
ABSTRACT
Store-operated Ca2+ entry (SOCE) plays an important role in regulating Ca2+ influx, which participates in tumor cell survival and motility. We aim to elucidate the role of SOCE in the behavior of C6 glioma cells. Lentiviral vector inserted with the Orai1-targeting shRNA was used to inhibit SOCE in C6 glioma cells. The down-regulation of Orai1 was confirmed by western blot. The ability of shOrai1 or SOCE inhibitor (SKF96365) in regulating SOCE inhibition was evaluated by measuring Ca2+ concentration. Additionally, its effect on cell behavior was assessed using methyl thiazolyl tetrazolium (MTT) assay, wound healing assay, transwell assay, and adhesion assay. Focal adhesions were visualized by immunofluorescence assay. Further, the expression of proline-rich tyrosine kinase 2 (Pyk2) and phosphorylated Pyk2 (p-Pyk2) was analyzed using western blot. Both, SKF96365 treatment and the Orai1 down-regulation inhibited SOCE by perturbing Ca2+ influx. The inhibitory effects of shOrai1 on C6 cell proliferation, migration, and invasion were similar to that of SKF96365. Moreover, Orai1 inhibition enhanced C6 cell adhesion by increasing the size of focal adhesion plaques. The down-regulation of Pyk2 was observed in both SKF96365-treated and Orai1-silenced C6 cells. Additionally, Orai1 inhibition blocked AKT/mTOR, NFAT, and NF-κB pathways. The silencing of Orai1 inhibited the C6 glioma cell migration, invasion and contributed to focal adhesion.
Introduction
Glioma, the most common tumor derived from the Central Nervous System (CNS) [Citation1,Citation2], is characterized by rapid growth, malignant invasion, and angiogenesis, resulting in the high recurrence rate of postoperative patients [Citation3,Citation4]. Despite a promising improvement in the malignant glioma patients, where surgery or chemoradiotherapy results in potent anti-tumor effect, the conventional treatment in the clinics for many glioma types only temporarily retards the glioma tumor progression with a lack in curative effect on the prolonged median survival of glioblastoma patients (GBM) [Citation4]. This discrepancy or the glioma recurrence may be partly due to the infiltrative or invasive growth of glioma cells [Citation5,Citation6]. Further, the glioma cell migration in the extracellular matrix (ECM) depends on the focal adhesion [Citation7]. Hence, finding an effective anti-invasive targeting strategy calls for further studies on biological behavior and molecular mechanism of malignant glioma cell adhesion.
Intracellular Ca2+, an important second messenger, is involved in the regulation of physiological and pathophysiological activities, such as metabolism and secretion [Citation8]. Store-operated calcium entry (SOCE), mediated by store-operated Ca2+ channel (SOCC), is an important intracellular Ca2+ flow pathway activated by the interaction of inositol 1,4,5-triphosphate (IP3) and its receptor in the endoplasmic reticulum, leading to depletion of intracellular Ca2+ [Citation9]. SOCE, which is abnormally enhanced in a variety of tumor cells, not only plays an important role in the maintenance of intracellular Ca [Citation2]+ homeostasis but is also closely related to the malignant phenotype of tumor [Citation10]. Thus, SOCE demonstrates a disparate activity where, though, it inhibits glioma cell proliferation and promotes cell apoptosis [Citation11], it might also contribute to glioma cell adhesion activity within the local tumor microenvironment.
Stromal interaction molecule 1 (STIM1) belongs to the family of single transmembrane proteins containing calcium-binding domains [Citation12]. When Ca2+ concentration in the endoplasmic reticulum decreases, STIM1 activates and bonds to another targeting protein of SOCC, known as calcium release-activated calcium channel protein1 (Orai1) [Citation13]. Being a kind of tetraspanin, Orai1 rearranges around the membrane, forming SOCC [Citation14]. In hepatoma carcinoma Huh-7 cells, knockout of Orai1 causes the down-regulation of cyclinD1 expression, thereby inhibiting the cell proliferation [Citation15]. Apart from hepatoma carcinoma cells, Orai1 is also important in breast cancer, as illustrated by the Orai1 knock-down in rats, resulting in abnormal cell adhesion and tumor metastasis [Citation16]. However, a systematic understanding of how the silencing of Orai1 contributes to glioma cell behavior is still lacking.
SKF96365 is an important SOCC inhibitor, which significantly inhibits the SOCE process in platelets and neutrophils [Citation17]. Traditionally, SKF96365 is used for assessing the physiological and pathological effects of SOCE [Citation18,Citation19]. Except for STIM1, another component of SOCE, known as the transient receptor potential canonical 1 (TRPC1), is also described as a crucial targeting molecule of SKF96365 [Citation12]. A report shows that SKF96365 significantly inhibits SOCE process in gastric cancer, thereby inhibiting tumor cell growth [Citation20]. SKF96365 is an effective inhibitor of SOCE. We used SKF96365 as a positive control to explore the effects of silenced Orai1 on C6cell migration, invasion, and adhesion. Our investigation will provide an opportunity to advance the knowledge of SOCE in glioma.
