
- Sharma S, Sommers JA, Wu L, et al. Stimulation of flap endonuclease-1 by the Bloom’s syndrome protein. J Biol Chem. 2004 Mar 12;279(11):9847–9856. PubMed PMID: 14688284.
- Bharti SK, Sommers JA, George F, et al. Specialization among iron-sulfur cluster helicases to resolve g-quadruplex DNA structures that threaten genomic stability. J Biol Chem. 2013 September 27;288(39):28217–28229. PubMed PMID: 23935105.
- Brosh RM Jr., Li JL, Kenny MK, et al. Replication protein A physically interacts with the Bloom’s syndrome protein and stimulates its helicase activity. J Biol Chem. 2000 Aug 4;275(31):23500–23508. PubMed PMID: 10825162.
- Doherty KM, Sommers JA, Gray MD, et al. Physical and functional mapping of the replication protein a interaction domain of the werner and bloom syndrome helicases. J Biol Chem. 2005 Aug 19;280(33):29494–29505. PubMed PMID: 15965237.
- Wu L, Davies SL, North PS, et al. The Bloom’s syndrome gene product interacts with topoisomerase III. J Biol Chem. 2000 Mar 31;275(13):9636–9644. PubMed PMID: 10734115
- Johnson FB, Lombard DB, Neff NF, et al. Association of the Bloom syndrome protein with topoisomerase IIIalpha in somatic and meiotic cells. Cancer Res. 2000 Mar 1;60(5):1162–1167. PubMed PMID: 10728666
- Sharma S, Sommers JA, Gary RK, et al. The interaction site of Flap Endonuclease-1 with WRN helicase suggests a coordination of WRN and PCNA. Nucleic Acids Res. 2005;33(21):6769–6781. . PubMed PMID: 16326861; PubMed Central PMCID: PMCPMC1301591.
- Wu L, Davies SL, Levitt NC, et al. Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51. J Biol Chem. 2001 Jun 1;276(22):19375–19381. PubMed PMID: 11278509.
- Nimonkar AV, Ozsoy AZ, Genschel J, et al. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc Natl Acad Sci U S A. 2008 Nov 4;105(44):16906–16911. PubMed PMID: 18971343; PubMed Central PMCID: PMCPMC2579351.
- Gong Z, Kim JE, Leung CC, et al. BACH1/FANCJ acts with TopBP1 and participates early in DNA replication checkpoint control. Mol Cell. 2010 Feb 12;37(3):438–446. PubMed PMID: 20159562; PubMed Central PMCID: PMCPMC3695484.
- Peng M, Litman R, Xie J, et al. The FANCJ/MutLalpha interaction is required for correction of the cross-link response in FA-J cells. EMBO J. 2007 Jul 11;26(13):3238–3249. PubMed PMID: 17581638; PubMed Central PMCID: PMCPMC1914102.
- Traverso G, Bettegowda C, Kraus J, et al. Hyper-recombination and genetic instability in BLM-deficient epithelial cells. Cancer Res. 2003 Dec 15;63(24):8578–8581. PubMed PMID: 14695165