Materials and methods
Cell culture
C6 glioma cell line was obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in DMEM (Gibco, Waltham, MA, USA) with 5% fetal bovine serum (FBS, Gibco) and other added supplements. C6 cells were maintained at 37 °C in humidified air of 5% CO2. For the SKF96365 treatment, firstly, SKF96365 (Sigma-Aldrich, St Louis, MO, USA) was dissolved in sterile deionized water. Then, the cell culture medium containing a final concentration (20 µM) of SKF96365 [Citation21] was prepared and used for cell cultivation.
RNA interference targeting Orai1
The hU6-MCS-CMV-based lentiviral vector with the Orai1 small hairpin RNA (shOrai1, 5©-TGG TCG CCA AGT ACT CGA GTG T-3©) was designed and synthesized by GeneChem (Shanghai, China). Scramble shRNA was used as a control. C6 cells were plated in a 6-well plate and incubated at 37 °C in a 5% CO2 incubator till 80% confluence was achieved. Next, the lentivirus expressing either shOrai1 or shScramble was added to the cell plate (MOI = 5). After culturing them for 2–5 days, lentiviral infection efficiency was determined using Western blot analysis. Further, stable Orai1-silenced C6 cells were selected using puromycin (Solarbio, Beijing, China).
Western blot analysis
C6 cells were lysed in RIPA lysis solution (Solarbio) containing protease and phosphatase inhibitor (phenylmethylsulfonyl fluoride, PMSF, Solarbio). After quantifying the protein, 20 µL of the samples was separated on an SDS-PAGE gel, transferred onto PVDF membranes, and blocked with 5% bovine albumin (BSA, Solarbio) for 1 h at room temperature. The PVDF membranes were then incubated with primary antibodies as follows: rabbit monoclonal anti-Orai1 (ab11196, Abcam, Cambridge, MA, USA, 1:1000), mouse monoclonal anti-proline-rich tyrosine kinase 2 (Pyk2, sc-393,181, Santa Cruz Biotechnology, Santa Cruz, CA, USA, 1:500), mouse monoclonal anti-phosphorylated Pyk2 (p-Pyk2, MAB6210-SP, R&D Systems, Minneapolis, MN, USA, 0.5 µg/mL), rabbit monoclonal anti-p21(ab109520, Abcam, 1:2000), rabbit monoclonal anti-Cyclin D1 (ab40754, Abcam, 1:2000), rabbit monoclonal anti-CDK4 (ab108357, Abcam, 1:2000), rabbit monoclonal anti-E-cadherin (ab133597, Abcam, 1:2000), rabbit polyclonal anti-N-cadherin (ab76057, Abcam, 1:1000), rabbit polyclonal anti-Vimentin (ab137321, Abcam, 1:2000), rabbit polyclonal anti-p-AKT (ab38449, Abcam, 1:1000), rabbit polyclonal anti-p-4EBP1 (ab47365, Abcam, 1:1000), rabbit polyclonal anti-NFAT (ab3447, Abcam, 1:1000), rabbit polyclonal anti-NF-κB (ab16502, Abcam, 0.5 µg/mL), rabbit polyclonal anti-p-S6K (ab59208, Abcam, 1:1000), and rabbit monoclonal anti-GAPDH (Abcam). After 12 h of incubation, the membranes were further incubated for 1 h with HRP-conjugated goat anti-rabbit IgG H&L and HRP-conjugated goat anti-mouse IgG H&L. PVDF membranes were then rinsed with Tris-Buffered Saline Tween-20 (TBST) solution twice and visualized on a ChemiDoc™ XRS system (Bio-Rad, Hercules, CA, USA) using ECL reagent (ThermoFisher, Carlsbad, CA, USA). The protein expression observed in shControl cells was used as a control.
Ca2+ measurement
C6 cells were plated onto the confocal dish at a density of 5 × 103 cells/mL and cultured for 48 h. Then, the cells were incubated with Fluo-4/AM (Becton Dickinson, San Diego, CA, USA) and 0.1% pluronic F-127 (Sigma-Aldrich) for 30 min. The final concentration of Fluo-4/AM was 5 µM in Hank’s Balanced Salt Solution (HBSS, Gibco). The Fluo-4-loaded cells were then rinsed thrice using Ca2+-free HBSS and immobilized on the Petri dish for 10 min. The fluorescence units were measured using a laser scanning confocal microscope (Olympus, Tokyo, Japan) at 485 nm excitation and 525 nm emission. After initially incubating for 3 min, Ca2+ ATPase inhibitor thapsigargin (TG, 5 µM, Sigma-Aldrich) was added to the cells in the Petri dish and maintained in HBSS for 5 min. The change in fluorescence was monitored, which reflected the intracellular Ca2+.
Cell proliferation assay
The proliferation of Orai1-silenced C6 cells was detected using methyl thiazolyl tetrazolium (MTT, Sigma-Aldrich) assay. Orai1-downregulated C6 cells were plated onto a 96-well plate at a density of 5 × 103 cells/well and cultured for 12 h. MTT reagent (20 µL, 5 mg/mL) in 180 µL DMEM medium was then added on the 1st, 2nd, 3rd, 4th, 5th, 6th, and 7th days and further incubated for another 2 h at 37 °C. Next, the supernatant was discarded, and 200 µL of dimethyl sulfoxide (DMSO) was added into each well to dissolve the formed formazan, followed by shaking for 10 min on a constant temperature shaker. Optical density at 570 nm was then detected on a microplate reader (Bio-Rad).
Cell cycle analysis
shOrai1-transfected C6 cells or shScramble-transfected cells were treated with 0.25% trypsin-EDTA solution (Solarbio) and fixed in cold 70% ethanol. Nucleic acids were stained with 100 µg/mL propidium iodide (PI, Thermo fisher) in 2 × SSC (0.3 M NaCl, 0.03 M sodium citrate, pH 7.0) and 100 µg/mL of DNase-free RNase (ThermoFisher). Stained DNA was then analyzed for PI fluorescence using a CytoFLEX flow cytometer (Beckman Coulter, Kraemer Boulevard Brea, CA, USA). Resulting DNA distribution in G0/G1, S, and G2/M phase of the cell cycle was analyzed using CytExpert analysis software (Beckman Coulter).
Wound healing assay
Cell migration was assessed by wound healing assay. C6 cells were seeded in a 6-well plate at a density of 2 × 105 cells/well and incubated for 24 h in 10% FBS containing DMEM. The culture medium was then removed and replaced with 5% FBS-containing DMEM. A wound or a denuded region was created using a 200 µL sterile micropipette tip at a consistent angle with consistent pressure. C6 cells were washed thrice with phosphate buffered saline (PBS, Solarbio) to remove extra cells. PBS was then removed and replaced with medium containing 5% FBS. Migrated cells were monitored for 48 h. The test images of each well, before and after the invasion, were captured by an inverted microscope (ThermoFisher) at a magnification of 40x. Wound closure was re-calculated and expressed as a ratio of the original width.
Cell invasion assay
Invasiveness of C6 cells was detected using a 24-well transwell plate inserted with 8-µm pore size filters. 100 µL of Matrigel (Corning, Corning, NY, USA) was diluted to 1 mg/mL solution using serum-free DMEM, and this solution was added in each of the upper compartments of the transwell plate. After coagulation at 37 °C, C6 cells were seeded on the upper chamber at a density of 5 × 104 cells/well in serum-free DMEM, followed by the addition of 250 µL DMEM containing 5% FBS in the lower chambers. After 16 h of incubation at 37 °C, the cells from the inner wall of the compartments were removed, while the cells from the outer wall of the compartment were washed with PBS and stained with Giemsa (Solarbio) for 10 min. Finally, after air-drying the membrane, the invaded cells were counted under an inverted microscope (ThermoFisher).
Cell adhesion assay
The 96-well plate was first pre-coated with 50 µL of fibronectin (30 µg/mL, Sigma-Aldrich), then washed and blocked with 2% BSA solution for 1 h at room temperature. Next, the cells (5 × 104 per well) were seeded in this fibronectin-coated plate and maintained in FBS-free DMEM. After incubating for 40 min, cells were rinsed twice with cold PBS and then immobilized with 4% paraformaldehyde (Solarbio). The attached cells were finally stained with Giemsa (Solarbio) and counted under an inverted microscope (ThermoFisher).
Immunofluorescence assay
After plating and growing C6 cells in a confocal dish for 24 h in DMEM, the cells were fixed with 4% paraformaldehyde (Solarbio), followed by cell lysis in 1% Triton X-100 for 10 min. BSA (5%) was used to reduce nonspecific background. Then, the cells were incubated in rabbit monoclonal anti-vinculin (Cell signaling technology, CST, Boston, MA, USA) overnight at 4 °C, followed by rinsing with PBS. Subsequently, the samples were incubated with Texas Red-conjugated goat anti-rabbit IgG (H + L) (CST). 4©,6-Diamidino-2-phenylindole (DAPI) stain was used to dye the cell nuclei. Finally, the image was captured using a laser scanning confocal microscope (Olympus).
Statistical analysis
All the results were expressed as the means ± SD of independent experiments in triplicate. Non-parametric statistical analysis was carried out (Analysis of Variance, ANOVA) using GraphPad Software (San Diego, CA, USA). *P < 0.05 and **P < 0.01 are indicated in the figures.
Result
Effect of Orai1 down-regulation on SOCE in C6 glioma cells
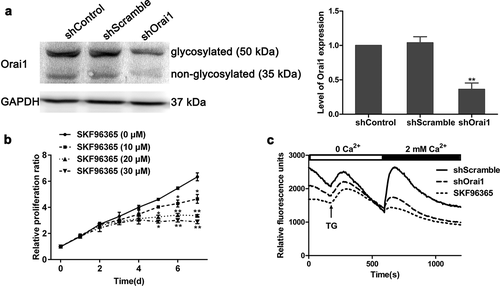
Considering that Orai1 was overexpressed in glioma tissues [Citation21], we constructed shRNA for Orai1 to inhibit its expression in C6 glioma cells. This down-regulated level of Orai1 was determined by western blot analysis. As displayed in ), the non-glycosylated protein level of Orai1 decreased by 64% after 48 h of transfection with shOrai1, compared to the shControl group (P < 0.01), whereas transfection with scramble shRNA practically did not change the expression level of Orai1. To investigate the effect of silenced Orai1 on SOCE in C6 glioma cells, we used SKF96365, a kind of pharmacological SOCE inhibitor [Citation22], as a positive control to measure the intracellular Ca2+ concentration. ) shows that 10–30 μM of SKF96365 significantly inhibited glioma C6 cell proliferation. According to the previous literature [Citation21], we utilized a concentration of 20 μM SKF96365 to carry out the next experiments. ) shows that the down-regulation of Orai1 significantly reduces the baseline level of Ca2+ influx regardless of the presence of Ca2+, compared to the shControl group. Interestingly, shOrai1 had a similar inhibitory effect as SKF96365 on SOCE. This suggests that the down-regulation of Orai1 inhibits SOCE in C6 glioma cells.
Figure 1. Effect of Orai1 suppression on SOCE in C6 glioma cells. (a) The glycosylated and non-glycosylated protein expression of Orai1 was measured in C6 glioma cells transfected with shControl, shScramble or shOrai1 by western blot analysis. Data are shown as means ± SDs from thrice independent experiments. ** P< 0.01 vs. the shControl group. (b) C6 glioma cells were treated with 0–30 μM of SKF96365 and the proliferation ratio was detected by MTT assay. (c) The Ca2+ influx curves, prepared with 25–45 single cells transfected with shOrai1/shScramble or treated with SKF96365 (20 µM) by using Fluo-4/AM-based Ca2+ measurements. TG, thapsigargin
Effect of Orai1 down-regulation on C6 glioma cell proliferation
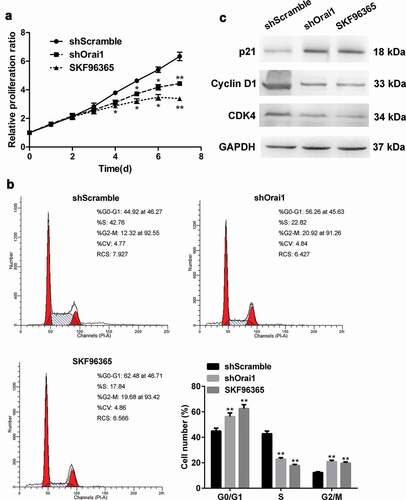
C6 cells were transfected with shOrai1/shScramble or treated with SKF96365 (20 µM). Cell proliferation was detected at day 1–7 using MTT assay. As revealed in ), the relative proliferation ratio of C6 cells did not show remarkable change within the first three days of shOrai1 transfection, but the proliferation ratio presented a significant reduction in the next four days of downregulating Orai1(P < 0.05 or P < 0.01, )). It was also noted that C6 cell proliferation was restrained by SKF96365 treatment (P < 0.05 or P < 0.01, )). The results of cell cycle analysis are shown in ). In the SKF96365-treated group and Orai1-downregulated group, a significant increase in G0/G1 and G2/M phase was detected while there was a reduction in S phase (P < 0.01, )). Next, we analyzed the protein levels of p21, Cyclin D1, and CDK4 by western blot after the shOrai1 transfection. Down-regulation of Orai1 strongly enhanced p21 expression but reduced the expression of Cyclin D1 and CDK4 ()), suggesting that Orai1 expression is needed to maintain cell cycle-related protein expression and the cell cycle progression. These data demonstrate that inhibiting Orai1 suppresses the proliferation of C6 cells.
Figure 2. Effect of Orai1 suppression on C6 glioma cell proliferation. C6 glioma cells were transfected with shScramble/shOrai1 or treated with SKF96365 (20 µM). (a) The relative proliferation ratio of C6 cells was determined by MTT assay. (b) Cell cycle distribution was determined by flow cytometry. (c) Cells were analyzed for expression of p21, Cyclin D1, and CDK4 protein with Western blot. Data are shown as means ± SDs from thrice independent experiments. *P < 0.05 or ** P< 0.01 vs. the shScramble group
Effect of Orai1 down-regulation on C6 glioma cell migration and invasion
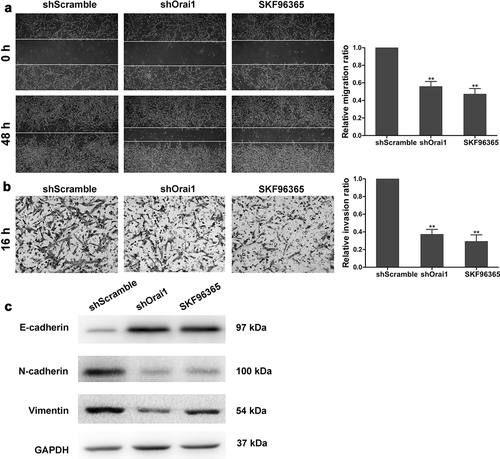
To further clarify the influence of SOCE on C6 glioma cell motility, the wound healing assay and transwell assay were performed to detect the effects of Orai1inhibition on the migration and invasion of C6 cells. The wound of shScramble group healed after 48 h of being wounded, and the relative migration ratio was normalized as 1.0. However, the shOrai1 group and SKF96365 group had only slight signs of healing. Compared to the shScramble group, the relative migratory ratio in the shOrai1 group and the SKF96365 group decreased by 44% and 53%, respectively (P < 0.01, )). Coincidentally, the results from Matrigel-coated transwell assay displayed that both Orai1 inhibition and SKF96365 treatment reduced the invasiveness of C6 cells by 63% and 71%, respectively (P < 0.01, )). Further, western blot analysis showed that Orai1 inhibition significantly increased the expression of E-cadherin, whereas an opposite trend was seen in the protein levels of N-cadherin and Vimentin after the Orai1 inhibition or the SKF96365 treatment ()). These findings suggest that Orai1-regulated SOCE inhibition suppresses the migration and invasion of C6 glioma cells.
Figure 3. Effect of Orai1 suppression on C6 glioma cell migration and invasion. C6 glioma cells were transfected with shScramble/shOrai1 or treated with SKF96365 (20 µM). (a) The migratory areas of three groups of C6 cells were marked and measured by utilizing the wound healing assay (magnification, ×40). (b) The invasive ability of C6 cells was determined by utilizing Matrigel-coated transwell assay (magnification, ×100). (c) Cells were analyzed for expression of E-cadherin, N-cadherin and Vimentin protein with Western blot. Data are shown as means ± SDs from thrice independent experiments. * P < 0.05 or ** P < 0.01 vs. the shScramble group
Effect of Orai1 down-regulation on C6 glioma cell adhesion and Pyk2 phosphorylation
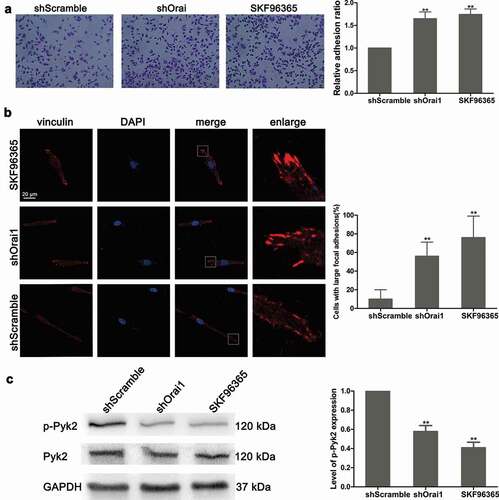
Subsequently, we also evaluated the effect of SOCE on cell adhesion ability. As shown in ), not only Orai1 inhibition but also SKF96365 treatment promoted C6 cell adhesion (P < 0.01). Additionally, in the shScramble group, C6 cells were characterized by a normal morphology of long and narrow shaped cells ()). Immunofluorescence assay showed a large amount of vinculin in the shOrai1 or SKF96365 group ()). According to the bar chart in ), transfection with shOrai1 or the SKF96365 treatment promoted large focal adhesions with 56% and 76% of the cells, respectively (P < 0.01). A recent study indicated that Pyk2 was located in the focal adhesions and modulated focal adhesion turnover [Citation21]. Next, we examined the protein expression and phosphorylation of Pyk2 by western blot assay. The phosphorylation of Pyk2 (p-Pyk2) was suppressed significantly after inhibition of Orai1 (P < 0.01, )). However, Pyk2 protein level did not change in shOrai1-transfected cells, compared to the shScramble group. Moreover, SKF96365 had a similar inhibitory effect on Pyk2 phosphorylation (P < 0.01, )). These data indicate that SOCE inhibition induced by the shOrai1 transfection may suppress cell adhesion and Pyk2 phosphorylation.
Figure 4. Effect of Orai1 suppression on C6 glioma cell adhesion and Pyk2 phosphorylation. shScramble or shOrai1 was introduced into C6 glioma cells, or cells were treated with SKF96365 (20 µM). (a) The relative cell adhesion was detected. (b) Cell morphology was visualized by immunofluorescence assay and the proportions of cells with large focal adhesions were calculated.(c) The protein levels of total Pyk2 and phosphorylated Pyk2 (p-Pyk2) were measured by western blot analysis. Data are shown as means ± SDs from thrice independent experiments. * P < 0.05 or ** P < 0.01 vs. the shScramble group
Effect of Orai1 down-regulation on signaling pathways in C6 glioma cells
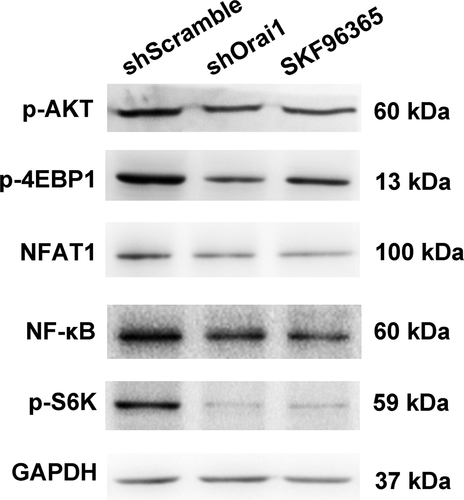
Based on the above data, we further investigated the molecular mechanism of tumor behavior inhibition by shOrai1 in C6 cells. Since the aberrant expression of molecules in AKT/mTOR, NFAT, and NF-κB pathways frequently occur in glioma, therapeutic approaches are targeted toward these signaling pathways [Citation23–25]. As shown in , transfection with shOrai1 decreased AKT phosphorylation and 4EBP1 phosphorylation in glioma C6 cells. Moreover, the protein expression levels of NFAT, NF-κB, and S6K phosphorylation were higher in shScramble-transfected cells compared to the shOrai1-transfected group (). Interestingly, both Orai1 inhibition and SKF96365 treatment blocked AKT/mTOR, NFAT, and NF-κB pathways.
Figure 5. Effect of Orai1 down-regulation on signal pathways in C6 glioma cells. C6 glioma cells were transfected with shScramble/shOrai1 or treated with SKF96365 (20 µM). Cells were analyzed for expression levels of AKT phosphorylated protein, 4EBP1 phosphorylated protein, NFAT protein, NF-κB protein and S6K phosphorylated protein with Western blot
Discussion
Recurrence and metastasis are the main reasons for the failure of a tumor treatment [Citation26]. Over the past decade, most research on SOCE emphasized on its close association with tumor metastasis [Citation27]. Our study disclosed that silencing Orai1 showed a similar performance as SKF96365 in inhibiting SOCE and malignant biological properties of C6 glioma.
As an important protein of SOCC, any abnormal expression levels and dysfunctioning of Orai1 can affect the intracellular Ca2+ homeostasis, participating in the pathophysiological process of many diseases [Citation13,Citation28]. Several recent investigations on Orai1 were carried out in the esophageal squamous cancer, revealing that the Orai1 mediated enhancement of Ca2+ influx resulted in decreased sensitivity of esophageal epithelial cells toward zinc, thereby promoting carcinogenesis in epithelial cells [Citation29,Citation30]. Additionally, it has been concluded that Orai1 expression was up-regulated in human astroglioma U373MG cells and closely related to histamine H1 receptor-mediated SOCE [Citation31]. Since SOCE is mainly mediated by STIM1 or the Orai1 proteins, down-regulating the expression of constituent proteins can also inhibit SOCE. RNAi technology was used to eliminate specific gene expression and explore gene function. In this study, the down-regulation of Orai1 caused a decline in Ca2+ influx and glioma cell proliferation. The results were consistent with the findings of other studies, where the association between SOCE inhibition and the GBM cell apoptotic potential was observed [Citation11,Citation32].
SKF96365 is an imidazole compound found in several cell lines, such as human platelets, neutrophils, and endothelial cells [Citation17]. SKF96365 plays an important role in inhibiting SOCE at a concentration of 20–30 µM [Citation33]. It was noted that some derivatives, such as calmidazolium and miconazole, could increase the basal level of Ca2+ in the cells and inhibit Ca2+ ATPase to regulate calmodulin [Citation34]. Moreover, a multitude of research papers has determined that SKF96365 can inhibit potassium channels [Citation35]. Although SKF96365 lacks specificity, it is widely used in various recent studies related to SOCE. Our results were consistent with other interesting data in the GBM literature where cell proliferation of C6 glioma was restrained by SKF96365-mediated SOCE inhibition [Citation11].
Recently, a lot of attention has been paid to tumor metastasis. An emerging report described that lung metastasis foci were significantly inhibited in the breast cancer mouse metastasis model with down-regulated Orai1 expression [Citation16,Citation36]. It is demonstrated that a high level of STIM1 is related to tumor size, depth of invasion, and lymphatic metastasis in colon cancer tissues [Citation37]. Moreover, Motiani et al. set up a series of cellular biological experiments in GBM1, GBM8, and U251 cell lines and stated that silencing STIM1 or Orai1 reduced the invasiveness of glioma, but hardly affected the cell proliferation [Citation32]. In line with the previous report, our preliminary observations demonstrated that shOrai1 transfection, as well as SKF96365 treatment, exhibited inhibitory influence on C6 glioma cell migration and invasion.
Previous studies on the regulatory mechanism of epithelial-mesenchymal transition (EMT) have focused on protein transcription factors, such as Snail, Slug, and ZEB1. However, a recent study by Hu J et al. [Citation38] in MCF-7 breast cancer cells found that SOCE was involved in EMT-related cell invasion and migration. Also, Davis et al. demonstrated that the expression of the calcium channel protein, TRPM7, can regulate EGF-induced phosphorylation of STAT3 and the expression of EMT marker Vimentin [Citation39]. EMT plays an important role in the invasion and metastasis of most epithelial tumors [Citation40]. However, its role in glioma is still controversial [Citation41]. Our results indicate that once the Orai1 gene is down-regulated, the expression levels of E-cadherin, N-cadherin, and Vimentin changes significantly, implying that inhibition of Orai1 may influence the EMT process. During cell migration, focal adhesion kinase (FAK) mediates the formation of focal adhesion plaques that degrade ECM proteins and produce energy to move the cells [Citation42]. The formation and disintegration of adhesive plaque is an important step in migration, which is regulated by FAK and also its subfamily member, calcium-activation dependent tyrosine kinase 2 (Pyk2) [Citation43]. In the rat C6 glioma model, the activation of Pyk2, after antiangiogenic treatment, enhanced the tumor’s aggressive behavior, hinting that Pyk2 played a promoting role in GBM aggressiveness [Citation44]. A study uncovered that SOCE could improve the intracellular Ca2+ concentration in cervical cancer to activate FAK and Ca2+-dependent protease, thus accelerating the turnover rate of adhesive plaques and promoting cell migration [Citation45]. In this research, the enhancement in relative adhesion ratio and large focal adhesion plaques was observed in both Orai1-silenced cells and the SKF96365-treated group. Therefore, intracellular SOCE may be involved in regulating the formation of adhesive plaques, accelerating their turnover, and promoting the migration of cells.
In conclusion, our research indicated that Orai1 silencing mediated-inhibition of SOCE might suppress C6 cell malignant behavior by enhancing cell adhesion. Further, Pyk2 expression was involved in C6 glioma cell motility. These findings warrant further investigations toward Orai1 as a therapeutic target in glioma oncology.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
The data used and/or analyzed during the current study are available with the corresponding author upon request.
Disclosure statement
The authors declare that they have no competing interests.
Additional information
Funding
References
- Chen R, Smith-Cohn M, Cohen AL, et al. Glioma subclassifications and their clinical significance. Neurotherapeutics. 2017;14:284–297.
- Heiss W-D. Contribution of PET imaging to clinical management of gliomas. OBM Neurobiol. 2018;2: 1–1.
- Wirsching HG, Galanis E, Weller M. Glioblastoma. In: Handbook of clinical neurology. Vol. 134; 2016. p. 381–397.
- Batash R, Asna N, Schaffer P, et al. Glioblastoma multiforme, diagnosis and treatment; Recent literature review. Curr Med Chem. 2017;24:3002–3009.
- Le AP, Huang Y, Pingle SC, et al. Plexin-B2 promotes invasive growth of malignant glioma. Oncotarget. 2015;6:7293–7304.
- Prayson RA. Angiocentric glioma: a review of clinicopathologic features. OBM Neurobiol. 2018;2. DOI:10.21926/obm.neurobiol.1804015
- Berrier AL, Yamada KM. Cell-matrix adhesion. J Cell Physiol. 2007;213:565–573.
- Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529.
- Putney JW, Steinckwich-Besancon N, Numaga-Tomita T, et al. The functions of store-operated calcium channels. Biochim Biophys Acta Mol Cell Res. 2017;1864:900–906.
- Xie J, Pan H, Yao J, et al. SOCE and cancer: recent progress and new perspectives. Int J Cancer. 2016;138:2067–2077.
- Liu H, Hughes JD, Rollins S, et al. Calcium entry via ORAI1 regulates glioblastoma cell proliferation and apoptosis. Exp Mol Pathol. 2011;91:753–760.
- Ambudkar IS, de Souza LB, Ong HL. TRPC1, Orai1, and STIM1 in SOCE: friends in tight spaces. Cell Calcium. 2017;63:33–39.
- Lacruz RS, Feske S. Diseases caused by mutations in ORAI1 and STIM1. Ann N Y Acad Sci. 2015;1356:45–79.
- Jha A, The MS. CAR that drives Ca2+ to Orai1. Sci Signal. 2016;9:fs5.
- Karacicek B, Erac Y, Tosun M. Functional consequences of enhanced expression of STIM1 and Orai1 in Huh-7 hepatocellular carcinoma tumor-initiating cells. BMC Cancer. 2019;19:751.
- Yang S, Zhang JJ, Huang XY. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 2009;15:124–134.
- Merritt JE, Armstrong WP, Benham CD, et al. SK&F 96365, a novel inhibitor of receptor-mediated calcium entry. Biochem J. 1990;271:515–522.
- Zhang J, Wei J, He Q, et al. SKF95365 induces apoptosis and cell-cycle arrest by disturbing oncogenic Ca(2+) signaling in nasopharyngeal carcinoma cells. Onco Targets Ther. 2015;8:3123–3133.
- Tanahashi Y, Wang B, Murakami Y, et al. Inhibitory effects of SKF96365 on the activities of K(+) channels in mouse small intestinal smooth muscle cells. J Vet Med Sci. 2016;78:203–211.
- Cai R, Ding X, Zhou K, et al. Blockade of TRPC6 channels induced G2/M phase arrest and suppressed growth in human gastric cancer cells. Int J Cancer. 2009;125:2281–2287.
- Zhu M, Chen L, Zhao P, et al. Store-operated Ca(2+) entry regulates glioma cell migration and invasion via modulation of Pyk2 phosphorylation. J Exp Clin Cancer Res. 2014;33:98.
- Singh A, Hildebrand ME, Garcia E, et al. The transient receptor potential channel antagonist SKF96365 is a potent blocker of low-voltage-activated T-type calcium channels. Br J Pharmacol. 2010;160:1464–1475.
- Jiang Y, Zhou J, Luo P, et al. Prosaposin promotes the proliferation and tumorigenesis of glioma through toll-like receptor 4 (TLR4)-mediated NF-κB signaling pathway. EBioMedicine. 2018;37:78–90.
- Lee JV, Berry CT, Kim K, et al. Acetyl-CoA promotes glioblastoma cell adhesion and migration through Ca(2+)-NFAT signaling. Genes Dev. 2018;32:497–511.
- Liu X, Zhao P, Wang X, et al. Celastrol mediates autophagy and apoptosis via the ROS/JNK and Akt/mTOR signaling pathways in glioma cells. J Exp Clin Cancer Res. 2019;38:184.
- Chistiakov DA, Chekhonin VP. Circulating tumor cells and their advances to promote cancer metastasis and relapse, with focus on glioblastoma multiforme. Exp Mol Pathol. 2018;105:166–174.
- Mo P, Yang S. The store-operated calcium channels in cancer metastasis: from cell migration, invasion to metastatic colonization. Front Biosci (Landmark Ed). 2018;23:1241–1256.
- Zhou Y, Cai X, Nwokonko RM, et al. The STIM-Orai coupling interface and gating of the Orai1 channel. Cell Calcium. 2017;63:8–13.
- Cui C, Chang Y, Zhang X, et al. Targeting Orai1-mediated store-operated calcium entry by RP4010 for anti-tumor activity in esophagus squamous cell carcinoma. Cancer Lett. 2018;432:169–179.
- Choi S, Cui C, Luo Y, et al. Selective inhibitory effects of zinc on cell proliferation in esophageal squamous cell carcinoma through Orai1. Faseb J. 2018;32:404–416.
- Barajas M, Andrade A, Hernandez-Hernandez O, et al. Histamine-induced Ca2+ entry in human astrocytoma U373 MG cells: evidence for involvement of store-operated channels. J Neurosci Res. 2008;86:3456–3468.
- Motiani RK, Hyzinski-Garcia MC, Zhang X, et al. STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflugers Arch. 2013;465:1249–1260.
- Franzius D, Hoth M, Penner R. Non-specific effects of calcium entry antagonists in mast cells. Pflugers Arch. 1994;428:433–438.
- Derler I, Fritsch R, Schindl R, et al. CRAC inhibitors: identification and potential. Expert Opin Drug Discov. 2008;3:787–800.
- Schwarz G, Droogmans G, Nilius B. Multiple effects of SK&F 96365 on ionic currents and intracellular calcium in human endothelial cells. Cell Calcium. 1994;15:45–54.
- Liu X, Wang T, Wang Y, et al. Orai1 is critical for notch-driven aggressiveness under hypoxic conditions in triple-negative breast cancers. Biochimica Et Biophysica Acta Mol Basis Dis. 2018;1864:975–986.
- Wang JY, Sun J, Huang MY, et al. STIM1 overexpression promotes colorectal cancer progression, cell motility and COX-2 expression. Oncogene. 2015;34:4358–4367.
- Hu J, Qin K, Zhang Y, et al. Downregulation of transcription factor Oct4 induces an epithelial-to-mesenchymal transition via enhancement of Ca2+ influx in breast cancer cells. Biochem Biophys Res Commun. 2011;411:786–791.
- Davis FM, Azimi I, Faville RA, et al. Induction of epithelial-mesenchymal transition (EMT) in breast cancer cells is calcium signal dependent. Oncogene. 2014;33:2307–2316.
- Mittal V. Epithelial mesenchymal transition in tumor metastasis. Annu Rev Pathol. 2018;13:395–412.
- Oh SJ, Ahn EJ, Kim O, et al. The Role played by SLUG, an epithelial-mesenchymal transition factor, in invasion and therapeutic resistance of malignant glioma. Cell Mol Neurobiol. 2019;39:769–782.
- Wolf K, Mazo I, Leung H, et al. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J Cell Biol. 2003;160:267–277.
- Torigoe G, Nagao M, Tanabe N, et al. PYK2 mediates BzATP-induced extracellular matrix proteins synthesis. Biochem Biophys Res Commun. 2017;494:663–667.
- Xu CS, Wang ZF, Dai LM, et al. Induction of proline-rich tyrosine kinase 2 activation-mediated C6 glioma cell invasion after anti-vascular endothelial growth factor therapy. J Transl Med. 2014;12:148.
- Chen YF, Chiu WT, Chen YT, et al. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc Natl Acad Sci U S A. 2011;108:15225–15230.